

Abstract
Immunotherapy is an innovative treatment approach that stimulates a patient’s immune system to fight cancer. It demonstrates characteristics distinct from conventional chemotherapy and stands to revolutionize cancer treatment. We propose a Bayesian phase I/II dosefinding design that incorporates the unique features of immunotherapy by simultaneously considering three outcomes: immune response, toxicity and efficacy. The objective is to identify the biologically optimal dose, defined as the dose with the highest desirability in the risk-benefit tradeoff. An Emax model is utilized to describe the marginal distribution of the immune response. Conditional on the immune response, we jointly model toxicity and efficacy using a latent variable approach. Using the accumulating data, we adaptively randomize patients to experimental doses based on the continuously updated model estimates. A simulation study shows that our proposed design has good operating characteristics in terms of selecting the target dose and allocating patients to the target dose.
Keywords: Immunotherapy, phase I/II trial, dose finding, immune response, risk-benefit tradeoff, Bayesian adaptive design
1. Introduction
Cancer immunotherapy — treatments that harness and enhance the innate power of the immune system to fight cancer — represents the most promising new cancer treatment approach since the first chemotherapies were developed in the late 1940s (Couzin-Frankel, 2013; Topalian, Weiner, and Pardoll, 2011; Makkouk and Weiner, 2015). Immunotherapeutic approaches include the use of antitumor monoclonal antibodies, cancer vaccines, and nonspecific immunotherapies. These approaches stand to revolutionize the treatment of almost every kind of cancer (Couzin-Frankel, 2013; Kaufman, 2015).
Because of a vastly different functional mechanism, immunotherapy behaves differently from conventional chemotherapies. For conventional chemotherapies, it is reasonable to assume that efficacy and toxicity monotonically increase with the dose; however, this assumption may not hold for immunotherapy agents (IAs). As a result, traditional dose-finding designs that aim to identify the maximum tolerated dose (MTD) are not suitable for immunotherapy. To achieve optimal treatment effects, IAs are not necessarily administered at the MTD. In addition, immunotherapy often involves multiple endpoints (Topalian, Weiner, and Pardoll, 2011; Brody et al., 2011; Cha and Fong, 2011). Besides toxicity and efficacy (i.e., tumor response) outcomes, immune response is a unique and important outcome that is essential for the assessment of immunotherapy. Immune response measures the biological efficacy of IAs in activating the immune system, manifested by the proliferation of CD8+ T-cells, CD4+ T-cells and various cytokines (e.g., IFN-α, IL-1β, IL-6, IL-8). As immunotherapy achieves its therapeutic effect by activating the immune system, it is critical to incorporate the immune response in the trial design and leverage its close relationship with clinical endpoints (i.e., efficacy and toxicity) for efficient and practical decision making. Pardoll (2012) described several studies that showed that post-treatment immune responses correlate with clinical outcomes.
Our research is motivated by an immunotherapy trial that aims to find the optimal dose of a novel anti-programmed death 1 (PD-1) immune checkpoint inhibitor for treating patients with recurrent, chemoresistant ovarian cancer. The PD-1 pathway is a negative feedback system that represses Th1 cytotoxic immune responses. This pathway is up-regulated in many tumors and in their surrounding microenvironment. Blocking this pathway with antibodies to PD-1 or its ligands has led to remarkable clinical responses in patients with many different types of cancer, including melanomas and non-small-cell lung cancer. Five dose levels (0.1, 0.3, 0.5, 0.7, 0.9 mg/kg) of the inhibitor will be investigated and the prepared doses will be administered by slow injection over 10 minutes. A maximum of 60 patients will be accrued to the trial. Patient efficacy response is characterized as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD) based on the Response Evaluation Criteria in Solid Tumors. CR is defined as the disappearance of all target lesions. PR is defined as a decrease of at least 30% in the sum of the diameters of target lesions, taking as a reference the baseline sum of the diameters. PD is defined as an increase of at least 20% in the sum of the diameters of target lesions, taking as a reference the smallest sum measured during the study. SD is defined as having neither a sufficient decrease in lesion sizes to qualify as PR, nor sufficient increase to qualify as PD. The immune response of primary interest is the number of CD8+ T-cells measured in the tumor biopsy at the end of the first cycle (28 days) of treatment. Previous studies suggest that the immune response is expected to be associated with the efficacy of the treatment (Sato, et al., 2005; Ercolini, et al., 2005; Hamanishi, et al., 2007; Bachmayr-Heyda, et al., 2013). Dose-limiting toxicity is defined as grade 3 or higher toxicity as scored using the NCI Common Toxicity Criteria for Adverse Events.
We developed a novel phase I/II trial design to find the biologically optimal dose (BOD) for immunotherapy, where BOD is defined as the dose yielding the highest risk-benefit tradeoff, which is formally defined in Section 2.4. In the design, we simultaneously consider three endpoints, including immune response, tumor response and toxicity. To capture the distinct features and relationships among the three endpoints, we model the marginal distribution of the immune response using the Emax model; and conditional on the immune response, we model the joint distribution of the binary toxicity outcome and the ordinal efficacy outcome through a latent variable approach. We elicit the numerical utility to quantify the desirability of the dose based on the risk-benefit tradeoff. During the trial, based on the accumulating data, we update the model estimates and assign a new patient to the dose with the highest desirability through adaptive randomization.
There is a rich body of literature on phase I/II trial designs that integrate the conventional phase I and II segments of clinical drug development trials by simultaneously considering toxicity and efficacy. Thall and Russell (1998) developed a phase I/II trial design that characterizes patient outcomes using a trinary ordinal variable to account for both toxicity and efficacy. Gooley et al. (1994) discussed a phase I/II design in bone marrow transplantation trials to determine a dose that balances the risks of two complications. Braun (2002) proposed the bivariate continual reassessment method, in which the MTD is based jointly on toxicity and disease progression. Thall and Cook (2004) described a Bayesian design based on tradeoffs between toxicity and efficacy probabilities. Yin et al. (2006) proposed a Bayesian phase I/II design based on the odds ratio of efficacy and toxicity. Yuan and Yin (2009, 2011) developed a time-to-event phase I/II design to accommodate late-onset toxicity and efficacy, and a Bayesian phase I/II design for drug-combination trials. Jin et al. (2014) proposed a general strategy to handle delayed toxicity and efficacy outcomes for phase I/II trials using Bayesian data augmentation. Guo and Yuan (2015) proposed a phase I/II design that accommodates informative dropouts. Liu and Johnson (2016) developed a phase I/II design without assuming parametric dose-toxicity and dose-efficacy curves. Comprehensive coverage of phase I/II designs is provided in the book of Yuan, Nguyen and Thall (2016). To the best of our knowledge, this article provides the first phase I/II design for immunotherapy trials that jointly accounts for immune response, toxicity, and efficacy.
The remainder of this article is organized as follows. In Section 2, we present the joint probability model for the continuous immune response, binary toxicity and ordinal efficacy outcomes, and the dose-finding algorithm. In Section 3, we examine the operating characteristics of the proposed design through simulation studies. We provide concluding remarks in Section 4.
2. Method
Probability Models
Consider a phase I-II trial with J prespecified doses, d < · · · < dj, under investigation. Let YT denote the binary toxicity outcome, with YT = 1 indicating toxicity (or severe adverse events), and = 0 otherwise. Let YE denote the tumor response, which is often classified as CR, PR, SD, or PD. Although CR and PR are generally more desirable, in immunotherapy, SD is often regarded as a positive response because some immunotherapies prolong survival by achieving durable SD without notable tumor shrinkage. Thus, we define YE as a trinary ordinal outcome, with YE = 0,1, and 2 indicating PD, SD and PR/CR, respectively. As described previously, besides YT and YE, an essential endpoint for immunotherapy is immune response. Let 17 denote a measure of the immune response (e.g., the count of CD8+ T-cells or the concentration of cytokine), which takes a real value after appropriate transformation. The outcome used for dose finding in our approach is a trinary vector Y = (YI,YT,YE). In contrast, most existing phase I/II designs are based on only (YT,YE). Thall et al. (2014) proposed a phase I/II design to optimize the sedative dose given to preterm infants using three clinical outcomes.
Adaptive decisions in the trial (e.g., dose assignment and selection) are based on the behavior of Y as a function of dose d. To reflect the fact that in immunotherapy, clinical responses rely on the activation of the immune system, we factorize the joint distribution [Yi,YT,YE | d] into the product of the marginal distribution of Yi and the conditional distributions of YT and YE as follows,
where θ is the vector of the parameters, and θ1 and θ2 are subvectors of θ. For notational brevity, we suppress arguments θ1 and θ2 when it will not cause confusion.
We model the marginal distribution [YI | d] using an Emax model,
where a0 is the baseline immune activity in the absence of the IA; α1 is the maximum immune activity that is possibly achieved by the IA above the baseline activity, often known as Emax; α2 is the dose that produces half of the maximum immune activity (i.e., ED50); α3 is the Hill factor that controls the steepness of the dose-response curve; and ε is the random error, which is normally distributed with a mean of 0 and variance σ2, i.e., ε ~ N(0, σ2).
Modeling the joint distribution of [YT,YE | d,YI] is more complicated because YT and YE are different types of variables, i.e., YT is a binary variable whereas YE is an ordinal variable, and they are correlated. To this end, we take the latent variable approach. Specifically, let ZT and ZE denote two continuous latent variables that are related to YT and YE, respectively, as follows,
where ζ1, ξ1 and ξ2 are unknown cutpoints. ZT and ZE can be interpreted as the patient’s latent traits, and YT and YE are the clinical manifestations of unobserved ZT and ZE. When ZT and ZE pass certain thresholds, certain clinical outcomes (e. g., toxicity, CR/PR) are observed. We assume that [ZT, ZE| d,YI] follows a bivariate normal distribution
Where μK(YI, d) = E(Zk|YI, d), k = E or T, is the conditional mean of Zk.
Specification of μT(YI, d) and μΕ(YI, d) requires some consideration. Immune activity is a normal biological phenomenon consistently occurring in the human body; thus, it is typically expected that a low or normal level of immune activity will not cause any immune-related toxicity and that severe immune-related toxicity will occur only when the therapy-induced immune response exceeds a certain threshold. To account for such a threshold effect, we model the relationship between mT (YI, d) and YI and d as
where β0,β1,β2 and β3 are unknown parameters, and the indicator function I(YI > β3) = 1 when YI > β3, and 0 otherwise. Under this model, YI induces toxicity only when it passes threshold β3. Because we do not expect the immune response to be the sole cause of toxicity, in (5), we include dose d as a covariate to capture other possible treatment-related toxicity.
To model the mean structure μΕ(YI, d) for efficacy, we assume a quadratic model,
where the quadratic term is used to accommodate the possibility that efficacy may not monotonically increase with the immune response. In practice, μΕ(YI,d) may first increase with YI and then plateau after YI reaches a certain value. Although the quadratic model cannot directly take an increasing-then-plateau shape, it works reasonably well in that case in our numerical study (i.e., scenarios 6 and 7 in Table 1). This may be because our goal is not to accurately estimate the whole immune-response curve, but to use (6) as a “working” model to obtain a reasonable local fit to guide the dose escalation and deescalation. As the quadratic model can provide good approximation to the plateau (e.g., by taking a slowly increasing shape) locally around the current dose, it leads to appropriate dose transition and selection. In addition, as the Emax model (2) allows YI to plateau with the dose d, the efficacy model (6) indeed accommodates the case that efficacy YE plateaus with d.
Table 1:
Utility based on toxicity, efficacy and immune response.
| Toxicity | Immune response | Efficacy | ||
|---|---|---|---|---|
| PD (YE = 0) |
SD (YE = 1) |
CR/PR (YE = 2) |
||
| No (YT = 0) | Desirable (ỸI = 1) | 5 | 70 | 100 |
| Undesirable (ỸI = 0) | 0 | 50 | 80 | |
| Yes (Yt =1) | Desirable (ỸI = 1) | 0 | 20 | 45 |
| Undesirable (ỸI = 0) | 0 | 10 | 35 | |
In equation (6), we assume that conditional on YI, YE is independent of dose d to reflect the consideration that the treatment effect of immunotherapy is mostly mediated by the immune response. For cases in which such an assumption may not be true, we can add d as a covariate in the model. Because latent variables ZT and ZE are never observed, to identify the model, we set ζ1 = ξι = 0, σ11 = 𝜎22 = 1 and accordingly constrain 0 < σ12 < 1 in (4).
Prior Specification
The prior specification of the parameters in [YI | d] is facilitated by their intuitive interpretations described previously. We elicit prior estimates of (α0,1,α2,α3) from clinicians, denoted as , j = 0, · · ·, 3, and assign αj an independent Gamma distribution with mean and variance . We set Tj at a relatively large value (e.g., ) to obtain a vague prior. Because of taking a vague prior approach, we do not require the prior estimate to be accurately specified. The primary objective of eliciting is to obtain a ballpark estimate of these parameters so that the prior is appropriately centered to avoid extreme (e.g., very small or large) estimates that may lead to inappropriate actions (e.g., terminate the trial too early or escalate the dose too quickly) at the beginning of the trial when data are sparse. As the trial proceeds, the accumulating data will dominate the vague prior and guide dose transition. The simulation described later shows that our design is not sensitive to the specification of . We assign σ2 a vague inverse Gamma prior distribution, e.g., σ2 ~ IG(0.1, 0.1).
To specify the prior distribution for the parameters that appear in [YT,YE| d,YI], we take the regularized vague prior approach (Gelman et al., 2008; Guo and Yuan, 2017). Conventional noninformative priors with huge variances work well for moderate and large samples, but are often problematic for small samples, such as in early phase trials, causing numerical instability and pathological posterior inference (Yuan, Nguyen and Thall, 2016). To obtain reliable inference, the prior should be vague enough to cover the plausible values of the parameter, but not too vague to cause stability issues. Gelman et al. (2008) proposed regularizing the prior using the fact that in practice, a typical change in an input variable is unlikely to lead to a dramatic change in the probability of the response variable. In our case, YT and YE marginally follow probit models after integrating out latent variables ZT and ZE. A change of 2.5 on the probit scale moves the probability of the outcome variable from 0.01 to 0.5 or from 0.5 to 0.99, which is considered unlikely for a typical change in a covariate. Therefore, we scale the input variables (i.e., d and 17) to have mean 0 and standard deviation 0.5, and assign each of the regression coefficients (i.e., β1,2,γ1,γ2) an independent normal prior N(0,1.252), such that a change in any of these covariates from one standard deviation below the mean to one standard deviation above the mean most likely results in a difference of less than 2.5 on the probit scale. The same normal prior is used for the intercepts β0 and γ0, under which a two-standard-deviation change in these parameters moves the outcome probability from 1% and 99% when covariates are set at their mean values. As toxicity is typically non-decreasing with the dose and immune response, it might seem more sensible to use a positive-valued prior, e.g., a gamma or truncated normal prior, to restrict the values of β1 and β2 to be positive. However, when done, this actually hurts the performance of the design, especially for the immunotherapy agents for which toxicity increases slowly with the dose. For these agents, the true values of β1 and β2 are close to 0. Because the gamma or truncated normal prior has most of its mass spanning the positive real line, using them tends to inflate the estimates of β1 and β2, especially at the beginning of the trial when data are sparse, which hinders dose escalation. In the case that toxicity increases rapidly with the dose, using the gamma or truncated normal prior does not have this issue because the true values of β1 and β2 are away from 0. The simulation results comparing the performance of the design under different priors are provided in the Supplementary Materials.
We assign β3 (i.e., the threshold of immune response for inducing toxicity) a uniform prior distribution β3 ~ Unif (a1, a2), with to cover the plausible range of immune response. We assign the correlation parameter σ12 a uniform prior Unif (0,1), and latent variable cutoff parameter ξ2 ~ Unif (0, b), where b is chosen to cover the practical range of Pr(YE < 2) (i.e., the probability of PD and SD).
Likelihood and Posterior
Let N denote the maximum trial sample size. For the ith patient, denote the observed outcome by yi = (yi,i, yT,i, yE,i) and the assigned dose by d[i], where i = 1,..., N. Integrating over (ZT,i, ZE,i) and defining (ζ0 = ξ0 = —œ, ζ2 = ξ3 = ∞, the likelihood for the observables of the ith patient is given by
Let n = 1, …, N denote an interim sample size when an adaptive decision is to be made during the trial, and denote the observed data from the first n patients. The likelihood for the first n patients in the trial is
Let (θ) denote the joint prior distribution of θ. The joint posterior distribution based on the data from the first n patients is . We sample from this posterior distribution using the Markov chain Monte Carlo algorithm with Gibbs sampler (Robert and Casella, 2004).
Desirability of Dose
For each individual endpoint YI, YT or YE, the evaluation of the desirability of a dose is straightfoward. We prefer a dose that has low toxicity, strong immune response and high objective response. However, when we consider (YI,YT,YE) simultaneously, the evaluation of the desirability of a dose becomes more complicated. We need to consider the risk-benefit tradeoffs between the undesirable and desirable clinical outcomes, as physicians routinely do in almost all medical decisions when selecting a treatment for a patient. A convenient tool to formalize such a process is to use a utility function U(YI,YT,YE) to map the multidimensional outcomes into a single index to measure the desirability of a dose in terms of the risk-benefit tradeoffs. The utility should be elicited from physicians and/or patients to reflect medical practice. This approach has been used in previous trial designs (Houede, et al., 2010; Thall, et al., 2013; Thall, et al., 2014; Yuan, Nguyen and Thall, 2016; Guo and Yuan, 2017; Murray, et al., 2017).
Based on our experience, a convenient way of eliciting U(YI,YT,YE) that works well in practice is as follows: we first dichotomize the immune response YI as desirable ( = 1) or undesirable ( = 0) based on a cutoff Cr specified by clinicians (i.e., = 1 if YI ≥ CI, and 0 otherwise), and fix the score of the most desirable outcome (i.e., desirable immune response, no toxicity and CR/PR) asU( = 1, YT = 0, YE = 2) = 100 and the least desirable outcome (i.e., undesirable immune response, toxicity and PD) as U( = 0, YT = 1,YE = 0) = 0. Using these two boundary cases as the reference, we then elicit the scores for other possible outcomes from clinicians, which must be located between 0 and 100. An example of elicited utility is given in Table 1. Note that the purpose of dichotomizing Yr here is to simplify the elicitation of utilities from clinicians. Our model and inference are based on the original scale of YI. If desirable, YI can be categorized into more than two levels, which allows us to account for the desirability of YI at a finer scale, but at the cost of slightly increasing the logistic burden for utility elicitation. For example, if we categorize Y1 into three levels (e.g., low, median, or high), a total of 18 utility values are required to be elicited from clinicians.
Although YE and YI are generally positively correlated, there are several benefits to considering both of them when constructing the utility. First, immunotherapy achieves its therapeutic effect of killing cancer cells by activating the immune response, and the tumor response YE (i.e., a short-term endpoint) may not be a perfect surrogate of the longterm treatment effect of the immunotherapy, e.g., progression-free survival (PFS) or overall survival time. Thus, when two doses have similar YE and YI, we often prefer the dose that has higher potency to activate the immune response, which is potentially translated into better long-term treatment efficacy. Second, using YE and YI simultaneously improves the power to identify the optimal dose. For example, given two doses with (Pr(YE > 0) = 0.3, E(YI) = 20) and (Pr(YE > 0) = 0.4, E(YI) = 60), respectively, the second dose is more likely to be identified as more desirable when we use (YI, YE) rather than YE only, because the difference in the value of YI is much larger than that of YE between the two doses.
Constructing the utility requires close collaboration between statisticians and clinicians, and should be customized for each trial to best reflect the clinical needs and practice. For example, if YE is the long-term efficacy endpoint of interest (e.g., PFS) or Yj is believed to have little impact on the clinical desirability of the dose (after considering YE), we may prefer to define the utility using only (YE,YT), while ignoring YI. Although the elicitation of utility seems rather involved, in our experience, the process actually is quite natural and straightforward. For many trials, this may be done by simply explaining what the utilities represent to the principal investigator (PI) during the design process, and asking the PI to specify all necessary values of U(YI,YT,YE) after fixing the scores for the best and worst elementary outcomes as described previously. After the initial values of utility are specified, comparing outcomes that have the same or similar numerical utilities often motivates the PI to modify the initial specification. In our experience, clinicians quickly understand what the utilities mean, since they reflect actual clinical practice. After completing this process and simulating the trial design, it then may be examined by the PI. In some cases, the simulation results may motivate slight modification of some of the numerical utility values, although such modification typically has little or no effect on the design’s operating characteristics. One possible criticism for using the utility values is that they require subjective input. However, we are inclined to view this as a strength rather than a weakness. This is because the utilities must be elicited from the physicians planning the trial, and thus their numerical values are based on the physician’s experience in treating the disease and observing the good and bad effects that the treatment has on the patients. The process of specifying the utility requires physicians to carefully consider the potential risks and benefits of the treatment that underlie their clinical decision making in a more formal way and incorporate that into the trial. In addition, our simulation study and previous studies show that the design is generally not sensitive to the numerical values of the utility as long as it reflects a similar trend.
For a given dose d, its true utility is given by
Since θ is not known, the utility of dose d must be estimated. Given interim data Dn collected from the first n patients at a decision-making point in the trial, the utility of dose d is estimated by its posterior mean
This posterior mean utility will be used to measure the desirability of a dose and guide dose escalation and selection.
Let πT = Pr(YT = 1|d) denote the toxicity rate and nE = Pr(YE > 0|d) denote the response rate of SD/PR/CR. Let ϕT denote the upper limit of the toxicity rate, and ϕΕ denote the lower limit of the response rate, specified by physicians. We define the BOD as the dose with the highest utility while satisfying πT < ϕT and πΕ > ϕΕ.
Dose Admissibility Criteria
A practical issue is that a dose that is “optimal” in terms of the utility alone may be unacceptable in terms of either safety or the response rate. To ensure that any administered dose has both an acceptably high success rate and an acceptably low adverse event rate, based on interim data Dn, we define a dose d as admissible if it satisfies both the safety requirement
and the efficacy requirement
where CT and CE are prespecified toxicity and efficacy cutoffs. We denote the set of admissible doses by An. Because the objective of the admissible rules (9) and (10) is to rule out doses that are excessively toxic or inefficacious, in practice we should set CT and CE at small values, such as CT = CE = 0.05, which could be further calibrated through simulation. To see this point, it is useful to state the two rules in the following equivalent forms: a dose is unacceptable or inadmissible if Pr(πT > ϕT|Dn) > 1 — CT = 0.95 or Ρr(πΕ < ϕΕ|Dn) > 1 — CE = 0.95. This says that the dose is unacceptable if it is either very likely to be inefficacious or very likely to be too toxic. If we set CT and CE at large values, then the design is very likely to stop the trial early with all doses declared inadmissible due to the large estimation uncertainty at the beginning of the trial; see page 62 of the book by Yuan, Nguyen and Thall (2016) for more discussion on this issue. In the Supplementary Materials, we report the results from a simulation study we conducted that confirmed this issue.
Dose-finding Algorithm
Based on the above considerations, our dose-finding algorithm is described formally as follows. Assume that patients are treated in cohorts of size m with the maximum sample size of N = m x R. We allow m =1 such that patients are treated one by one. The first cohort of patients is treated at the lowest dose d1. Assume that r cohort(s) of patients have been enrolled in the trial, where r =1, · · ·, R — 1. Let dh denote the current highest tried dose, Ces denote the probability for escalation based on toxicity, and n = m x r. To assign a dose to the (r + 1)th cohort of patients:
If the posterior probability of toxicity at dh satisfies Pr(πT(dh) < ϕT|Dn) > Ces and dh ≠ dJ, then we treat the (r + 1)th cohort of patients at dh+1. In other words, if the current data show that the highest tried dose is safe, we want to continue to explore the dose space by treating the next cohort of patients at the next higher new dose.
Otherwise, we identify the admissible set An and adaptively randomize the (r + 1)th patient or cohort of patients to dose dj ∈ An with probability
which is the posterior probability that dose j is the optimal dose having the highest posterior mean utility. We restrict the randomization in admissible dose set An to avoid treating patients at doses that are futile or overly toxic. If An is empty, the trial is terminated.
Once the maximum sample size of N is exhausted, the dose in AN with the largest posterior mean utility E(U(d)|DN) is recommended.
In step 2, to assign a patient to a dose, we use adaptive randomization rather than the greedy algorithm that always assigns the patient to the dose with the currently highest estimate of utility. This is because the latter method tends to become stuck at the local optima and leads to poor precision for identifying the BOD. Adaptive randomization provides a coherent mechanism to avoid that issue and improve the operating characteristics of the design (Yuan, Nguyen, and Thall, 2016).
3. Simulation
We assessed the performance of our proposed design using simulation studies. Taking the setting of the motivating trial, we considered five doses (0.1, 0.3, 0.5, 0.7, 0.9), with a maximum sample size of 60 in a cohort size of 3. The toxicity upper bound ϕT= 0.3 and efficacy lower bound ϕE= 0.3. Rescaled by the prior estimate of the baseline immune response, we set based on the prior estimates of Emax, ED50, and the steepness of the dose-response relationship elicited from clinicians. We set to obtain vague priors forαj, j = 0, 3, so that the prior standard deviation was 4 times the prior mean. Since the estimates of the baseline and the maximum immune response were respectively, we assigned β3 (i.e., the threshold of immune response for inducing toxicity) a uniform prior β3 ~ Unif (0, 9) to cover the whole plausible range, and ξ2 a uniform prior Unif (0, 6) to cover a reasonable range of Pr(YE < 2) that may be encountered in practice. For example, when Pr(YE = 0) = 0.1, the range for Pr(YE < 2) is (0.1, 0.999) under this prior. Calibrated by simulation, we took the probability cutoffs CT = CE = 0.05 for defining admissible doses and Ces = 0.5 for dose escalation. The utility elicited from physicians is displayed in Table 1. The same prior distribution, probability cutoffs, and utility were used throughout the simulation. We designed 8 scenarios that varied in the number of target doses, location of the target doses, as well as the patterns of toxicity, efficacy, and immune response (see Table 2). Figure 1 shows the true dose-response curves for immune response, toxicity and efficacy for these scenarios. Under each scenario, we simulated 1,000 trials.
Table 2:
True immune response, toxicity, efficacy probabilities, and utility at each dose, and selection percentage and the average number of patients treated at each dose level under the proposed design and the EffTox design. The boldface numbers are the target doses.
| dose level | |||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Scenario 1 | |||||
| E(YI ) | 2.0 | 2.9 | 4.5 | 5.7 | 6.3 |
| πT | 0.03 | 0.04 | 0.1 | 0.17 | 0.23 |
| (πE,1, πE,2)† | (0.46, 0.11) | (0.50, 0.13) | (0.35, 0.07) | (0.16, 0.02) | (0.08, 0.01) |
| Utility | 30.9 | 34.6 | 26.2 | 13.1 | 8 |
| Selection % (proposed) | 0.282 | 0.649 | 0.069 | 0 | 0 |
| # of patients | 16.0 | 16.9 | 13.9 | 9.0 | 4.8 |
| Selection % (EffTox) | 0.476 | 0.521 | 0.001 | 0 | 0.002 |
| # of patients | 17.4 | 16.3 | 13.1 | 8.2 | 4.4 |
| Scenario 2 | |||||
| E(YI) | 2.0 | 2.9 | 4.5 | 5.7 | 6.3 |
| πT | 0.03 | 0.07 | 0.19 | 0.35 | 0.49 |
| (πE,1, πE,2) | (0.28, 0.14) | (0.31, 0.17) | (0.31, 0.18) | (0.28, 0.14) | (0.25, 0.11) |
| Utility | 24.2 | 27.8 | 30.6 | 23.8 | 17.7 |
| Selection % (proposed) | 0.117 | 0.094 | 0.736 | 0.047 | 0.006 |
| # of patients | 13.0 | 14.0 | 14.6 | 10.9 | 7.4 |
| Selection % (EffTox) | 0.212 | 0.602 | 0.183 | 0.001 | 0.002 |
| # of patients | 14.2 | 15.2 | 14.4 | 10.1 | 6.2 |
| Scenario 3 | |||||
| E(YI ) | 2.0 | 2.9 | 4.5 | 5.7 | 6.3 |
| πT | 0.07 | 0.09 | 0.12 | 0.3 | 0.49 |
| (πE,1, πE,2) | (0.16, 0.04) | (0.17, 0.05) | (0.22, 0.07) | (0.25, 0.10) | (0.27, 0.12) |
| Utility | 9.8 | 11.9 | 19.7 | 22.1 | 20.1 |
| Selection % (proposed) | 0.028 | 0.005 | 0.29 | 0.572 | 0.057 |
| # of patients | 9.5 | 9.8 | 15.0 | 15.1 | 9.2 |
| Selection % (EffTox) | 0.143 | 0.103 | 0.254 | 0.255 | 0.203 |
| # of patients | 11.1 | 12.0 | 13.0 | 12.9 | 9.8 |
| Scenario 4 | |||||
| E(YI ) | 2.0 | 2.9 | 4.5 | 5.7 | 6.3 |
| πT | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 |
| (πE,1, πE,2) | (0.21, 0.06) | (0.26, 0.09) | (0.34, 0.17) | (0.36, 0.20) | (0.37, 0.22) |
| Utility | 12.0 | 17.7 | 32.8 | 40.2 | 42.3 |
| Selection % (proposed) | 0.000 | 0.000 | 0.006 | 0.257 | 0.735 |
| # of patients | 7.9 | 8.7 | 12.4 | 15.3 | 15.6 |
| Selection % (EffTox) | 0.008 | 0.004 | 0.046 | 0.207 | 0.733 |
| # of patients | 9.3 | 10.6 | 12.4 | 13.4 | 14.3 |
| Scenario 5 | |||||
| E(YI ) | 2.6 | 3.4 | 3.9 | 4.3 | 4.6 |
| πT | 0.05 | 0.12 | 0.2 | 0.28 | 0.36 |
| (πE,1, πE,2) | (0.15, 0.09) | (0.22, 0.15) | (0.24, 0.17) | (0.23, 0.16) | (0.21, 0.15) |
| Utility | 13.8 | 21.9 | 24.5 | 23.7 | 20.6 |
| Selection % (proposed) | 0.024 | 0.036 | 0.431 | 0.42 | 0.077 |
| # of patients | 11.6 | 12.8 | 13.0 | 12.3 | 10.0 |
| Selection % (EffTox) | 0.002 | 0.258 | 0.711 | 0.017 | 0.002 |
| # of patients | 11.4 | 14.7 | 14.4 | 11.2 | 7.8 |
| Scenario 6 | |||||
| E(YI ) | 2.6 | 3.4 | 3.9 | 4.3 | 4.6 |
| πT | 0.01 | 0.04 | 0.09 | 0.14 | 0.2 |
| (πE,1, πE,2) | (0.24, 0.06) | (0.31, 0.10) | (0.34, 0.12) | (0.35, 0.13) | (0.36, 0.13) |
| Utility | 17.3 | 24.9 | 28.3 | 29.6 | 29.4 |
| Selection % (proposed) | 0.016 | 0.001 | 0.098 | 0.401 | 0.483 |
| # of patients | 10.4 | 11.1 | 12.1 | 13.1 | 13.2 |
| Selection % (EffTox) | 0.024 | 0.094 | 0.43 | 0.321 | 0.129 |
| # of patients | 10.3 | 12.1 | 12.9 | 12.7 | 11.8 |
| Scenario 7 | |||||
| E(YI ) | 2.6 | 3.4 | 3.9 | 4.3 | 4.6 |
| πT | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 |
| (πE,1, πE,2) | (0.16, 0.37) | (0.16, 0.37) | (0.16, 0.37) | (0.16, 0.37) | (0.16, 0.37) |
| Utility | 32.5 | 34.3 | 36.9 | 38.5 | 39.7 |
| Selection % (proposed) | 0.175 | 0.021 | 0.038 | 0.091 | 0.675 |
| # of patients | 11.9 | 11.8 | 12.2 | 11.9 | 12.2 |
| Selection % (EffTox) | 0.351 | 0.086 | 0.146 | 0.07 | 0.347 |
| # of patients | 13.2 | 12.0 | 11.4 | 11.5 | 11.9 |
| Scenario 8 | |||||
| E(YI ) | 2.5 | 3.1 | 3.5 | 3.8 | 3.9 |
| πT | 0.72 | 0.78 | 0.84 | 0.88 | 0.91 |
| (πE,1, πE,2) | (0.06, 0.04) | (0.08, 0.06) | (0.09, 0.06) | (0.09, 0.07) | (0.10, 0.08) |
| Utility | 2.4 | 3.5 | 4.1 | 4.7 | 5.2 |
| Selection % (proposed) | 0 | 0 | 0 | 0 | 0 |
| # of patients | 4.3 | 3.2 | 3.1 | 1.7 | 0.7 |
| Selection % (EffTox) | 0 | 0 | 0 | 0 | 0 |
| # of patients | 4.1 | 3.0 | 2.7 | 2.0 | 0.8 |
πE,1 = Pr(YE = 1), and πE,2 = Pr(YE = 2). 24
Figure 1: Dose-response curves for the 8 scenarios in the simulation study.
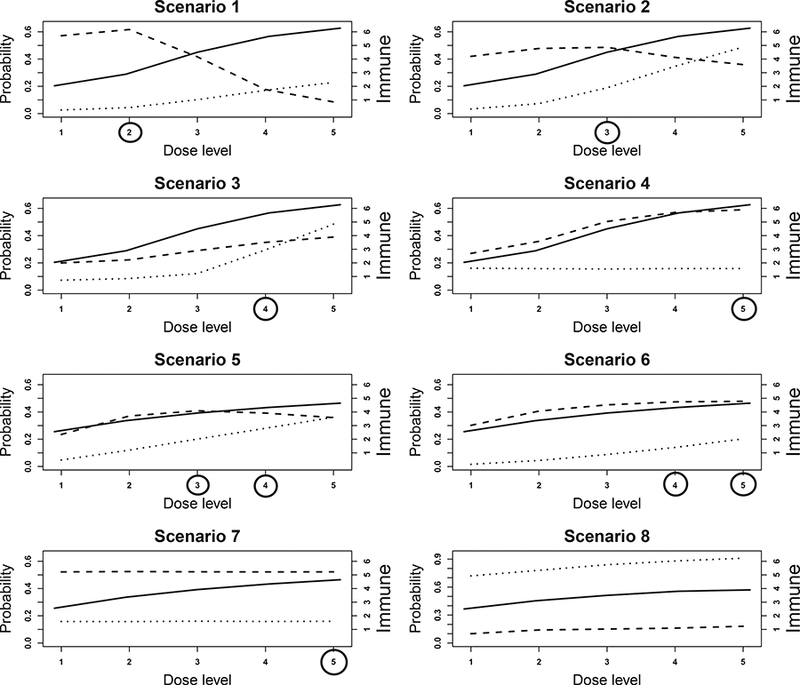
The dotted, dashed, and solid lines are the toxicity (πt), efficacy (πE), and immune response (E(YI)) curves, respectively. Toxicity and efficacy are plotted against the left y-axis, and the immune response is plotted against the right y-axis. Target doses are indicated by circles.
We compared our design with a design that considers only efficacy and toxicity (denoted as the EffTox design), as in most existing phase I/II designs such as that of Thall and Cook (2004). To make the comparison more meaningful, we used the same toxicity and efficacy models as the proposed design, but with the immune response term dropped such that
The risk-benefit utility used in the EffTox design was obtained by averaging U(Yj,YT,YE) in Table 1 over Yj.
Table 2 summarizes the operating characteristics of our proposed design and the EffTox design. Scenarios 1 to 4 consider the case with one target dose and different shapes of dose- toxicity and dose-efficacy curves. In scenarios 1 and 2, the efficacy probabilities first increase and then decrease with the dose; in scenario 3, both toxicity and efficacy increase with the dose; and in scenario 4, toxicity remains constant across the doses, and efficacy increases with the dose.
In scenario 1, the target dose is dose level 2. Dose level 1 has similar toxicity probability as dose level 2, but lower efficacy probabilities. By taking advantage of the immune response data, the proposed design has higher power to distinguish these two doses. The percentage of correct selection of the target dose under the proposed design is 12.8% higher than that under the EffTox design. The number of patients allocated to the target dose is similar between the two designs. In scenario 2, dose level 3 is the target dose that is safe and has the highest utility. Our proposed design correctly identified the target dose 73.6% of the time, and allocated the largest number of patients to the target dose (i.e., 14.6) among the 5 doses. In contrast, the EffTox design selected the target dose only 18.3% of the time because it ignored the immune response. Dose levels 2 and 3 have similar efficacy, but level 3 has a much higher immune response and thus is more desirable. Because of ignoring the immune response, the EffTox design failed to recognize that dose level 3 is better. For a similar reason, the proposed design also outperformed the EffTox design in scenario 3, under which the target dose is level 4. The percentage of correct selection of the target was 57.2% under the proposed design, and only 25.5% under the EffTox design. The proposed design also assigned more patients to the target dose. In scenario 4, the dose-toxicity curve is flat and the two designs performed comparably.
Scenarios 5 and 6 were designed to have two target doses. In scenario 5, efficacy first increases and then decreases, whereas in scenario 6, efficacy first increases then plateaus. In these two scenarios, the proposed design performed well, with the combined percentage of correct selection of the two target doses exceeding 85%. In contrast, the percentage of correct selection of the target doses under the EffTox design was 72.8% and 45%, respectively. Scenario 7 considers a special case in which five doses have the same toxicity and efficacy probabilities, but higher doses induce a stronger immune response and thus have higher utility or desirability. The target dose is level 5. The proposed design selected the target dose 67.5% of the time, whereas the EffTox design selected the target dose 34.7% of the time. In scenario 8, the toxicity is higher than the toxicity upper bound ϕE = 0.3 and the efficacy is lower than the efficacy lower bound ϕΕ = 0.3 at all dose levels. Across 1000 simulations, the trial was terminated early 100% of the time under both designs.
3.1. Sensitivity Analyses
We carried out sensitivity analyses to assess the robustness of the performance of our proposed design by using 1) another set of utility values, and 2) a smaller sample size. Compared to the utility in Table 1, the new utility (see Table 3) assigns higher scores (i.e., less penalty) to YT = 1 (toxicity), that is, patients are willing to tolerate higher toxicity to attain higher efficacy. The simulation results (see Table 4) show that the proposed design performed well, with high percentages of correct selection of the target doses. When the maximum sample size dropped from 60 to 42, the performance of our design was slightly worse, as summarized in Table 5, but the selection percentage of the target dose was still the highest among all doses.
Table 3:
Utility for sensitivity analysis.
| Toxicity | Immune response | Tumor response | ||
|---|---|---|---|---|
| PD (YE = 0) |
SD (YE = 1) |
CR/PR (YE = 2) |
||
| No (YT = 0) | Desirable (ỸI = 1) | 10 | 70 | 100 |
| Undesirable (ỸI = 0) | 5 | 50 | 80 | |
| Yes (Yt =1) | Desirable (ỸI = 1) | 5 | 40 | 60 |
| Undesirable (ỸI = 0) | 0 | 30 | 55 | |
Table 4:
Simulation results with an alternative utility.
| dose level | dose level | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 | |
| scenario 1 | scenario 2 | |||||||||
| Selection % | 0.23 | 0.67 | 0.1 | 0 | 0 | 0.081 | 0.05 | 0.785 | 0.079 | 0.005 |
| # of patients | 15.6 | 15.5 | 14.0 | 9.6 | 5.4 | 12.6 | 13.3 | 14.6 | 12.0 | 7.6 |
| scenario 3 | scenario 4 | |||||||||
| Selection % | 0.011 | 0.004 | 0.194 | 0.616 | 0.135 | 0.008 | 0 | 0.009 | 0.261 | 0.722 |
| # of patients | 9.6 | 10.1 | 14.2 | 15.0 | 9.8 | 8.7 | 9.2 | 12.5 | 14.3 | 15.4 |
| scenario 5 | scenario 6 | |||||||||
| Selection % | 0.013 | 0.017 | 0.291 | 0.533 | 0.117 | 0.002 | 0.001 | 0.031 | 0.266 | 0.695 |
| # of patients | 11.3 | 12.2 | 12.6 | 12.3 | 10.7 | 10.4 | 11.2 | 12.0 | 13.0 | 13.2 |
| scenario 7 | scenario 8 | |||||||||
| Selection % | 0.126 | 0.013 | 0.038 | 0.106 | 0.717 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| # of patients | 11.9 | 11.9 | 11.7 | 11.9 | 12.6 | 4.3 | 3.2 | 2.9 | 2.1 | 0.8 |
Table 5:
Simulation results with a maximum sample size of 42.
| dose level | dose level | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 | |
| scenario 1 | scenario 2 | |||||||||
| Selection % | 0.375 | 0.491 | 0.132 | 0 | 0 | 0.178 | 0.105 | 0.633 | 0.078 | 0.006 |
| # of patients | 10.6 | 10.5 | 9.6 | 6.8 | 4.4 | 9.5 | 9.5 | 9.6 | 7.9 | 5.5 |
| scenario 3 | scenario 4 | |||||||||
| Selection % | 0.042 | 0.008 | 0.309 | 0.489 | 0.108 | 0.004 | 0.005 | 0.014 | 0.258 | 0.719 |
| # of patients | 7.1 | 7.4 | 9.7 | 10.0 | 7.2 | 6.5 | 6.7 | 8.5 | 9.8 | 10.5 |
| scenario 5 | scenario 6 | |||||||||
| Selection % | 0.067 | 0.041 | 0.305 | 0.452 | 0.117 | 0.034 | 0.016 | 0.088 | 0.321 | 0.539 |
| # of patients | 8.6 | 8.7 | 8.9 | 8.3 | 7.3 | 7.2 | 7.9 | 8.6 | 9.0 | 9.2 |
| scenario 5 | scenario 6 | |||||||||
| Selection % | 0.185 | 0.031 | 0.036 | 0.093 | 0.655 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| # of patients | 9.0 | 8.2 | 8.0 | 8.0 | 8.8 | 4.3 | 3.2 | 2.9 | 2.1 | 0.8 |
Our prior specification requires elicitation of prior estimates of άs from clinicians. Given the incipient stage of research in immunotherapy, these estimates may not be very reliable. To evaluate the robustness of our proposed design to different values of these prior estimates, we performed sensitivity analysis with two alternative prior estimates of α’s: or As shown in Figure 2, the results are similar across different prior estimates of α′s, suggesting that our design is not sensitive to the prior estimates of α′s. Detailed results are provided in the Supplementary Materials.
Figure 2: Sensitivity analysis with three different prior estimates of α′s in scenarios 1–4.
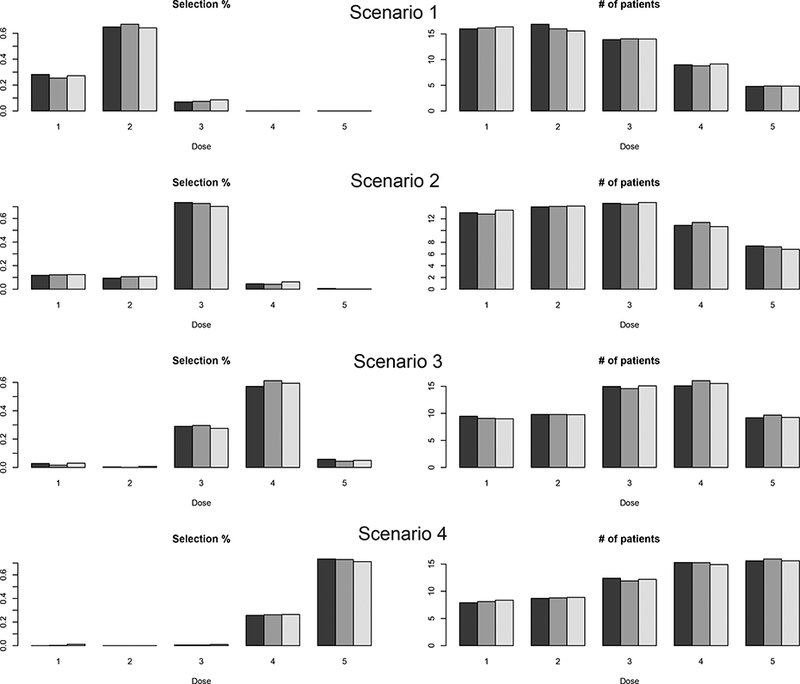
In each plot, at each dose level, the three bars from left to right correspond to the results with prior estimates = ( 1, 5, 0.5, 2), (2, 4, 0.4, 3), and (1.5, 6, 0.6, 2.5), respectively.
Finally, we evaluated the sensitivity of the proposed design to different prior distributions. We made all the priors more non-informative. Specifically, for α′s, we set the prior standard deviation to five times the prior mean, i.e., To σ2, we assigned an inverse Gamma prior with parameters 0.01, i.e., σ2 ~ 1G(0.01,0.01). The regression coefficients β1,2, γ1, γ 2 were assigned normal prior N(0, 2.52) so that the prior standard deviation was twice the previous value. The simulation results are very similar to the original results (see Figure 3), suggesting that our design is not sensitive to the prior distributions.
Figure 3: Sensitivity analysis with two different prior distributions of model parameters under scenarios 1–4.
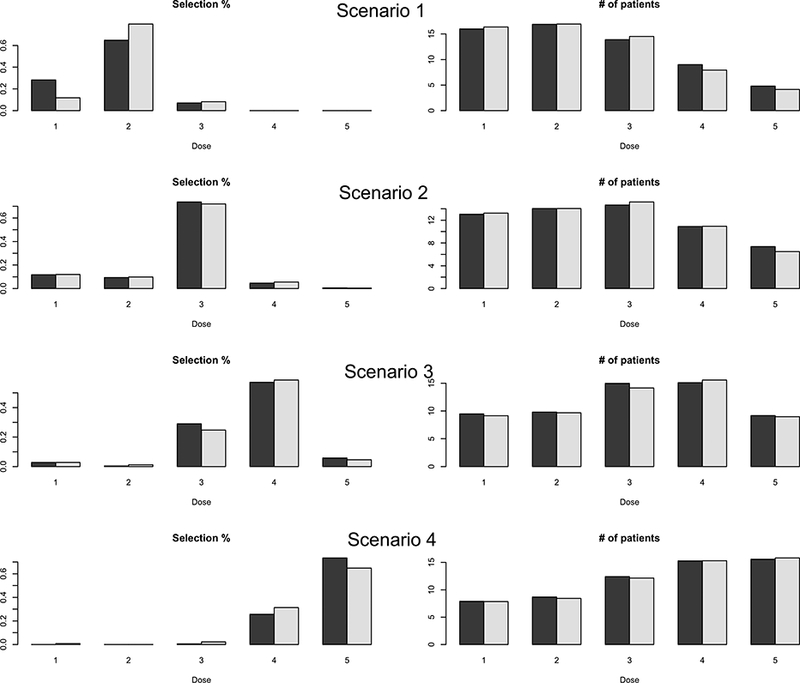
In each plot, at each dose level, the two bars represent the results under two different prior distributions.
4. Discussion
We have proposed a Bayesian phase I/II clinical trial design for immunotherapy by simultaneously considering immune response, toxicity and efficacy. We use an Emax model for the marginal distribution of the immune response and a latent variable approach to model the joint distribution of the binary toxicity and ordinal efficacy outcomes conditional on the immune response. Based on these three outcomes, utility is used to quantify the desirability of the dose and make the decision of dose assignment and selection. Our simulation study shows that the proposed design has desirable operating characteristics.
In order to capture the important features of immune response, toxicity, and efficacy, and the interplay among the three endpoints, our model has a relatively large number of parameters. One concern may be that at the inception of the trial, data are sparse and the parameter estimates are highly variable and mainly driven by the prior. However, this does not cause issues because the number of investigational doses is typically small (e.g., < 8 doses) and our dose-finding algorithm does not allow for skipping untried doses for dose escalation. At the beginning of the trial when the parameter estimates are highly variable, the dose-finding algorithm acts somewhat “semi-randomly” by trying the doses sequentially from low to high, guided largely by the priors. Actually, in some anti-intuitive sense, such uncertainty and “randomness” are helpful because it provides the design freedom to move around and explore the dose space, and to avoid being stuck at a local dose. When the trial proceeds and data accumulate, we obtain more reliable estimates and the dose assignment becomes more stable and converges to the target dose. Therefore, as long as at the middle or late stage of the trial, we have adequate data to make reasonable estimates, we are likely to make the correct dose assignment and select the target dose at the end of the trial. In addition, the primary objective of the phase I/II trial is to identify the optimal dose among a set of prespecified doses, not to obtain accurate estimates. This also renders the design higher tolerance to the variability of parameter estimates. As long as the method obtains the rank of estimated desirability correctly, it will correctly select the target dose.
In this article, ordinal tumor response is used as the efficacy endpoint. In some immunotherapy trials, the PFS time may be a more appropriate endpoint to quantify the therapeutic efficacy of the treatment. To accommodate these cases, we can model the joint distribution of the immune response, toxicity and PFS as follows: first model the marginal distribution of immune response using the Emax model; conditional on the immune response, model the conditional distribution of a binary (or ordinal) toxicity outcome using a logistic (or multinomial) model; and then conditional on both the immune response and toxicity, model the conditional distribution of PFS using a survival regression model, e.g., proportional hazards model (Cox, 1972).
Supplementary Material
Acknowledgments
Yuan’s research was supported by NCI grant R01 CA 154591 and 5P50CA098258. The authors thank the editor, the associate editor and two anonymous referees for their insightful and constructive comments which substantially improved the paper.
Contributor Information
Suyu Liu, Assistant Professor, Department of Biostatistics, University of Texas MD Anderson Cancer Center, Houston, TX 77030-4009.
Beibei Guo, Assistant Professor, Department of Experimental Statistics, Louisiana State University, Baton Rouge, LA 70803.
Ying Yuan, Professor, Department of Biostatistics, University of Texas MD Anderson Cancer Center, Houston, TX 77030-4009, (yyuan@mdanderson.org)..
References
- Bachmayr-Heyda A, Aust S, Heinze G, Polterauer S, Grimm C, Braicu EI, Sehouli J, Lambrechts S, Vergote I, Mahner S, et al. (2013) Prognostic impact of tumor infiltrating CD8+ T cells in association with cell proliferation in ovarian cancer patients-a study of the OV- CAD consortium. BMC Cancer 13:422, doi: 10.1186/1471-2407-13-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun TM. The bivariate continual reassessment method: extending the CRM to phase I trials of two competing outcomes. Controlled clinical trials 2002; 23: 240–256. [DOI] [PubMed] [Google Scholar]
- Brody J, Kohrt H, Marabelle A and Levy R (2011), Active and passive immunotherapy for lymphoma: proving principles and improving results. Journal of Clinical Oncology, 29, 1864–1875 [DOI] [PubMed] [Google Scholar]
- Cha E and Fong L (2011), Immunotherapy for prostate cancer: biology and therapeutic approaches Journal of Clinical Oncology, 27, 3677–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DR (1972) Regression models and life-tables (with discussion). Journal of the Royal Statistical Society, Series Bb 34: 187–220. [Google Scholar]
- Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JP, Bieler JG, Emens LA, Reilly RT, Jaffee EM (2005) Recruitment of latent pools of high-avidity CD8+ T cells to the antitumor immune response, Journal of Experimental Medicine, 201, 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin-Frankel J (2013), Cancer immunotherapy. Science, 324, 1432–1433. [DOI] [PubMed] [Google Scholar]
- Gelman A, Jakulin A, Pittau MG, Su YS (2008) A weakly informative default prior distribution for logistic and other regression models. The Annals of Applied Statistics, 2: 1360–1383. [Google Scholar]
- Gooley TA., Martin PJ, Fisher LD, Pettinger M (1994) Simulating as a design tool for phase I/II clinical trials: an example from bone marrow transplantation. Controlled Clinical Trials, 15, 450–462. [DOI] [PubMed] [Google Scholar]
- Guo B, Yuan Y (2015) A Bayesian Design for Phase I/II Clinical Trials with Nonignorable Dropout. Statistics in Medicine, 34, 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Yuan Y (2017) Bayesian phase I/II biomarker-based dose finding for precision medicine with molecularly targeted agents. Journal of the American Statistical Association, 112, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, Higuchi T, Yagi H, Takakura K, Minato N, et al. (2007) Programmed cell death 1 ligand 1 and tumor- infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer, Proceedings of the National Academy of Sciences of the United States of America, 104, 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houede N, Thall PF, Nguyen H, et al. (2010) Utility-based optimization of combination therapy using ordinal toxicity and efficacy in phase I/II trials. Biometrics 66, 532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin IH, Liu S, Thall P and Yuan Y (2014) Using Data Augmentation to Facilitate Conduct of Phase I/II Clinical Trials with Delayed Outcomes. Journal of American Statistical Association, 109, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman HL (2015), Precision immunology: the promise of immunotherapy for the treatment of cancer. Journal of Clinical Oncology, 33, 1315–1317. [DOI] [PubMed] [Google Scholar]
- Liu S, Johnson VE (2016) A robust Bayesian dose-finding design for phase I/II clinical trials. Biostatistics 2016; 17: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkouk A, and Weiner GJ (2015), Cancer immunotherapy and breaking immune tolerance: new approaches to an old challenge. Cancer Research, 75, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D (2012), The blockade of immune checkpoints in cancer immunotherapy. Nature Review Cancer, 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C Casella G (2004). Monte Carlo Statistical Methods. New York: Springer-Verlag, 2nd edition. [Google Scholar]
- Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America, 102, 18538–18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thall P, Russell K (1998) A strategy for dose-finding and safety monitoring based on efficacy and adverse outcomes in phase I/II clinical trials. Statistics in Medicine, 27, 4895–4913. [PubMed] [Google Scholar]
- Thall P, Cook J (2004) Dose-finding based on efficacy-toxicity trade-offs. Biometrics, 60, 684–693. [DOI] [PubMed] [Google Scholar]
- Thall PF, Nguyen HQ, Braun TM, et al. (2013) Using joint utilities of the times to response and toxicity to adaptively optimize schedule-dose regimes. Biometrics 69, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thall PF, Nguyen HQ, Zohar S and Maton P (2014) Optimizing sedative dose in preterm infants undergoing treatment for respiratory distress syndrome. Journal of the American Statistical Association, 109, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Weiner GJ and Pardoll DM (2011), Cancer immunotherapy comes of age. Journal of Clinical Oncology, 23, 4828–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G, Li Y, Ji Y (2006) Bayesian dose-finding in phase I/II clinical trials using toxicity and efficacy odds ratios. Biometrics, 62, 777–784. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Nguyen H, Thall P (2016). Bayesian designs for phase I-II clinical trials. Chapman & Hall/CRC Biostatistics Series. [Google Scholar]
- Yuan Y, Yin G (2009) Bayesian dose finding by jointly modeling toxicity and efficacy as time-to-event outcomes. Journal of the Royal Statistical Society, Series C, 58, 719–736. [Google Scholar]
- Yuan Y and Yin G (2011) Bayesian phase I/II drug-combination trial design in oncology. Annals of Applied Statistics, 5, 924–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray TA, Thall PF, Yuan Y, McAvoy S and Gomez DR (2017) Robust treatment comparison based on utilities of semi-competing risks in non-small-cell lung cancer. Journal of the American Statistical Association, 112, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.