

Abstract
Plasmodium falciparum and Plasmodium vivax, the two protozoan parasite species that cause the majority of cases of human malaria, have developed resistance to nearly all known antimalarials. The ability of malaria parasites to develop resistance is primarily due to the high numbers of parasites in the infected person’s bloodstream during the asexual blood stage of infection in conjunction with the mutability of their genomes. Identifying the genetic mutations that mediate antimalarial resistance has deepened our understanding of how the parasites evade our treatments and reveals molecular markers that can be used to track the emergence of resistance in clinical samples. In this review, we examine known genetic mutations that lead to resistance to the major classes of antimalarial medications: the 4-aminoquinolines (chloroquine, amodiaquine and piperaquine), antifolate drugs, aryl amino-alcohols (quinine, lumefantrine and mefloquine), artemisinin compounds, antibiotics (clindamycin and doxycycline) and a napthoquinone (atovaquone). We discuss how the evolution of antimalarial resistance informs strategies to design the next generation of antimalarial therapies.
Keywords: malaria, drug resistance, Plasmodium falciparum, Plasmodium vivax, artemisinin
Article
Malaria, a protozoan infection caused by Plasmodium parasites, remains a major cause of morbidity and mortality worldwide primarily among children less than 5 years old. It caused an estimated 219 million cases and 435 000 deaths in 2017, with 92% of cases and 93% of deaths in Africa (2017 #885; 2018 #1580). Plasmodium falciparum and Plasmodium vivax, the two species that cause the majority of cases of human malaria, have demonstrated resistance to nearly all known antimalarials, with the highest levels of resistance found in P. falciparum in Southeast Asia. When parasite resistance to chloroquine (CQ) and antifolate medications, former first line medications, emerged, there were enormous increases in morbidity and mortality [1]. More recently, delayed parasite clearance times following artemisinin combination therapy (ACT), the current first line treatment for uncomplicated P. falciparum infections, have been reported in the Greater Mekong sub-region and represent a major threat to the ability to control and treat malaria [2, 3].
The ability of malaria parasites to develop resistance is primarily due to the high burden of parasites in an infected person’s bloodstream during the asexual blood stage of infection in conjunction with the mutability of the parasites’ genomes [4]. Identifying the genetic mutations that mediate antimalarial resistance is key to understanding how the parasites evade our treatments. Tracking these molecular markers in clinical samples can help evaluate the emergence of resistance in a particular region and inform recommendations for first line therapy. This is especially useful since empirical testing for drug resistance, either in patients or after taking parasites into short-term culture, can be very expensive and requires resources that are not available in many malaria-endemic regions. Our understanding of the mechanisms of antimalarial resistance is primarily focused on P. falciparum, for which there is a robust in vitro culture system. Major mechanisms of resistance include point mutations in or amplification of genes encoding transporters that mediate transport of a drug to or from the parasite’s digestive vacuole (DV) and point mutations in the target of the antimalarial that disrupt binding. Whole genome scans of P. falciparum and P. vivax using technologies such as microarrays and whole genome sequencing (WGS) have revealed insights into mechanisms of resistance in both in vitro and clinical studies. Genome-wide association studies (GWASs) have helped identify genes associated with resistance. In this review, we examine the genetic mechanisms that underlie resistance to the major classes of antimalarial medications and discuss how this knowledge has contributed to our understanding of developing more effective, ‘irresistible’ malaria treatments.
The emergence and spread of antimalarial resistance
Resistance is defined as the ability of a parasite to survive or multiply despite properly administered and dosed medication [5]. Currently, antimalarials are administered as combination therapy with two drugs to prevent the rapid emergence of resistance. As levels of resistance increase, there is an increased number of patients presenting with late recrudescence, or persistent parasitemia [4]. In addition, patients present with recrudescence earlier following treatment. High-grade resistance is evident when there is failure to clear parasitemia or there is an increase in parasitemia despite appropriate therapy. An important marker of resistance is delayed parasite clearance times. A major challenge with assessing antimalarial efficacy in the era of combination therapy is that failure may not be observed even when the parasites are resistant to one of the partner drugs.
The first step in the development of resistance is the initial genetic event, which is thought to be spontaneous and rare [4]. Since an average human infection can comprise 109–1013 parasites in the blood stream during the asexual blood stage (Figure 1) with an estimated 1.0–9.7 × 10−9 mutations per base pair per generation [6], there is a high likelihood that a random mutation can occur that leads to antimalarial resistance within a few cycles of replication. Subsequent selection for that mutation occurs due to a survival advantage in the presence of drug pressure. Factors that favor selection of resistant parasites are higher levels of parasitemia, decreased blood levels of antimalarials and decreased patient immunity [4, 7]. Drugs with a longer drug half-life such as mefloquine (MFQ), piperaquine (PPQ) and CQ may be more likely to select for resistance [8]. The level of malaria transmission also can affect the development of resistance since persons in low transmission settings are more likely to be symptomatic and receive treatment compared to those in high transmission settings [4]. Individuals in lower transmission areas also have lower acquired immunity, which can result in increased transmission of resistant parasites. In high transmission settings, there are more likely to be multiple genotypes present in a single infection and thus resistant parasites have to compete with wild-type parasites. In areas with seasonal malaria transmission, however, persons with asymptomatic parasitemia can serve as a reservoir for sensitive parasites [9]. The transmissibility of the allele is also an important consideration and may determine whether resistance can spread from patient to patient. For example, some alleles that confer resistance to atovaquone cause parasites to die in the mosquitos so that they should, in principle, not spread from one person to the next [10].
Figure 1.
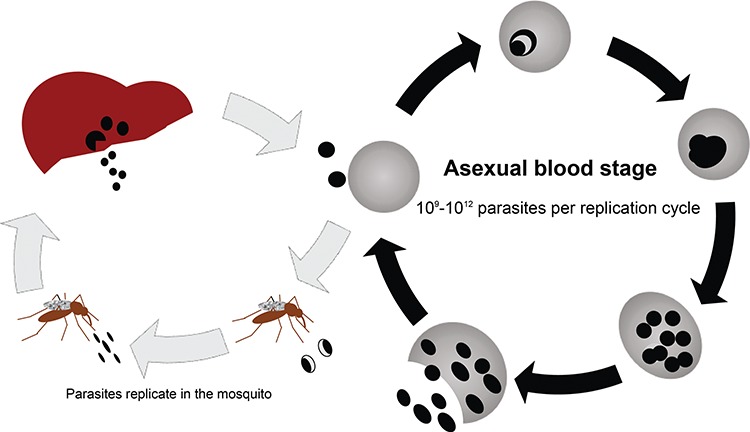
The P. falciparum life cycle highlighting the asexual blood stage of infection where antimalarial resistance mutations arise. Infection begins with inoculation of sporozoites by an infected mosquito. Sporozoites infect liver cells, and merozoites are released into the bloodstream, which invade red blood cells (RBCs). During the asexual blood stage of infection, which is responsible for the clinical manifestations of disease, the parasites undergo maturation and replication with an average of 109–1012 parasites per replication cycle. The infected RBCs rupture, releasing new merozoites into the bloodstream to begin another cycle of replication. A subset of parasites becomes gametocytes which can be ingested by another mosquito to continue malaria transmission.
Known genetic mediators of resistance
4-Aminoquinolines
The 4-aminoquinolines include CQ, amodiaquine (AQ) and PPQ (Table 1). CQ was previously the first-line treatment for uncomplicated P. falciparum infections, while AQ and PPQ are currently used as partner drugs for artemisinin derivatives. Hemoglobin catabolism in the DV of the parasite is important as a source of amino acids (Figure 2). The breakdown of hemoglobin releases Fe2+ iron-containing reactive heme moieties that undergo oxidation in the DV into ferriprotoporphyrin IX (FPIX) [11]. This process causes oxidative stress, and thus FPIX undergoes detoxification by becoming incorporated into hemozoin [12]. Medications from this class bind to the reactive heme and interfere with its detoxification. CQ is a weak base at a neutral pH that can diffuse across membranes into the erythrocyte and DV in its uncharged form. Once it is in the acidic DV, becomes protonated and accumulates in the DV [13, 14].
Table 1.
Commonly used antimalarials and their known genetic mediators of resistance in P. falciparum and P. vivax. SNVs known to be essential to resistance are highlighted with an asterisk
| Antimalarial drug class | Mechanism of action | Specific drugs | Genetic mediator(s) of resistance | |
|---|---|---|---|---|
| P. falciparum | P. vivax | |||
| 4-aminoquinolines | Interfere with heme detoxification | chloroquine (CQ) | SNVs in pfcrt (K76 T*); SNVs in pfmdr1 (N86Y*) | Not well understood; pvcrt-o amplification |
| amodiaquine(AQ) | ||||
| piperaquine (PPQ) | SNVs in pfcrt (C101F, H97Y, F145I, M343 L, G353 V); Plasmepsin 2 and 3 amplifications; pfmdr1 single copy | |||
| 4-aminoquinolines | Unknown | Primaquine | Unknown | Unknown |
| Tafenaquine | ||||
| Antifolate drugs | Inhibition of folate synthesis | DHFR inhibitors (proguanil, pyrimethamine) | SNVs in pfdhfr (S108 N, N51I, C59R, I164L); amplification of gtp cyclohydrolase 1 | SNVs in pvdhfr |
| Sulfa drugs (sulfamethoxazole, sulfadoxine) | SNVs in SNVs in pfdhps | Inherently resistant due to SNV in pvdhps (V585) | ||
| Aryl amino-alcohols | Unclear; thought to interfere with heme detoxification | lumefantrine (LMF) | Amplification of pfmdr1 | Amplification of pvmdr1 |
| mefloquine (MFQ) | ||||
| Quinine | Not clear, involves mediators of LMF and MQ resistance; ms4760 microsatellites in pfnhe-1 | Not reported | ||
| Antibiotics | Inhibition of protein synthesis | Doxycycline | Unknown | Not reported |
| Clindamycin | SNV in 23S rRNA (A1875C) | |||
| Napthoquinones | Inhibits cytochrome bc1 complex | Atovaquone | SNV in cyt-b (Y268S/C/N) | Not reported |
| Artemisinin compounds | Causes oxidative stress | Artemisinin, artemether, DHA | SNVs in kelch13 (C580Y) | Not reported |
Figure 2.
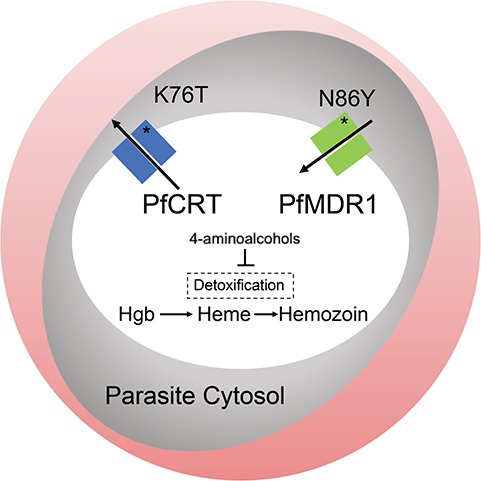
The parasite DV and the role of the P. falciparum CQ resistance transporter (PfCRT) and the P. falciparum multidrug resistance protein 1 (PfMDR1). The parasite (gray oval) is shown within an RBC. The DV (white oval) is a compartment within the parasite where the catabolism of hemoglobin (Hgb) from the host RBC occurs. The breakdown of Hgb results in reactive heme which undergoes detoxification to hemozoin. Medications from the 4-aminoquinoline class bind heme and interfere with detoxification. PfCRT and PfMDR1 are DV membrane proteins. It is thought that PfCRT transports drugs out of the DV while PfMDR1 transports them into the DV [19, 36]. The T mutation in pfcrt is essential to CQ resistance, while the N86Y mutations in pfmdr1 augment CQ resistance. Mutations in these transporters have also been found to mediate resistance to the aryl-amino alcohols and artemisinin.
CQ was the most widely used antimalarial in this class prior to the development of widespread resistance in P. falciparum and P. vivax in certain areas. It was introduced in the 1950s and 1960s and was used as the basis of the World Health Organization (WHO) Global Malaria Eradication Program. P. falciparum resistance subsequently appeared in Southeast Asia in the late 1950s, then emerged in other countries in Asia, South America and finally Africa over the course of 30 years [15, 16]. Resistance is primarily determined by mutations in pfcrt, a gene that encodes the CQ resistance transporter (PfCRT), a 424 amino acid protein that localizes to the DV membrane [17–19]. This is a highly polymorphic protein with over 20 different point mutations described [20, 21]. However, the K76T mutation was found to be essential for in vitro CQ resistance [17, 19, 22] (Table 1). Reversal of the K76T mutation resulted in wild-type CQ susceptibility and led to increased binding of CQ to FPIX [23]. Parasites harboring the K76T mutation demonstrate an increased leak of H+ from the DV in the presence of CQ compared to sensitive parasites [24]. The loss of the positively charged lysine 76 leads to increased efflux of protonated CQ from the DV [24, 25]. One study which expressed wild-type and mutant PfCRT on Xenopus laevis oocytes elegantly demonstrated that CQ resistance is due to direct transport of a protonated form of CQ through the K76T pfcrt mutant [19]. In CQR-resistant parasites that have arisen independently around the world, there are at least 4 and up to 10 additional mutations in pfcrt that are seen [26]. A follow-up study using the Xenopus system to express over 100 variants of PfCRT found that although there were many mutational routes that could confer CQ transport, the overall process was rigid, requiring that mutations were in a specific order [27]. Clinical studies have demonstrated that there was a selective sweep at the pfcrt locus worldwide due to CQ pressure [28–33]. The K76T mutation was also found to be associated with clinical failures [34].
The pfmdr1 gene encodes the p-glycoprotein transmembrane pump multidrug resistance protein 1, which localizes to the DV membrane [35]. This pump functions as a transporter of antimalarial drugs, with studies demonstrating that it imports solutes into the DV [36, 37]. An N86Y mutation has been associated with decreased CQ susceptibility in vitro [38, 39]; however mutations in pfmdr1 alone are not adequate to cause CQ resistance [28, 40]. Introduction of the pfmdr1 N86Y mutation into parasites with a CQ-resistant genetic background leads to increased resistance to CQ and monodesethylamodiaquine (md-ADQ; the primary metabolite of AQ) [41].
Interestingly, after CQ was no longer used as therapy for P. falciparum due to widespread resistance, the K76T mutation reverted to the wild-type pfcrt allele in parts of Africa [42–45], suggesting that the mutation confers a loss of fitness. However, in Southeast Asia and South America this has not been the case, with the mutation persisting [46–48]. One likely reason for this is the continued use of CQ for treatment of P. vivax in these regions. Another potential reason is that resistance-conferring mutations are fixed in certain populations and thus there are no sensitive parasites to emerge following withdrawal of drug pressure. However, widespread CQ pressure has led to many variants of PfCRT throughout the world. In one study, researchers genetically engineered several sets of pfcrt mutations found at different sites around the world into P. falciparum parasites. They found that each PfCRT variant conferred varying degrees of CQ resistance and affected growth in vitro. One highly mutated pfcrt variant of Cambodian origin actually demonstrated enhanced growth compared to wild-type parasites [49]. Interestingly, a GWAS with CQ sensitive and resistant isolates in French Guiana found that a C350R PfCRT variant was associated with the restoration of CQ susceptibility [50]. This C350R variant was also associated with PPQ resistance in vitro, which likely explains the failure of PPQ in the region.
In contrast to P. falciparum, CQ resistance in P. vivax was not reported until 1989 in Papua New Guinea [51] and is now found throughout Southeast Asia and some countries in South America [52]. CQ resistance is more challenging to detect with this species since parasitemia is generally lower relative to P. falciparum. Additionally, it is difficult to distinguish recrudescence (parasites returning after incomplete or ineffective treatment) from relapses due to reactivation of dormant liver parasites in endemic settings. There is also no robust in vitro culture system as there is with P. falciparum, so confirmation with in vitro susceptibility testing is even more challenging than with P. falciparum. There are no clear molecular markers of CQ resistance in P. vivax. Although pvcrt-o is orthologous to pfcrt, there is no clear direct association between CQ resistance and mutations in pvcrt-o [53–55]. There is also no clear association between pvmdr1, the homologue of pfmdr1 and CQ resistance. Although some studies have detected point mutations in pvmdr1 in resistant parasite populations, such as a Y976F substitution in Indonesia and an F1076L mutation in Southeast Asia [54], the polymorphisms are not consistent across different parasite populations. In addition, there are CQ-resistant parasites that have the wild-type pvmdr1 gene [56]. One recent study of patients with recurrent P. vivax infections in the Brazilian Amazon found that CQ resistance was associated with increased copies of pvcrt-o [57].
There is currently evidence of PPQ resistance in Western Cambodia, where dihydroartemisinin–piperaquine has been the frontline treatment for uncomplicated P. falciparum malaria [58]. A GWAS study of 297 P. falciparum clinical isolates from Cambodia found that a nonsynonymous SNP on chromosome 13, a single copy of pfmdr1 and amplifications of plasmepsin 2 and 3 were associated with increased in vitro PPQ resistance and decreased clinical efficacy [59]. Another study of culture-adapted parasites from clinical isolates from Cambodia found that ex vivo PPQ survival assay profiles correlated with plasmepsin 2 copy number [60]. In addition, multicopy plasmepsin 2 was significantly associated with DHA-PPQ treatment failure. The plasmepsin genes encode aspartic proteases that function as hemoglobinases in the DV. The mechanism of resistance is not clearly identified; however one hypothesis is that increased hemoglobin digestion due to the amplification decreases concentrations of the reactive heme species that PPQ binds, thereby overcoming the inhibition of heme detoxification by PPQ [60].
There is also growing evidence that mutations in pfcrt can mediate resistance to PPQ independent from amplifications of plasmepsin genes. PPQ-resistant strains evolved in vitro were analyzed with microarrays and were found to have a C101F mutation in pfcrt in addition to an amplification of pfmdr1 [61]. Subsequently, the introduction of the C101F pfcrt mutation with zinc finger nuclease-based gene editing into CQ-resistant parasites resulted in significantly higher PPQ resistance and also reversed CQ resistance [62]. Three independent pfcrt mutations were associated with ex vivo PPQ resistance in culture-adapted parasites from Cambodia [63]. A GWAS study of samples primarily from Cambodia identified a point mutation in pfcrt (F145I), which was associated with DHA-PPQ treatment failure even after adjustment for amplification in plasmepsin 2 and 3 [64]. A subsequent analysis of pfcrt allelic diversity from clinical isolates from Southeast Asia found a rapid rise in novel mutations following the introduction of DHA-PPQ treatment [65]. Introduction of the H97Y, F145I, M343L and G353V mutations into PPQ sensitive parasites resulted in PPQ resistance.
8-Aminoquinolines
The 8-aminoquinolines have a similar structure to the 4-aminoquinolines, with the exception of the amino group at the 8-position of the quinoline. Their mechanism of action is not well understood. Primaquine and tafenoquine are two agents that are used for malaria treatment and prophylaxis. Primaquine is given along with CQ to treat the liver-stage parasites in P. vivax and P. ovale infections to prevent relapses [66]. It also has potent activity against stage V gametocytes of P. falciparum and is used to reduce malaria transmission [67]. Tafenoquine was recently FDA-approved for the prevention of P. vivax relapses administered as a single dose. Interestingly, primaquine appears to increase the activity of CQ against CQ-resistant P. falciparum [68]. Primaquine resistance in P. vivax is difficult to determine as it is confounded by reinfections in malaria-endemic regions [69]. A study that performed WGS of P. vivax from known relapses that occurred despite primaquine treatment found polymorphisms in several putative resistance genes [70]. However, there are currently no known genetic markers of primaquine resistance.
Antifolate drugs
Antifolate drugs disrupt parasite folate synthesis (Figure 3) and include dihydrofolate reductase (DHFR) inhibitors (proguanil, pyrimethamine, trimethoprim) and sulfa drugs (sulfamethoxazole, sulfadoxine; Table 1). Sulfadoxine–pyrimethamine (Fansidar; SP) was deployed in the 1960s in areas where P. falciparum CQ resistance had developed, with the emergence of resistant parasites in the 1970s and 1980s [71]. Antifolates are now used most commonly as combination therapy such as atovaquone-proguanil, which is used for prophylaxis, and SP which is used in combination with artemisinin for treatment of P. falciparum or as part of intermittent preventive treatment in pregnant women and children.
Figure 3.
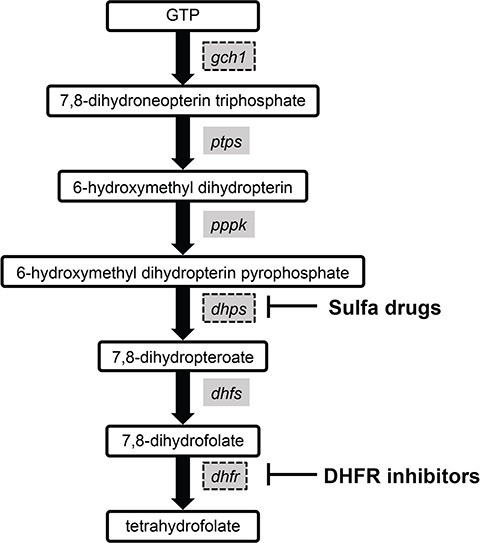
The P. falciparum folate biosynthesis pathway. Enzymes inhibited by the antifolate drugs are shown. Point mutations in the dhps and dhfr mediate resistance to sulfa drugs and DHFR inhibitors, respectively. Increased copy number of the gch1 gene has been detected in clinical isolates from Southeast Asia and likely represents an adaptive evolutionary response to antifolate pressure [95]. Other abbreviations: pyruvoyltetrahydropterin synthase (ptps), hydroxymethyldihydropterin pyrophospholkinase (pppk) and dihydrofolate synthase (dhfs). Adapted from [94].
In contrast with CQ resistance, which took many years to develop, resistance to antifolates developed much faster. The genetic mechanism of resistance to antifolates is more straightforward in comparison to CQ resistance, with single point mutations in the genes encoding either DHFR or dihydropteroate synthase (DHPS) in response to sulfa drugs (Table 1; Figure 3). These mutations cause resistance by altering binding of the drug to the enzyme’s active site [72, 73]. Three studies of microsatellites flanking the pfdhfr gene in P. falciparum clinical isolates from Thailand, South America and Southeast Africa found that in each of the locations there was a common origin of emergence [74–77]. The pfdhfr mutations appear in a particular order in the setting of drug pressure: S108 N, followed by N51I, C59R and finally I164L, with increasing resistance seen when going from two to three mutations in both in vitro and field isolates [78–82]. S108N appears to be a necessary first mutation in DHFR [78]. A C50R mutation was identified in samples from Latin America [72], with genetic transformation studies demonstrating that it likely has an analogous role to the C59R mutation in African isolates [83].
Mutations in dhfr decrease the overall enzyme efficacy and result in a fitness cost for the parasite [84, 85]. After changes in first line malaria treatment from sulfa drugs to ACTs, a decline in triple and quadruple dhfr mutants has been seen in certain areas [86, 87]. However, in countries where SP is part of the ACT or is used as intermittent preventive therapy, these mutants remain prevalent [88–91]. In addition, the persistence of parasites carrying dhfr mutations may be attributed to the use of trimethroprim-sulfamethoxazole for prophylaxis or treatment for opportunistic infections in HIV-positive persons [92].
Interestingly, P. falciparum parasites in Southeast Asia are able to develop a compensatory mutation for the fitness cost incurred by the mutant dhfr. A genome scanning study of 14 field and laboratory-adapted P. falciparum strains first identified an amplification surrounding GTP-cyclohydrolase 1 (gch1), which encodes an enzyme in the folate biosynthesis pathway that is upstream from DHFR and DHPS [93, 94] (Figure 3). A later population genetic study with a focus on the role of copy number variations (CNVs) in P. falciparum compared parasites from Thailand, where antifolate medications were commonly used, to those from Laos, where antifolates were rarely used [95]. They found extensive CNV surrounding gch1 in the Thai isolates with the amplicon structure demonstrating multiple sites of origin in addition to a strong association between copy number and the dhfr I164L mutation, supporting how the amplification is likely an adaptive evolutionary response to antifolate therapy. The amplification reduces the cost of acquiring the drug-resistance mutations further downstream in the folate synthesis pathway [96]. A WGS study of parasites in Malawi, which experienced prolonged use of SP, found a gene duplication in a gch1 promotor, which was also detected in parasites from West Africa and the Democratic Republic of Congo [97]. This duplication was not found as frequently in other African populations, where antifolate medications were not as extensively used and is different from the whole gene amplification found in Southeast Asia.
In P. vivax, the enzymes in the folate synthesis pathway are the same, and thus similar mutations in the P. vivax dhfr and dhps have been suggested to mediate resistance to the antifolate medications [98–100]. However, sulfa drugs were found to be inherently less effective against P. vivax parasites [101, 102]. A study that cloned and sequenced the pvdhps gene in multiple P. vivax and P. falciparum isolates from around the world and modeled and compared the 3-D structure of the P. vivax DHPS to that of P. falciparum. The predicted sulfadoxine-binding sites differed by one residue between the species: a valine at position 585 in P. vivax (probable wild type, seen in all 14 isolates examined), which is equivalent to an alanine at position 613 in P. falciparum. The larger valine residue of P. vivax at this site was predicted to reduce binding of sulfadoxine compared to the smaller alanine residue in P. falciparum, thus demonstrating a possible mechanism for increased inherent resistance [103]. Epidemiological studies have identified several point mutations in dhfr and dhps associated with resistance in Southeast Asia [104, 105]. A study in which recombinant, variant PvDHPS proteins were expressed showed that the mutated enzymes had reduced sulfadoxine sensitivity which correlated with higher resistance [106].
Aryl amino-alcohols
The aryl amino-alcohols include lumefantrine (LMF) and MFQ, which are aryl amino alcohol derivatives of quinine (Table 1). Their mechanism of action is not well understood; however, they likely interfere with the detoxification of the toxic byproducts of heme degradation [107]. Quinine is a natural compound found in bark from the Cinchona tree that has been used for the treatment of malaria for centuries [108]. It is currently used for treatment of severe malaria and for uncomplicated malaria in the first trimester of pregnancy. LMF and MFQ were introduced shortly after the antifolate medications in the mid-1970s. However, resistance to MFQ emerged rapidly and was first reported in 1982 [109]. LMF resistance has been reported in a clinical study [110]; however this has not been confirmed [111]. LMF and MFQ are now used as partner drugs for artemisinin derivatives, and MFQ is used alone as a prophylaxis.
Resistance to MFQ was found to be primarily mediated by increased pfmdr1 copy number [112, 113], rather than via point mutations as described for CQ and antifolate medications. Amplification of the pfmdr1 gene has also been associated with increased risk for treatment failure with artemether-LMF [114]. The pfmdr1 CNV is a large tandem amplification of up to 100 kb which includes several genes [115, 116]. Of note, in P. falciparum amplicon break points in CNVs are primarily found in monomeric tracts of A or T in intergenic regions [117]. Since the P. falciparum genome is highly AT-rich and has common AT monomeric tracts [118], CNVs are an important mechanism of evolutionary adaptation [117, 119, 120]. It has been found in clinical isolates throughout the world, with evidence of nonidentical chromosomal breakpoint sequences from different regions, providing evidence of independent origins [116]. This does not apply to Africa, where this amplification is rare [121]. A study of 618 samples of patients from the Thai border found that an increased copy number of pfmdr1 was the major determinant of both in vitro and in vivo MFQ resistance [122]. The number of copies of the gene has been shown to increase the degree of resistance [123]. A study of microsatellite markers flanking pfmdr1 and mapping of breakpoint sequences and amplicon size in clinical isolates from the Thai border found an estimated 5–15 independent origins of the amplification [117]. In contrast to the point mutations that caused CQ and antifolate resistance, which had a common origin within a population, the findings demonstrate how pfmdr1 amplification occurs much more frequently and thus multiple independent origins can be found within a single population. The mechanism of MFQ resistance appears to be similar in P. vivax, with studies of clinical isolates demonstrating a correlation with in vivo and in vitro MFQ resistance and increased pvmdr1 copy number [124, 125].
The mechanism of resistance to quinine appears to be more complex. Although there are reports of decreased sensitivity in Asia [126–128] and South America [129], high-grade resistance in the treatment of severe malaria appears to be rare [130]. In vitro cross resistance between quinine, the other aryl amino-alcohols and the 4-aminoquinolines is observed [123, 131–133], suggesting that there may be a common genetic mechanism of resistance. Mutations in pfmdr1 and pfcrt have been found to confer decreased parasite susceptibility to quinine [18, 22, 113, 134–136]. However, they are not sufficient to cause resistance, implying that there are additional genes involved. Researchers used quantitative trait loci analysis to detect genes associated with quinine resistance in 71 P. falciparum isolates from diverse locations and identified pfmdr1, pfcrt and pfnhe-1, which encodes P. falciparum Na+/H+ exchanger 1 and is on chromosome 13 [137]. One of the microsatellite markers detected in pfnhe-1 (ms4760) was significantly associated with in vitro response to quinine. More than two DNNND repeat motifs in block 2 of ms4760 were associated with decreased quinine response. Subsequent studies showed that an increased number of DNNND repeats were associated with in vitro quinine resistance [138–140]. A comprehensive analysis of pfnhe-1 ms4760 alleles from P. falciparum isolates from diverse geographic locations found significant polymorphisms in these alleles, with a higher number of DNNND repeats found in Southeast Asian parasites [141].
Artemisinin compounds
After CQ and antifolates were lost to resistance, artemisinin compounds became vital for effective malaria treatment. Artemisinin compounds are sesquiterpene lactone compounds that were discovered in China as the active ingredient in traditional medicine (extracts of the sweet wormwood plant, Artemisia annua) with fever-reducing properties that had been known for millennia. Related derivatives include artesunate, artemether and dihydroartemisinin (DHA) as well as the synthetic artemisinin compounds, such as OZ439. They are highly effective at rapidly clearing parasites from a person’s bloodstream. Since some have a short half-life, they typically have been combined with long-lasting drugs. These medications are currently first-line therapy as a component of ACTs. Intravenous artemisinin is used to treat severe malaria. Although their mechanism is not completely defined, within parasites these compounds undergo activation via disruption of their endoperoxide bridge, leading to oxidative stress [142] (Table 1). The precise target of artemisinin compounds is not completely defined, although current studies suggest that they cause significant stress which overpowers the parasite’s protein repair system and inactivates important housekeeping functions [142]. The phosphatidylinositol-3-kinase (PfPI3K) has been proposed as a potential target of the artemisinin compounds [143]; however the overall mechanism appears to be more complex, involving the general stress response. Treatment of P. falciparum with artemisinin compounds results in slowed parasite growth, decreased uptake of hemoglobin and increased oxidative damage [144]. Increased protein ubiquitination is seen in parasite following treatment with artemisinin compounds, which is likely due to substantive cellular damage [145]. One study examining the proteins covalently modified by an alkyne-tagged biotinylated artemisinin analogue identified 124 binding targets and demonstrated that heme is primarily responsible for its activation [146]. The 124 targets identified are involved in a wide variety of cellular processes and may indicate the breakdown of the general stress response rather than a specific target.
Decreased sensitivity to artemisinin compounds, as demonstrated by delayed parasite clearance (observed during clinical trials), was first reported in Cambodia in 2008 and has since emerged in other countries in the Greater Mekong region [147, 148]. One study of 91 parasites from Cambodia, Thailand and Laos used 6969 polymorphic SNPs to identify genomic regions under selection. Within these regions, analysis of SNPs and microsatellites identified a selective sweep on chromosome 13 that was associated with delayed parasite clearance following treatment with artemisinin compounds [149]. A subsequent study identified four SNPs on chromosomes 10, 13 and 14 that were associated with delayed parasite clearance time [150]. The two SNPs detected on chromosome 13 were under strong selection in the parasite population. A major breakthrough in identifying a molecular marker of artemisinin resistance was obtained in a WGS study of clinical P. falciparum isolates from Cambodia and a parasite line originally from Africa and selected for artemisinin resistance in vitro. This led to the identification of mutations in the propeller domain of the kelch 13 gene as a mediator of artemisinin resistance [151]. The association between kelch 13 mutations and delayed parasite clearance was subsequently confirmed with a large clinical trial [152] as well as gene editing [153, 154].
The Kelch 13 protein is thought to be involved in the cellular response to oxidative stress [142]. It is not entirely clear what specific functional changes the mutations in Kelch 13 impart; however artemisinin-resistant parasites have an enhanced stress response during the early ring stage where artemisinin is especially active [145]. Studies implicate that protein degradation or ubiquitination pathways are likely involved in this enhanced response. Transcriptional profiling of resistant parasites from patients found that proteins involved in the unfolded protein response were associated with delayed parasite clearance time [155]. The Kelch 13 C580Y variant was found to decrease interactions between the P. falciparum PfPI3K and artemisinin, leading to a decrease in polyubiquitination by PfPI3K and subsequent decrease in PI3P, which participates in phospholipid signaling [143]. In addition, proteasome inhibitors, which inhibit a complex that degrades unfolded proteins, were found to increase activity of artemisinin against sensitive and resistant P. falciparum strains [145, 156].
Epidemiologic studies demonstrate that Kelch 13 mutations have arisen independently in multiple locations in Southeast Asia, with initial soft sweeps leading to a hard sweep at this locus in parasites in Southeast Asia [157–160]. Although at least 20 mutations in K13 were identified, most parasites in the region were found to harbor a C580Y variation [151, 157]. Introduction of several kelch 13 mutations found in field strains into isogenic parasite lines in vitro demonstrated different degrees of resistance, with R539T and I543T variants resulting in higher levels of resistance compared to the C580Y variant [154]. There are thus likely other factors that contribute to the widespread prevalence of a particular mutation. A GWAS study of Southeast Asian parasites showed that mutations in pffd (ferredoxin), pfarps10 (apicoplast ribosomal protein S10), pfmdr2 (multidrug resistance protein 2) and pfcrt were strongly associated with artemisinin resistance [159]. These mutations are thought to represent a background upon which the kelch 13 mutations are especially likely to occur.
It is unclear what the significance of kelch 13 mutations is in Africa where polymorphisms have been detected [161–163], but there is no clear association with artemisinin resistance. A comparison of kelch 13 mutations between Southeast Asian and African parasites found that there was a low frequency of resistance-conferring mutations in the African parasites [164]. In addition, Asian parasites harbored an excessive number of non-synonymous mutations, while African parasites demonstrated a normal variation pattern. This suggests that these resistance-conferring mutations are not currently undergoing selection in Africa. There was one report of a returned traveler from Guinea with delayed parasite clearance with WGS showing that the strain was indigenous to Guinea and harbored a previously unreported M579I variation in Kelch 13 [165].
Antibiotics
Antibiotics that have been used for treatment or prevention of malaria include clindamycin [166, 167] and doxycycline [168], whose mechanism of action is the interruption of protein synthesis in the parasite (Table 1). Mutations in apicoplast ribosomal RNA mediate P. falciparum resistance. For clindamycin, an A1875C mutation in the gene encoding the apicoplast 23S rRNA has been found in resistant field isolates that were taken into culture [169]. When tested, these parasites show resistance to clindamycin with a classic ‘delayed death’ phenotype [169]. There are no clear markers of doxycycline resistance that have been identified thus far.
Napthoquinones
Atovaquone was developed in the 1990s and is currently used in combination with proguanil as malaria prophylaxis under the brand name malarone. Its mechanism of action is through inhibition of the electron transport chain at the cytochrome bc1 complex (Table 1). This system provides electrons for dihydroorotate dehydrogenase (DHODH), an enzyme that is responsible for de novo pyrimidine synthesis, which is very important for asexual blood stage parasites [170]. During clinical trials, high rates of recrudescence were seen in patients treated with atovaquone alone for P. falciparum malaria [171]. Resistance to atovaquone monotherapy develops rapidly and is associated with single point mutations in the gene encoding cytochrome-b [172]. Y268S/C/N mutations are found in resistant field isolates [173]. These mutations result in a significant fitness cost, since parasites harboring cytb mutations are unable to produce sporozoites in mosquitos rendering them untransmissible [10]. A recent study found that P. falciparum lines which harbor cryptic Y268S alleles in the ~22 copy mitochondrial genome can more readily evolve resistance to atovaquone in vitro [174]. In addition, the resistant lines demonstrated >3-fold copy number amplification of the mitochondrial genome. This suggests that the mechanism of atovaquone resistance is related to mitochondrial diversity rather than de novo selection of resistance mutations.
Multidrug resistance mediators to inform ACT partner drug selection
As resistance to artemisinin emerges in Southeast Asia, there is also an increasing risk of resistance developing to the artemisinin partner drugs as parasites are effectively exposed to monotherapy. There are five partner drugs recommended by the WHO: LMF, AQ, MFQ, SP and PPQ. There are now reports of PPQ resistance emerging rapidly in Cambodia, where DHA-PPQ is a first line treatment for P. falciparum [58, 160, 175]. Mutations in pfcrt and pfmdr1 influence P. falciparum sensitivity to a wide variety of antimalarial drugs, which includes quinine, MFQ, md-ADQ and artemisinin [18, 22, 23, 113, 134–136]. Clinical studies have demonstrated linkage disequilibrium between these two genes [176, 177]. The interrelatedness of mutations in these two DV membrane transporters likely reflects compensatory mutations for fitness losses or may be a mechanism to maximize drug resistance. For example, a study in which the pfmdr1 N86Y mutation was introduced via genetic engineering into CQ-resistant and CQ-sensitive genetic backgrounds demonstrated that the mutation increased susceptibility to LMF, MFQ and DHA [41]. However, the mutation decreased susceptibility to CQ and m-ADQ in both CQ-resistant and CQ-sensitive parasites, although the decrease was more pronounced in the CQ-resistant strains. Another study that genetically edited a C101F pfcrt mutation into CQ-resistant P. falciparum found that it reversed CQ resistance and increased susceptibility to AQ, quinine and artemisinin [62]. As previously discussed, mutations in pfcrt are also associated with PPQ resistance. Knowledge of the mutations already present in pfmdr1 or pfcrt in particular regions can inform optimal partner drug use in the setting of increasing artemisinin resistance.
A systematic analysis of the genetic changes that arose in response to 37 compounds with potent antimalarial activity detected mutations in pfmdr1 and pfcrt in response to structurally diverse compounds as would be expected for pleiotropic drug transporters [178]. In addition, pfabcI3 and pfaat1, genes that encode an ABC transporter and an amino acid transporter, respectively, were mutated in response to a variety of diverse compounds and likely represent multidrug resistance mediators. Interestingly, CNVs were found to contribute to one-third of the resistance acquisition events in this study.
Parasite genetics that determine the geographic origins of resistance
The emergence of antimalarial resistance has most frequently been detected in the Greater Mekong region of Southeast Asia, as evidenced by CQ, MFQ and artemisinin resistance [4, 179, 180]. It was originally thought that parasites from this region might have a hypermutable phenotype [181], with a parasite strain from Southeast Asia demonstrating a mutation rate in vitro that was over 100 times greater than other clones. However, subsequent in vitro studies have not found a higher mutation rate in parasites derived from Southeast Asian strains [6, 182, 183]. In Southeast Asia, malaria transmission is intermittent and seasonal, which results in decreased host immunity. This may contribute to an increased propensity for drug resistance to arise. There is also a significant amount of substandard medication and poor patient compliance found in areas such as the Thai–Cambodia border that are notorious for the emergence of drug resistance [180].
Designing new therapies
With resistance to all known antimalarial medications now apparent, the need for new medications and new approaches to treatment has become extremely urgent. In reviewing the history of antimalarial resistance and studies examining the evolution of resistance in vitro, there are clear lessons that can inform the design of future therapies. Resistance develops rapidly when monotherapy is employed, and thus combination therapy with at least two medications with different mechanisms of action helps mitigate this. Other important considerations to make when determining which partner drugs to use include the following: matching half-lives so that a drug with a long half-life does not persist as monotherapy, pairing drugs with synergistic mechanisms of action and avoiding a combination with antagonistic pharmacokinetics [184, 185]. Clinical trials of triple drug regimens with additional partner drugs added to established ACTs are currently underway in Southeast Asia [186, 187]. Fast acting compounds such as artemisinin are also less likely to generate resistance rapidly compared to slower-acting compounds like clindamycin or MFQ, as demonstrated by in vitro experiments [188, 189] and experience in the field. Drugs that have multiple cellular targets, such as artemisinin, have a higher barrier to resistance compared to drugs with a single target such as pyrimethamine.
In recent years, there has been a rapid increase in new antimalarial compounds advancing in development. Organizations such as the Medicines for Malaria Venture have partnered with academic and industrial laboratories to efficiently identify new promising antimalarial compounds for further development. There are several criteria for these compounds to fulfill, including high potency against clinical isolates from regions known for antimalarial resistance and no cross-resistance against laboratory-adapted strains that are resistant to antimalarials currently in use [190]. Another important step is determining how rapidly in vitro resistance occurs and what fitness cost the resistance mutations confer. Several promising compounds with new antimalarial targets have been identified, with many advancing to clinical trials [191].
The method of in vitro resistance evolution followed by whole genome analysis (sequencing or microarray) can identify the molecular basis of antimalarial resistance and can generate hypotheses about a new antimalarial compound’s target through comparison of SNPs acquired by the resistant clones compared to the compound sensitive parent [192]. Using this method, it was shown that resistance to Cipargamin (NITD609, KAE609), a spiroindolone drug that is the furthest along in development, is conferred by mutations in the gene encoding the plasma membrane P-type cation translocating ATPase (PfATP4) [193, 194] and this is also the likely target of the compound. Other promising compounds whose resistance mechanisms have been studied using genomic methods include the following: KAF156, an imidazolopiperazine that is active against all three parasite stages [195, 196]; DDD107498, which targets the eukaryotic elongation factor 2 [197]; bicyclic azetidines, which target phenylalanyl-tRNA synthetase [198]; imidazopyrazines, which target phosphatidylinositol 4-kinase [199], in addition to many others. Genomic analyses are also important in assessing the emergence of resistance during clinical trials for new antimalarials. DSM265 is a DHODH inhibitor that was designed using target-based drug discovery [200, 201]. During a phase 2a clinical trial, parasites from two of four recurrent P. falciparum infections demonstrated a resistance-associated mutation in dhodh [202].
Conclusions
To review the history of antimalarial therapy is to also examine the myriad ways that the malaria parasite can develop resistance via genetic mutations. The ability to readily culture P. falciparum along with advances in sequencing and gene editing technologies in P. falciparum has greatly increased our ability to understand the effect of these mutations and confirm the changes that these mutations impart. These findings have had a direct impact on evaluating and tracking antimalarial resistance in the field, as seen most recently with the discovery of kelch 13 mutations as a marker of artemisinin resistance. This knowledge also enables a detailed investigation into why particular treatments fail and the design of more effective antimalarial therapies.
Summary Key Points:
Malaria parasites have developed resistance to all major classes of antimalarial drugs.
Resistance to the 4-aminoquinolines and aryl-amino alcohols is primarily mediated by mutations in genes encoding transporters at the parasite’s DV membrane.
Resistance to the antifolate drugs and atovaquone is primarily due to point mutations in the genes encoding target enzymes causing decreased binding of the drug.
New antimalarial medications with novel drug targets are urgently needed to combat antimalarial resistance.
Annie N. Cowell is an Assistant Professor of Medicine at the University of California, San Diego in the Division of Infectious Diseases. Her research interests include using whole genome sequencing of Plasmodium parasites from laboratory and clinical samples to better understand drug resistance and malaria epidemiology.
Elizabeth A. Winzeler is a Professor of Pediatrics at the University of California, San Diego in the Division of Host Microbe Systems and Therapeutics where her group is known for (1) investigating microbial biology using genomic approaches and (2) using informatics and high throughput approaches to develop new drugs. With respect to drug development, she initiated work that resulted in two novel anti-malarial compounds that are in clinical trials (KAE609, Phase IIB and KAF156, Phase IIB) and developed high throughput assays for identifying inhibitors for exoerythrocytic stages. She is a fellow of the American Academy of Microbiology and a recipient of the ASTMH Trager award, the ASBMB Alice and CC Wang Award, the ASTMH Bailey K. Ashford Medal.
Funding
This work was supported by grants from the National Institutes of Health (5R01AI090141 and R01AI103058), Bill & Melinda Gates Foundation (OPP1086217, OPP1141300) and Medicines for Malaria Venture.
References
- 1. Korenromp EL, Williams BG, Gouws E, et al. . Measurement of trends in childhood malaria mortality in Africa: an assessment of progress toward targets based on verbal autopsy. Lancet Infect Dis 2003;3:349–358. [DOI] [PubMed] [Google Scholar]
- 2. World Health Organization Artemisinin and Artemisinin-Based Combination Therapy Resistance. Geneva, Switzerland, 2017. [Google Scholar]
- 3. Maxmen A. How to defuse malaria’s ticking time bomb. Nature 2018;559:458–465. [DOI] [PubMed] [Google Scholar]
- 4. White NJ. Antimalarial drug resistance. J Clin Invest 2004;113:1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. World Health Organization Guidelines for the Treatment of Malaria, 3rd edn Geneva, Switzerland, 2015. [Google Scholar]
- 6. Bopp SE, Manary MJ, Bright AT, et al. . Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet 2013;9:e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. ter Kuile FO, Luxemburger C, Nosten F, et al. . Predictors of mefloquine treatment failure: a prospective study of 1590 patients with uncomplicated falciparum malaria. Trans R Soc Trop Med Hyg 1995;89:660–664. [DOI] [PubMed] [Google Scholar]
- 8. Hastings IM, Watkins WM, White NJ. The evolution of drug-resistant malaria: the role of drug elimination half-life. Philos Trans R Soc Lond B Biol Sci 2002;357:505–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Babiker HA, Gadalla AA, Ranford-Cartwright LC. The role of asymptomatic P. falciparum parasitaemia in the evolution of antimalarial drug resistance in areas of seasonal transmission. Drug Resist Updat 2013;16:1–9. [DOI] [PubMed] [Google Scholar]
- 10. Goodman CD, Siregar JE, Mollard V, et al. . Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 2016;352:349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sigala PA, Goldberg DE. The peculiarities and paradoxes of Plasmodium heme metabolism. Annu Rev Microbiol 2014;68:259–278. [DOI] [PubMed] [Google Scholar]
- 12. Combrinck JM, Mabotha TE, Ncokazi KK, et al. . Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem Biol 2013;8:133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Homewood CA, Warhurst DC, Peters W, et al. . Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972;235:50–52. [DOI] [PubMed] [Google Scholar]
- 14. Yayon A, Cabantchik ZI, Ginsburg H. Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J 1984;3:2695–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis 2001;184:770–776. [DOI] [PubMed] [Google Scholar]
- 16. Duru V, Witkowski B, Menard D. Plasmodium falciparum resistance to artemisinin derivatives and piperaquine: a major challenge for malaria elimination in Cambodia. Am J Trop Med Hyg 2016;95:1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fidock DA, Nomura T, Talley AK, et al. . Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 2000;6:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cooper RA, Ferdig MT, Su XZ, et al. . Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol 2002;61:35–42. [DOI] [PubMed] [Google Scholar]
- 19. Martin RE, Marchetti RV, Cowan AI, et al. . Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science 2009;325:1680–1682. [DOI] [PubMed] [Google Scholar]
- 20. Baro NK, Callaghan PS, Roepe PD. Function of resistance conferring Plasmodium falciparum chloroquine resistance transporter isoforms. Biochemistry 2013;52:4242–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagesha HS, Casey GJ, Rieckmann KH, et al. . New haplotypes of the Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene among chloroquine-resistant parasite isolates. Am J Trop Med Hyg 2003;68:398–402. [PubMed] [Google Scholar]
- 22. Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 2002;298:210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lakshmanan V, Bray PG, Verdier-Pinard D, et al. . A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J 2005;24:2294–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lehane AM, Kirk K. Chloroquine resistance-conferring mutations in pfcrt give rise to a chloroquine-associated H+ leak from the malaria parasite’s digestive vacuole. Antimicrob Agents Chemother 2008;52:4374–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martin RE, Kirk K. The malaria parasite’s chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol Biol Evol 2004;21:1938–1949. [DOI] [PubMed] [Google Scholar]
- 26. Summers RL, Nash MN, Martin RE. Know your enemy: understanding the role of PfCRT in drug resistance could lead to new antimalarial tactics. Cell Mol Life Sci 2012;69:1967–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Summers RL, Dave A, Dolstra TJ, et al. . Diverse mutational pathways converge on saturable chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Proc Natl Acad Sci U S A 2014;111:E1759–E1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Djimde A, Doumbo OK, Cortese JF, et al. . A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med 2001;344:257–263. [DOI] [PubMed] [Google Scholar]
- 29. Pillai DR, Labbe AC, Vanisaveth V, et al. . Plasmodium falciparum malaria in Laos: chloroquine treatment outcome and predictive value of molecular markers. J Infect Dis 2001;183:789–795. [DOI] [PubMed] [Google Scholar]
- 30. Mayor AG, Gomez-Olive X, Aponte JJ, et al. . Prevalence of the K76T mutation in the putative Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene and its relation to chloroquine resistance in Mozambique. J Infect Dis 2001;183:1413–1416. [DOI] [PubMed] [Google Scholar]
- 31. Vieira PP, das Gracas Alecrim M, da Silva LH, et al. . Analysis of the PfCRT K76T mutation in Plasmodium falciparum isolates from the Amazon region of Brazil. J Infect Dis 2001;183:1832–1833. [DOI] [PubMed] [Google Scholar]
- 32. Dorsey G, Kamya MR, Singh A, et al. . Polymorphisms in the Plasmodium falciparum pfcrt and pfmdr-1 genes and clinical response to chloroquine in Kampala, Uganda. J Infect Dis 2001;183:1417–1420. [DOI] [PubMed] [Google Scholar]
- 33. Basco LK, Ringwald P. Analysis of the key pfcrt point mutation and in vitro and in vivo response to chloroquine in Yaounde, Cameroon. J Infect Dis 2001;183:1828–1831. [DOI] [PubMed] [Google Scholar]
- 34. Picot S, Olliaro P, de Monbrison F, et al. . A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar J 2009;8:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cowman AF, Karcz S, Galatis D, et al. . A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J Cell Biol 1991;113:1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rohrbach P, Sanchez CP, Hayton K, et al. . Genetic linkage of pfmdr1 with food vacuolar solute import in Plasmodium falciparum. EMBO J 2006;25:3000–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanchez CP, Rotmann A, Stein WD, et al. . Polymorphisms within PfMDR1 alter the substrate specificity for anti-malarial drugs in Plasmodium falciparum. Mol Microbiol 2008;70:786–798. [DOI] [PubMed] [Google Scholar]
- 38. Wellems TE, Panton LJ, Gluzman IY, et al. . Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature 1990;345:253–255. [DOI] [PubMed] [Google Scholar]
- 39. Foote SJ, Kyle DE, Martin RK, et al. . Several alleles of the multidrug-resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum. Nature 1990;345:255–258. [DOI] [PubMed] [Google Scholar]
- 40. Babiker HA, Pringle SJ, Abdel-Muhsin A, et al. . High-level chloroquine resistance in Sudanese isolates of Plasmodium falciparum is associated with mutations in the chloroquine resistance transporter gene pfcrt and the multidrug resistance gene pfmdr1. J Infect Dis 2001;183:1535–1538. [DOI] [PubMed] [Google Scholar]
- 41. Veiga MI, Dhingra SK, Henrich PP, et al. . Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun 2016;7:11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mekonnen SK, Aseffa A, Berhe N, et al. . Return of chloroquine-sensitive Plasmodium falciparum parasites and emergence of chloroquine-resistant Plasmodium vivax in Ethiopia. Malar J 2014;13:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mwanza S, Joshi S, Nambozi M, et al. . The return of chloroquine-susceptible Plasmodium falciparum malaria in Zambia. Malar J 2016;15:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohammed A, Ndaro A, Kalinga A, et al. . Trends in chloroquine resistance marker, Pfcrt-K76T mutation ten years after chloroquine withdrawal in Tanzania. Malar J 2013;12:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plowe CV. Monitoring antimalarial drug resistance: making the most of the tools at hand. J Exp Biol 2003;206:3745–3752. [DOI] [PubMed] [Google Scholar]
- 46. Srimuang K, Miotto O, Lim P, et al. . Analysis of anti-malarial resistance markers in pfmdr1 and pfcrt across Southeast Asia in the tracking resistance to artemisinin collaboration. Malar J 2016;15:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chenet SM, Okoth SA, Kelley J, et al. . Molecular profile of malaria drug resistance markers of Plasmodium falciparum in Suriname. Antimicrob Agents Chemother 2017;61:e02655-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vieira PP, Ferreira MU, Alecrim M, et al. . Zalis MG: pfcrt polymorphism and the spread of chloroquine resistance in Plasmodium falciparum populations across the Amazon Basin. J Infect Dis 2004;190:417–424. [DOI] [PubMed] [Google Scholar]
- 49. Petersen I, Gabryszewski SJ, Johnston GL, et al. . Balancing drug resistance and growth rates via compensatory mutations in the Plasmodium falciparum chloroquine resistance transporter. Mol Microbiol 2015;97:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pelleau S, Moss EL, Dhingra SK, et al. . Adaptive evolution of malaria parasites in French Guiana: reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc Natl Acad Sci U S A 2015;112:11672–11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rieckmann KH, Davis DR, Hutton DC. Plasmodium vivax resistance to chloroquine? Lancet 1989;2:1183–1184. [DOI] [PubMed] [Google Scholar]
- 52. Baird JK. Resistance to therapies for infection by Plasmodium vivax. Clin Microbiol Rev 2009;22:508–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nomura T, Carlton JM, Baird JK, et al. . Evidence for different mechanisms of chloroquine resistance in 2 Plasmodium species that cause human malaria. J Infect Dis 2001;183:1653–1661. [DOI] [PubMed] [Google Scholar]
- 54. Suwanarusk R, Russell B, Chavchich M, et al. . Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS One 2007;2:e1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Orjuela-Sanchez P, da Silva NS, da Silva-Nunes M, et al. . Recurrent parasitemias and population dynamics of Plasmodium vivax polymorphisms in rural Amazonia. Am J Trop Med Hyg 2009;81:961–968. [DOI] [PubMed] [Google Scholar]
- 56. Sa JM, Nomura T, Neves J, et al. . Plasmodium vivax: allele variants of the mdr1 gene do not associate with chloroquine resistance among isolates from Brazil, Papua, and monkey-adapted strains. Exp Parasitol 2005;109:256–259. [DOI] [PubMed] [Google Scholar]
- 57. Silva SR, Almeida ACG, da Silva GAV, et al. . Chloroquine resistance is associated to multi-copy pvcrt-o gene in Plasmodium vivax malaria in the Brazilian Amazon. Malar J 2018;17:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Amaratunga C, Lim P, Suon S, et al. . Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis 2016;16:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amato R, Lim P, Miotto O, et al. . Genetic markers associated with dihydroartemisinin–piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype–phenotype association study. Lancet Infect Dis 2017;17:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Witkowski B, Duru V, Khim N, et al. . A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype–genotype association study. Lancet Infect Dis 2017;17:174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eastman RT, Dharia NV, Winzeler EA, et al. . Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured Plasmodium falciparum parasites. Antimicrob Agents Chemother 2011;55:3908–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dhingra SK, Redhi D, Combrinck JM, et al. . A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. MBio 2017;8:e00303-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Duru V, Khim N, Leang R, et al. . Plasmodium falciparum dihydroartemisinin–piperaquine failures in Cambodia are associated with mutant K13 parasites presenting high survival rates in novel piperaquine in vitro assays: retrospective and prospective investigations. BMC Med 2015;13:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Agrawal S, Moser KA, Morton L, et al. . Association of a novel mutation in the Plasmodium falciparum chloroquine resistance transporter with decreased piperaquine sensitivity. J Infect Dis 2017;216:468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ross LS, Dhingra SK, Mok S, et al. . Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat Commun 2018;9:3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chu CS, White NJ. Management of relapsing Plasmodium vivax malaria. Expert Rev Anti Infect Ther 2016;14:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Butterworth AS, Skinner-Adams TS, Gardiner DL, et al. . Plasmodium falciparum gametocytes: with a view to a kill. Parasitology 2013;140:1718–1734. [DOI] [PubMed] [Google Scholar]
- 68. Bray PG, Deed S, Fox E, et al. . Primaquine synergises the activity of chloroquine against chloroquine-resistant P. falciparum. Biochem Pharmacol 2005;70:1158–1166. [DOI] [PubMed] [Google Scholar]
- 69. Thomas D, Tazerouni H, Sundararaj KG, et al. . Therapeutic failure of primaquine and need for new medicines in radical cure of Plasmodium vivax. Acta Trop 2016;160:35–38. [DOI] [PubMed] [Google Scholar]
- 70. Bright AT, Alenazi T, Shokoples S, et al. . Genetic analysis of primaquine tolerance in a patient with relapsing vivax malaria. Emerg Infect Dis 2013;19:802–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hurwitz ES, Johnson D, Campbell CC. Resistance of Plasmodium falciparum malaria to sulfadoxine–pyrimethamine (‘Fansidar’) in a refugee camp in Thailand. Lancet 1981;1:1068–1070. [DOI] [PubMed] [Google Scholar]
- 72. Plowe CV, Cortese JF, Djimde A, et al. . Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase and epidemiologic patterns of pyrimethamine–sulfadoxine use and resistance. J Infect Dis 1997;176:1590–1596. [DOI] [PubMed] [Google Scholar]
- 73. Wu Y, Kirkman LA, Wellems TE. Transformation of Plasmodium falciparum malaria parasites by homologous integration of plasmids that confer resistance to pyrimethamine. Proc Natl Acad Sci USA 1996;93:1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cortese JF, Caraballo A, Contreras CE, et al. . Origin and dissemination of Plasmodium falciparum drug-resistance mutations in South America. J Infect Dis 2002;186:999–1006. [DOI] [PubMed] [Google Scholar]
- 75. Nair S, Williams JT, Brockman A, et al. . A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol 2003;20:1526–1536. [DOI] [PubMed] [Google Scholar]
- 76. Roper C, Pearce R, Bredenkamp B, et al. . Antifolate antimalarial resistance in Southeast Africa: a population-based analysis. Lancet 2003;361:1174–1181. [DOI] [PubMed] [Google Scholar]
- 77. Roper C, Pearce R, Nair S, et al. . Intercontinental spread of pyrimethamine-resistant malaria. Science 2004;305:1124. [DOI] [PubMed] [Google Scholar]
- 78. Peterson DS, Milhous WK, Wellems TE. Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proc Natl Acad Sci USA 1990;87:3018–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Peterson DS, Walliker D, Wellems TE. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci USA 1988;85:9114–9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Basco LK, Eldin de Pecoulas P, Wilson CM, et al. . Point mutations in the dihydrofolate reductase-thymidylate synthase gene and pyrimethamine and cycloguanil resistance in Plasmodium falciparum. Mol Biochem Parasitol 1995;69:135–138. [DOI] [PubMed] [Google Scholar]
- 81. Nzila-Mounda A, Mberu EK, Sibley CH, et al. . Kenyan Plasmodium falciparum field isolates: correlation between pyrimethamine and chlorcycloguanil activity in vitro and point mutations in the dihydrofolate reductase domain. Antimicrob Agents Chemother 1998;42:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Plowe CV, Kublin JG, Doumbo OK. P. falciparum dihydrofolate reductase and dihydropteroate synthase mutations: epidemiology and role in clinical resistance to antifolates. Drug Resist Updat 1998;1:389–396. [DOI] [PubMed] [Google Scholar]
- 83. Cortese JF, Plowe CV. Antifolate resistance due to new and known Plasmodium falciparum dihydrofolate reductase mutations expressed in yeast. Mol Biochem Parasitol 1998;94:205–214. [DOI] [PubMed] [Google Scholar]
- 84. Fohl LM, Roos DS. Fitness effects of DHFR-TS mutations associated with pyrimethamine resistance in apicomplexan parasites. Mol Microbiol 2003;50:1319–1327. [DOI] [PubMed] [Google Scholar]
- 85. Sirawaraporn W, Sathitkul T, Sirawaraporn R, et al. . Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA 1997;94:1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tessema SK, Kassa M, Kebede A, et al. . Declining trend of Plasmodium falciparum dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) mutant alleles after the withdrawal of sulfadoxine–pyrimethamine in North Western Ethiopia. PLoS One 2015;10:e0126943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Patel P, Bharti PK, Bansal D, et al. . Prevalence of mutations linked to antimalarial resistance in Plasmodium falciparum from Chhattisgarh, Central India: a malaria elimination point of view. Sci Rep 2017;7:16690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cisse M, Awandare GA, Soulama A, et al. . Recent uptake of intermittent preventive treatment during pregnancy with sulfadoxine–pyrimethamine is associated with increased prevalence of Pfdhfr mutations in Bobo-Dioulasso, Burkina Faso. Malar J 2017;16:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Esu E, Tacoli C, Gai P, et al. . Prevalence of the Pfdhfr and Pfdhps mutations among asymptomatic pregnant women in Southeast Nigeria. Parasitol Res 2018;117:801–807. [DOI] [PubMed] [Google Scholar]
- 90. Ogouyemi-Hounto A, Ndam NT, Kinde Gazard D, et al. . Prevalence of the molecular marker of Plasmodium falciparum resistance to chloroquine and sulphadoxine/pyrimethamine in Benin seven years after the change of malaria treatment policy. Malar J 2013;12:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Voumbo-Matoumona DF, Kouna LC, Madamet M, et al. . Prevalence of Plasmodium falciparum antimalarial drug resistance genes in Southeastern Gabon from 2011 to 2014. Infect Drug Resist 2018;11:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kateera F, Nsobya SL, Tukwasibwe S, et al. . Molecular surveillance of Plasmodium falciparum drug resistance markers reveals partial recovery of chloroquine susceptibility but sustained sulfadoxine–pyrimethamine resistance at two sites of different malaria transmission intensities in Rwanda. Acta Trop 2016;164:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kidgell C, Volkman SK, Daily J, et al. . A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog 2006;2:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dittrich S, Mitchell SL, Blagborough AM, et al. . An atypical orthologue of 6-pyruvoyltetrahydropterin synthase can provide the missing link in the folate biosynthesis pathway of malaria parasites. Mol Microbiol 2008;67:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nair S, Miller B, Barends M, et al. . Adaptive copy number evolution in malaria parasites. PLoS Genet 2008;4:e1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kumpornsin K, Modchang C, Heinberg A, et al. . Origin of robustness in generating drug-resistant malaria parasites. Mol Biol Evol 2014;31:1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ravenhall M, Benavente ED, Mipando M, et al. . Characterizing the impact of sustained sulfadoxine/pyrimethamine use upon the Plasmodium falciparum population in Malawi. Malar J 2016;15:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Imwong M, Pukrittakayamee S, Looareesuwan S, et al. . Association of genetic mutations in Plasmodium vivax dhfr with resistance to sulfadoxine–pyrimethamine: geographical and clinical correlates. Antimicrob Agents Chemother 2001;45:3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Leartsakulpanich U, Imwong M, Pukrittayakamee S, et al. . Molecular characterization of dihydrofolate reductase in relation to antifolate resistance in Plasmodium vivax. Mol Biochem Parasitol 2002;119:63–73. [DOI] [PubMed] [Google Scholar]
- 100. Hastings MD, Porter KM, Maguire JD, et al. . Dihydrofolate reductase mutations in Plasmodium vivax from Indonesia and therapeutic response to sulfadoxine plus pyrimethamine. J Infect Dis 2004;189:744–750. [DOI] [PubMed] [Google Scholar]
- 101. Laing AB. Hospital and field traials of sulformethoxine with pyrimethamine against Malaysian strains of Plasmodium falciparum and P. vivax. Med J Malaya 1968;23:5–19. [PubMed] [Google Scholar]
- 102. Martin DC, Arnold JD. Trimethroprim and sulfalene therapy of Plasmodium vivax. J Clin Pharmacol J New Drugs 1969;9:155–159. [PubMed] [Google Scholar]
- 103. Korsinczky M, Fischer K, Chen N, et al. . Sulfadoxine resistance in Plasmodium vivax is associated with a specific amino acid in dihydropteroate synthase at the putative sulfadoxine-binding site. Antimicrob Agents Chemother 2004;48:2214–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Auliff A, Wilson DW, Russell B, et al. . Amino acid mutations in Plasmodium vivax DHFR and DHPS from several geographical regions and susceptibility to antifolate drugs. Am J Trop Med Hyg 2006;75:617–621. [PubMed] [Google Scholar]
- 105. Rungsihirunrat K, Sibley CH, Mungthin M, et al. . Geographical distribution of amino acid mutations in Plasmodium vivax DHFR and DHPS from malaria endemic areas of Thailand. Am J Trop Med Hyg 2008;78:462–467. [PubMed] [Google Scholar]
- 106. Pornthanakasem W, Riangrungroj P, Chitnumsub P, et al. . Role of Plasmodium vivax dihydropteroate synthase polymorphisms in sulfa drug resistance. Antimicrob Agents Chemother 2016;60:4453–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hawley SR, Bray PG, Mungthin M, et al. . Relationship between antimalarial drug activity, accumulation, and inhibition of heme polymerization in Plasmodium falciparum in vitro. Antimicrob Agents Chemother 1998;42:682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Baird JK. Effectiveness of antimalarial drugs. N Engl J Med 2005;352:1565–1577. [DOI] [PubMed] [Google Scholar]
- 109. Boudreau EF, Webster HK, Pavanand K, et al. . Type II mefloquine resistance in Thailand. Lancet 1982;2:1335. [DOI] [PubMed] [Google Scholar]
- 110. Plucinski MM, Talundzic E, Morton L, et al. . Efficacy of artemether-lumefantrine and dihydroartemisinin–piperaquine for treatment of uncomplicated malaria in children in Zaire and Uige provinces, Angola. Antimicrob Agents Chemother 2015;59:437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hamed K, Kuhen K. No robust evidence of lumefantrine resistance. Antimicrob Agents Chemother 2015;59:5865–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cowman AF, Galatis D, Thompson JK. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc Natl Acad Sci USA 1994;91:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Reed MB, Saliba KJ, Caruana SR, et al. . Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 2000;403:906–909. [DOI] [PubMed] [Google Scholar]
- 114. Price RN, Uhlemann AC, van Vugt M, et al. . Molecular and pharmacological determinants of the therapeutic response to artemether-lumefantrine in multidrug-resistant Plasmodium falciparum malaria. Clin Infect Dis 2006;42:1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Foote SJ, Thompson JK, Cowman AF, et al. . Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell 1989;57:921–930. [DOI] [PubMed] [Google Scholar]
- 116. Triglia T, Foote SJ, Kemp DJ, et al. . Amplification of the multidrug resistance gene pfmdr1 in Plasmodium falciparum has arisen as multiple independent events. Mol Cell Biol 1991;11:5244–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nair S, Nash D, Sudimack D, et al. . Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol Biol Evol 2007;24:562–573. [DOI] [PubMed] [Google Scholar]
- 118. Gardner MJ, Hall N, Fung E, et al. . Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002;419:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Guler JL, Freeman DL, Ahyong V, et al. . Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog 2013;9:e1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cheeseman IH, Miller B, Tan JC, et al. . Population structure shapes copy number variation in malaria parasites. Mol Biol Evol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cheeseman IH, Miller B, Tan JC, et al. . Population structure shapes copy number variation in malaria parasites. Mol Biol Evol 2016;33:603–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Price RN, Uhlemann AC, Brockman A, et al. . Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 2004;364:438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sidhu AB, Uhlemann AC, Valderramos SG, et al. . Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J Infect Dis 2006;194:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Khim N, Andrianaranjaka V, Popovici J, et al. . Effects of mefloquine use on Plasmodium vivax multidrug resistance. Emerg Infect Dis 2014;20:1637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Auburn S, Serre D, Pearson RD, et al. . Genomic analysis reveals a common breakpoint in amplifications of the Plasmodium vivax multidrug resistance 1 locus in Thailand. J Infect Dis 2016;214:1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chongsuphajaisiddhi T, Sabchareon A, Attanath P. Treatment of quinine resistant falciparum malaria in Thai children. Southeast Asian J Trop Med Public Health 1983;14:357–362. [PubMed] [Google Scholar]
- 127. Pukrittayakamee S, Chantra A, Vanijanonta S, et al. . Therapeutic responses to quinine and clindamycin in multidrug-resistant falciparum malaria. Antimicrob Agents Chemother 2000;44:2395–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mayxay M, Barends M, Brockman A, et al. . In vitro antimalarial drug susceptibility and pfcrt mutation among fresh Plasmodium falciparum isolates from the Lao PDR (Laos). Am J Trop Med Hyg 2007;76:245–250. [PMC free article] [PubMed] [Google Scholar]
- 129. Legrand E, Volney B, Meynard JB, et al. . In vitro monitoring of Plasmodium falciparum drug resistance in French Guiana: a synopsis of continuous assessment from 1994 to 2005. Antimicrob Agents Chemother 2008;52:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Achan J, Talisuna AO, Erhart A, et al. . Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J 2011;10:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Simon F, Le Bras J, Charmot G, et al. . Severe chloroquine-resistant falciparum malaria in Gabon with decreased sensitivity to quinine. Trans R Soc Trop Med Hyg 1986;80:996–997. [DOI] [PubMed] [Google Scholar]
- 132. Basco LK, Le Bras J. In vitro activity of halofantrine and its relationship to other standard antimalarial drugs against African isolates and clones of Plasmodium falciparum. Am J Trop Med Hyg 1992;47:521–527. [DOI] [PubMed] [Google Scholar]
- 133. Wurtz N, Fall B, Pascual A, et al. . Role of Pfmdr1 in in vitro Plasmodium falciparum susceptibility to chloroquine, quinine, monodesethylamodiaquine, mefloquine, lumefantrine, and dihydroartemisinin. Antimicrob Agents Chemother 2014;58:7032–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sidhu AB, Valderramos SG. Fidock DA: pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol 2005;57:913–926. [DOI] [PubMed] [Google Scholar]
- 135. Johnson DJ, Fidock DA, Mungthin M, et al. . Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents. Mol Cell 2004;15:867–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Duraisingh MT, Cowman AF. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop 2005;94:181–190. [DOI] [PubMed] [Google Scholar]
- 137. Ferdig MT, Cooper RA, Mu J, et al. . Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol 2004;52:985–997. [DOI] [PubMed] [Google Scholar]
- 138. Henry M, Briolant S, Zettor A, et al. . Plasmodium falciparumPlasmodium falciparum Na+/H+ exchanger 1 transporter is involved in reduced susceptibility to quinine. Antimicrob Agents Chemother 2009;53:1926–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pelleau S, Bertaux L, Briolant S, et al. . Differential association of Plasmodium falciparum Na+/H+ exchanger polymorphism and quinine responses in field- and culture-adapted isolates of Plasmodium falciparum. Antimicrob Agents Chemother 2011;55:5834–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sinou V, Quang le H, Pelleau S, et al. . Polymorphism of Plasmodium falciparum Na(+)/H(+) exchanger is indicative of a low in vitro quinine susceptibility in isolates from Viet Nam. Malar J 2011;10:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Menard D, Andriantsoanirina V, Khim N, et al. . Global analysis of Plasmodium falciparum Na(+)/H(+) exchanger (pfnhe-1) allele polymorphism and its usefulness as a marker of in vitro resistance to quinine. Int J Parasitol Drugs Drug Resist 2013;3:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tilley L, Straimer J, Gnadig NF, et al. . Artemisinin action and resistance in Plasmodium falciparum. Trends Parasitol 2016;32:682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Mbengue A, Bhattacharjee S, Pandharkar T, et al. . A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015;520:683–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Klonis N, Crespo-Ortiz MP, Bottova I, et al. . Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci USA 2011;108:11405–11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dogovski C, Xie SC, Burgio G, et al. . Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 2015;13:e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wang J, Zhang CJ, Chia WN, et al. . Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun 2015;6:10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Noedl H, Se Y, Schaecher K, et al. . Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med 2008;359:2619–2620. [DOI] [PubMed] [Google Scholar]
- 148. Dondorp AM, Nosten F, Yi P, et al. . Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2009;361:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Cheeseman IH, Miller BA, Nair S, et al. . A major genome region underlying artemisinin resistance in malaria. Science 2012;336:79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Takala-Harrison S, Clark TG, Jacob CG, et al. . Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci USA 2013;110:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ariey F, Witkowski B, Amaratunga C, et al. . A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014;505:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Ashley EA, Dhorda M, Fairhurst RM, et al. . Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014;371:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ghorbal M, Gorman M, Macpherson CR, et al. . Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 2014;32:819–821. [DOI] [PubMed] [Google Scholar]
- 154. Straimer J, Gnadig NF, Witkowski B, et al. . Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015;347:428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Mok S, Ashley EA, Ferreira PE, et al. . Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 2015;347:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Li H, Bogyo M, da Fonseca PC. The cryo-EM structure of the Plasmodium falciparum 20S proteasome and its use in the fight against malaria. FEBS J 2016;283:4238–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Takala-Harrison S, Jacob CG, Arze C, et al. . Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis 2015;211:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Miotto O, Almagro-Garcia J, Manske M, et al. . Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet 2013;45:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Miotto O, Amato R, Ashley EA, et al. . Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 2015;47:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Amato R, Pearson RD, Almagro-Garcia J, et al. . Origins of the current outbreak of multidrug-resistant malaria in Southeast Asia: a retrospective genetic study. Lancet Infect Dis 2018;18:337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Kamau E, Campino S, Amenga-Etego L, et al. . K13-propeller polymorphisms in Plasmodium falciparum parasites from sub-Saharan Africa. J Infect Dis 2015;211:1352–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Taylor SM, Parobek CM, DeConti DK, et al. . Absence of putative artemisinin resistance mutations among Plasmodium falciparum in sub-Saharan Africa: a molecular epidemiologic study. J Infect Dis 2015;211:680–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Menard D, Khim N, Beghain J, et al. . A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med 2016;374:2453–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. MalariaGEN Plasmodium falciparum Community Project Genomic epidemiology of artemisinin resistant malaria. eLife 2016;5:e08714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lu F, Culleton R, Zhang M, et al. . Emergence of indigenous artemisinin-resistant. Plasmodium falciparum in Africa. N Engl J Med 2017;376:991–993. [DOI] [PubMed] [Google Scholar]
- 166. Kremsner PG. Clindamycin in malaria treatment. J Antimicrob Chemother 1990;25:9–14. [DOI] [PubMed] [Google Scholar]
- 167. Lell B, Kremsner PG. Clindamycin as an antimalarial drug: review of clinical trials. Antimicrob Agents Chemother 2002;46:2315–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Gaillard T, Madamet M, Pradines B. Tetracyclines in malaria. Malar J 2015;14:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Dharia NV, Plouffe D, Bopp SE, et al. . Genome scanning of Amazonian Plasmodium falciparum shows subtelomeric instability and clindamycin-resistant parasites. Genome Res 2010;20:1534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets 2010;10:226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chiodini PL, Conlon CP, Hutchinson DB, et al. . Evaluation of atovaquone in the treatment of patients with uncomplicated Plasmodium falciparum malaria. J Antimicrob Chemother 1995;36:1073–1078. [DOI] [PubMed] [Google Scholar]
- 172. Korsinczky M, Chen N, Kotecka B, et al. . Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother 2000;44:2100–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sutherland CJ, Laundy M, Price N, et al. . Mutations in the Plasmodium falciparum cytochrome b gene are associated with delayed parasite recrudescence in malaria patients treated with atovaquone-proguanil. Malar J 2008;7:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Siegel SRA, Adapa SR, Wang C, et al. . Mitochondrial heteroplasmy is responsible for Atovaquone drug resistance in Plasmodium falciparum, bioRxiv 232033, 2017.
- 175. Spring MD, Lin JT, Manning JE, et al. . Dihydroartemisinin–piperaquine failure associated with a triple mutant including kelch13 C580Y in Cambodia: an observational cohort study. Lancet Infect Dis 2015;15:683–691. [DOI] [PubMed] [Google Scholar]
- 176. Sa JM, Twu O, Hayton K, et al. . Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci USA 2009;106:18883–18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Happi CT, Gbotosho GO, Folarin OA, et al. . Linkage disequilibrium between two distinct loci in chromosomes 5 and 7 of Plasmodium falciparum and in vivo chloroquine resistance in Southwest Nigeria. Parasitol Res 2006;100:141–148. [DOI] [PubMed] [Google Scholar]
- 178. Cowell AN, Istvan ES, Lukens AK, et al. . Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 2018;359:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Cui L, Mharakurwa S, Ndiaye D, et al. . Antimalarial drug resistance: literature review and activities and findings of the ICEMR network. Am J Trop Med Hyg 2015;93:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Packard RM. The origins of antimalarial-drug resistance. N Engl J Med 2014;371:397–399. [DOI] [PubMed] [Google Scholar]
- 181. Rathod PK, McErlean T, Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci USA 1997;94:9389–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Lee AH, Fidock DA. Evidence of a mild mutator phenotype in Cambodian Plasmodium falciparum malaria parasites. PLoS One 2016;11:e0154166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Brown TS, Jacob CG, Silva JC, et al. . Plasmodium falciparum field isolates from areas of repeated emergence of drug resistant malaria show no evidence of hypermutator phenotype. Infect Genet Evol 2015;30:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Kremsner PG, Krishna S. Antimalarial combinations. Lancet 2004;364:285–294. [DOI] [PubMed] [Google Scholar]
- 185. Hastings IM, Hodel EM. Pharmacological considerations in the design of anti-malarial drug combination therapies—is matching half-lives enough? Malar J 2014;13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Shanks GD, Edstein MD, Jacobus D. Evolution from double to triple-antimalarial drug combinations. Trans R Soc Trop Med Hyg 2015;109:182–188. [DOI] [PubMed] [Google Scholar]
- 187. Dini S, Zaloumis S, Cao P, et al. . Investigating the efficacy of triple artemisinin-based combination therapies (TACTs) for treating Plasmodium falciparum malaria patients using mathematical modelling. Antimicrob Agents Chemotherz 2018;62:11,e01068–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Witkowski B, Lelievre J, Barragan MJ, et al. . Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother 2010;54:1872–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Corey VC, Lukens AK, Istvan ES, et al. . A broad analysis of resistance development in the malaria parasite. Nat Commun 2016;7:11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Burrows JN, van Huijsduijnen RH, Mohrle JJ, et al. . Designing the next generation of medicines for malaria control and eradication. Malar J 2013;12:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Burrows JN, Duparc S, Gutteridge WE, et al. . New developments in anti-malarial target candidate and product profiles. Malar J 2017;16:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Luth MR, Gupta P, Ottilie S, et al. . Using in vitro evolution and whole genome analysis to discover next generation targets for antimalarial drug discovery. ACS Infect Dis 2018;4:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Rottmann M, McNamara C, Yeung BK, et al. . Spiroindolones, a potent compound class for the treatment of malaria. Science 2010;329:1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Spillman NJ, Kirk K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int J Parasitol Drugs Drug Resist 2015;5:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Meister S, Plouffe DM, Kuhen KL, et al. . Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011;334:1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Wu T, Nagle A, Kuhen K, et al. . Imidazolopiperazines: hit to lead optimization of new antimalarial agents. J Med Chem 2011;54:5116–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Baragana B, Hallyburton I, Lee MC, et al. . A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015;522:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Kato N, Comer E, Sakata-Kato T, et al. . Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016;538:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. McNamara CW, Lee MC, Lim CS, et al. . Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013;504:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Phillips MA, Lotharius J, Marsh K, et al. . A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med 2015;7:296ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. McCarthy JS, Lotharius J, Ruckle T, et al. . Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: a two-part first-in-human phase 1a/1b randomised study. Lancet Infect Dis 2017;17:626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Llanos-Cuentas A, Casapia M, Chuquiyauri R, et al. . Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: a proof-of-concept, open-label, phase 2a study. Lancet Infect Dis 2018;18:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]