

Abstract
Peptide agonists of GPCRs and other receptors are powerful signaling molecules with high potential as biological tools and therapeutics, but they are typically plagued by instability and short half-lives in vivo. Nature uses protein glycosylation to increase the serum stability of secreted proteins. However, these extracellular modifications are complex and heterogeneous in structure, making them an impractical solution. In contrast, intracellular proteins are subjected to a simple version of glycosylation termed O-GlcNAc modification. In our studies of this modification, we found that O-GlcNAcylation inhibits proteolysis, and strikingly, this stabilization occurs despite large distances in primary sequence (10–15 amino acids) between the O-GlcNAc and the site of cleavage. We therefore hypothesized that this “remote stabilization” concept could be useful to engineer the stability and potentially additional properties of peptide or protein therapeutics. Here, we describe the application of O-GlcNAcylation to two clinically important peptides: glucagon-like peptide-1 (GLP-1) and the parathyroid hormone (PTH), which respectively help control glucose and calcium levels in the blood. For both peptides, we found O-GlcNAcylated analogs that are equipotent to unmodified peptide in cell-based activation assays, while several GLP-1 analogs were biased agonists relative to GLP-1. As we predicted, O-GlcNAcylation can improve the stability of both GLP-1 and PTH in serum despite the fact that the O-GlcNAc can be quite remote from characterized sites of peptide cleavage. The O-GlcNAcylated GLP-1 and PTH also displayed significantly improved in vivo activity. Finally, we employed structure-based molecular modeling and receptor mutagenesis to predict how O-GlcNAcylation can be accommodated by the receptors and the potential interactions that contribute to peptide activity. This approach demonstrates the potential of O-GlcNAcylation for generating analogs of therapeutic peptides with enhanced proteolytic stability.
Graphical Abstract
INTRODUCTION
Peptide therapeutics have recently garnered significant attention from the pharmaceutical industry due to their specificity, potency, and diminished off-target effects.1 A prime example is the case of type-2 diabetes mellitus, where the insulin receptor and the GLP-1 receptor (GLP-1R) are well-established clinical targets.2,3 In fact, extensive efforts have focused on the development of small molecule modulators of GLP-1R with little to no success. In contrast, peptide agonists have proven to be effective as therapeutic agents. In a similar fashion, the PTH receptor (PTHR1)4 has been clinically targeted by PTH(1–34), marketed by Eli Lilly as Forteo, and a peptide analog of the parathyroid hormone-related protein, approved as Tymlos from Radius Health. One critical roadblock to this strategy is the typically poor pharmacokinetic profiles of peptides in vivo including through proteolytic degradation by endogenous enzymes. For example, GLP-1(7–37) and PTH(1–34) have respective half-lives of only ~2 and 10 min in the bloodstream in vivo.5,6 Accordingly, a great deal of effort has been expended to create stable analogs of these and similar peptides, including the incorporation of unnatural amino acids, polyethylene-glycol (PEG) modification, and lipidation.7,8 While successful, stable analogs of these peptides can have drawbacks. For example, while unnatural amino acid modifications at the N-terminus of GLP-1, such as N-pyroglutamyl modification and thioamide substitution, can block its major degradation pathway through dipeptidyl peptidase (DPP-4) cleavage at alanine 8, these analogs can have reduced potency, as the N-terminal segment of GLP-1 is largely responsible for activation and signaling.9,10 Additionally, PEG and lipid modifications introduced into peptides and proteins can often be immunogenic and must be tested on an individual basis. Therefore, new approaches to therapeutic peptide stabilization that circumvent some of these issues are still of great interest.
The stability of secreted proteins can be increased through endogenous glycosylation, but these N- and mucin O-linked structures are often large and complex, making them both difficult to prepare and likely to interfere with peptide-receptor interactions. In contrast, intracellular proteins can be modified by the single monosaccharide N-acetylglucosamine, termed O-GlcNAc modification (Figure 1a).11–13 Interestingly, O-GlcNAcylation is known to inhibit protein–protein interactions and protein aggregation.14–16 During the course of our studies on O-GlcNAcylation, we found that O-GlcNAcylation of the caspase-8 activation loop can prevent its cleavage during caspase activation.17 Additionally, when analyzing the Parkinson’s disease associated protein α-synuclein, which is modified by O-GlcNAc in vivo, we found that single modifications of this protein dramatically inhibited the proteolysis of α-synuclein by the protease calpain.18 Notably, this inhibition occurred despite the fact that O-GlcNAc modifications were very distant in primary sequence (10–15 amino acids) from certain cleavage sites. Therefore, we wondered if this property of “remote stabilization,” by O-GlcNAcylation could be repurposed to engineer improved properties of clinically important peptides and proteins. Here, we test this possibility on two different peptide hormones: the glucagon-like peptide-1 [GLP-1(7–37)] and the N-terminal domain of the parathyroid hormone [PTH(1–34)].
Figure 1.
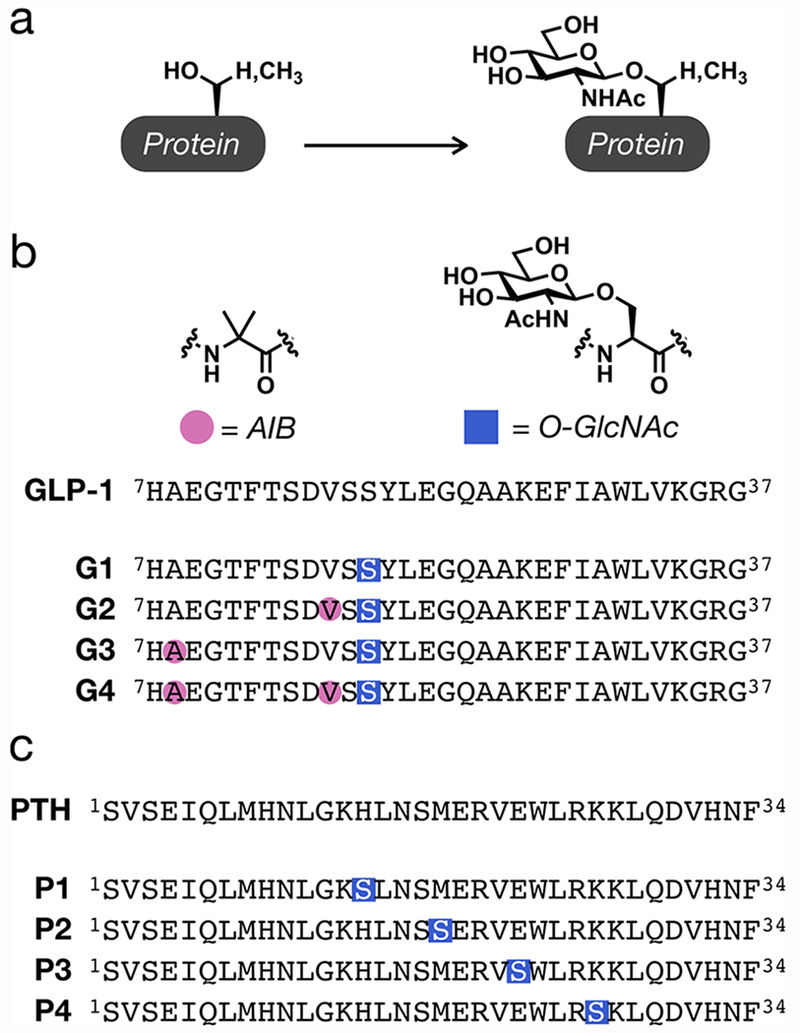
Design of O-GlcNAcylated peptides. (a) O-GlcNAcylation is an endogenous modification of serine and threonine residues of intracellular proteins. (b) Schematic of the GLP-1 sequence and peptides G1–G4, synthesized here, which contain either an O-GlcNAc modification at serine 18 or this modification in combination with AIB substitutions at residues 8 and/or 16. (c) Schematic of the PTH(1–34) sequence and peptides P1–P4 containing O-GlcNAcylated serines at residues 14, 18, 22, or 26.
To test whether O-GlcNAcylation can improve therapeutic peptides, we synthesized an analog of GLP-1(7–37) (Figure 1b), which contained a single O-GlcNAc at serine 18 with the expectation that this modification would stabilize the peptide. Additionally, we generated GLP-1 analogs containing this same O-GlcNAc modification in combination with one or two 2-aminoisobutyric acid (AIB) residues at 8 and/or 16, as they result in altered signaling profiles when combined with other modifications at serine 18.19,20 We then used a range of cell-based, in vitro, and in vivo assays to demonstrate that O-GlcNAcylation does not compromise the agonism of GLP-1R by the peptide but does indeed stabilize GLP-1 against cleavage by serum proteases, including DPP-4. Excitingly, O-GlcNAcylation also improved the in vivo activity of GLP-1 in a glucose clearance assay in mice. Consistent with previous results, the introduction of the AIB residues resulted in further stabilization of the peptide.19–21 Interestingly, we found that O-GlcNAc in combination with AIB substitutions resulted in biased agonism profiles that differed compared to the AIB substitutions alone.19,20 AIB substitutions reported by Hager et al. resulted in decreased efficacy for G-protein-mediated cAMP formation and to a lesser extent to β-arrestin recruitment, which manifested as bias toward recruitment of β-arrestins relative to G-protein-mediated cAMP signaling. In contrast, the O-GlcNAc peptides enhanced the maximal signal for cAMP signaling, while two of the peptides had reduced β-arrestin coupling. This resulted in biased agonism toward certain G-protein signaling pathways relative to recruitment of β-arrestins, when compared to GLP-1. To examine the generality of our O-GlcNAcylation approach, we also synthesized four analogs of PTH(1–34) (Figure 1c) bearing single O-GlcNAcylated serines at positions 14, 18, 22, or 26. Similar to GLP-1, O-GlcNAcylation did not adversely affect signaling through PTHR1 in cultured cells. Importantly, we found that O-GlcNAc engineering can indeed increase the serum stability of at least one PTH(1–34) analog. Additionally, as with GLP-1, we identify an O-GlcNAc analog that exhibits significantly improved in vivo activity as measured by peptide-induced increases in serum calcium levels. Finally, we used molecular modeling to rationalize how O-GlcNAc may be accommodated by the two receptors without affecting peptide agonism.
RESULTS
Design and Synthesis of O-GlcNAcylated Variants of GLP-1 and PTH.
As noted above, biochemical and structural studies have demonstrated that the N-terminal residues of GLP-1 are critical for making the appropriate interactions with the GLP-1R helical bundle that are essential for receptor activation. Nonetheless, the peptides exhibit an extended amphipathic α-helix that makes additional interactions with the extracellular domain (ECD) that are required for their high affinity.22,23 With these considerations in mind, we chose to place O-GlcNAc at serine 18 of GLP-1, as we hypothesized that this position would be solvent-exposed upon receptor binding, based on the active cryo-EM structure of GLP-1R bound to GLP-1.22 Accordingly, we prepared the suitably protected O-GlcNAcylated serine using published procedures24,25 and generated the peptides (G1–G4, Figure 1b) using standard Fmoc-based solid phase peptide synthesis. We then purified each peptide by RP-HPLC and confirmed their identities by ESI-MS (Figure S1). Unmodified GLP-1 has significant α-helical character in solution. Therefore, we next used circular dichroism (CD) spectroscopy to examine the secondary structure of all of our peptides and found no dramatic differences (Figure S2). Direct comparison of the normalized signals shows a major minimum for G1 at a slightly lower wavelength compared to GLP-1, indicating a small loss of structure, which was recovered through the addition of backbone modifications. In the case of PTH, we based our O-GlcNAcylation sites on the crystal structure of PTH(15–34) bound to the extracellular domain of PTHR1.26 More specifically, we chose to map the individual O-GlcNAc moieties along the α-helical face of PTH that points away from the extracellular domain of PTHR1, yielding peptides P1–P4 (Figure 1c). As with GLP-1, we employed Fmoc-based solid phase peptide synthesis, purified the peptides by RP-HPLC, and characterized them by ESI-MS (Figure S3). PTH also tends to form an α-helix in solution, so we again used CD spectroscopy to examine any effects of O-GlcNAcylation (Figure S4). Similar to GLP-1, modification on peptides P1 and P2 had little-to-no effect on the structure of PTH. However, peptides P3 and P4 were much more unstructured in solution with P4 having the largest degree of disorder.
O-GlcNAcylation Enhances Efficacy for Canonical Peptide-Mediated cAMP Signaling at the GLP-1R and Causes Biased Agonism in Combination with AIB.
Endogenous binding of GLP-1 to the GLP-1R results in recruitment of the Gαs G-protein, subsequently stimulating the production of cyclic AMP (cAMP) from ATP and leading to glucose-stimulated insulin secretion.27 To assess the ability of the O-GlcNAc modified GLP-1 analogs G1–G4 to activate human GLP-1R, cAMP accumulation was measured in FlpIn CHO cells stably expressing human GLP-1R.20 Cells were also treated with native GLP-1 as a reference agonist, which exhibited a pEC50 of 10.8 ± 0.1 (mean ± s.e.m., n = 4) and a maximal response of 129 ± 0.1% relative to 100 mM forskolin (mean ± s.e.m., n = 4) (Figure 2a and Table S1). O-GlcNAc peptide analogs G1–G4 showed very similar potencies that were not significantly different from unmodified GLP-1. Notably all four ligands displayed higher maximal responses compared to unmodified GLP-1, although only G1 and G3 were statistically significant (P < 0.05) (Table S1). We next measured the ability of peptides G1–G4 to promote intracellular calcium mobilization, an established assay that reports on Gαq activation by GLP-1R.20,28 In this assay, recruitment of Gαq activates phospholipase Cβ (PLCβ), catalyzing the formation of inositol-1,4,5-trisphosphate (IP3) that can bind and open the IP3-gated calcium channel, increasing calcium concentration in the cytoplasm.27 In this assay (Figure 2b and Table S1), the O-GlcNAcylated peptide analogs G1 and G2 showed comparable potencies and maximum response relative to GLP-1. In contrast, analogs G3 and G4 displayed lower maximal response, but with similar potencies, indicating that G3 and G4 could be biased agonists. Operational modeling of the concentration response data was performed to determine transduction ratios that, when compared between pathways and to the reference agonist, revealed a small degree of bias that did not reach statistical significance (Figure S5 and Table S2).
Figure 2.
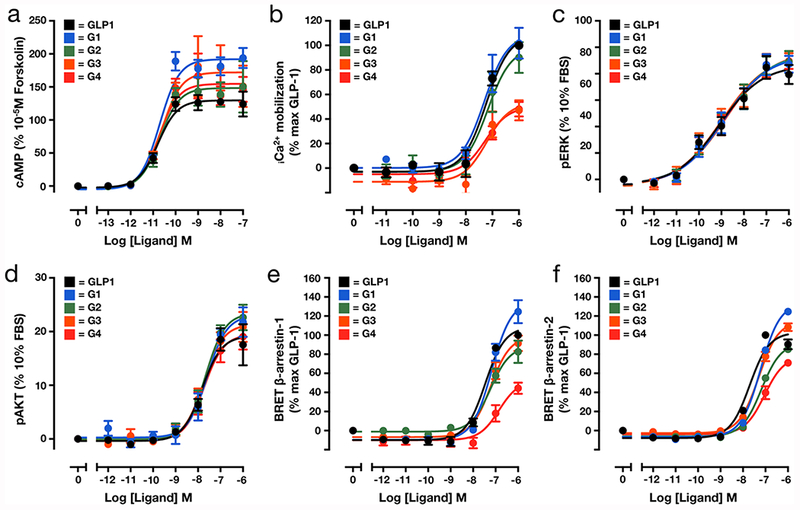
Cell-based characterization of GLP-1 peptide analogs. (a) O-GlcNAcylation alone or with AIB substitution does not affect the EC50 of cAMP production when compared to unmodified GLP-1 (n = 4). (b) O-GlcNAcylation of GLP-1 alone does not alter Ca2+ accumulation (n = 4), but in combination with AIB at residue 4 attenuated signaling. (c,d) O-GlcNAcylation alone or in combination with AIB substitution does not affect phosphorylation of ERK (c) (n = 4) or AKT (d) (n = 4), measured by peak response. (e,f) β-Arrestin-1 (e) or β-Arrestin-2 (f) recruitment to GLP-1R is unaffected by O-GlcNAcylation but is reduced by O-GlcNAcylation in combination with two AIB substitutions (n = 4). All experiments were performed in CHO cells expressing the human GLP-1R. Quantitative analysis of the data is presented in Table S1.
In addition to these G-protein coupled pathways, GLP-1 activates kinases through both canonical and noncanonical pathways; therefore, analogues G1–G4 were assessed for their ability to promote ERK1/2 and AKT phosphorylation.29,30 As expected, unmodified GLP-1 resulted in concentration-dependent phosphorylation/activation of both ERK and AKT (Figure 2c,d and Tables S1 and S2). In these assays, all of the O-GlcNAcylated analogs induced phosphorylation of both kinases in a manner that was indistinguishable from the unmodified peptide (Figure 2c,d and Table S1). Like most GPCRs, GLP-1R activation can also result in recruitment of β-arrestin proteins that play a central role in receptor desensitization31 and initiation of alternate signaling cascades. To evaluate β-arrestin recruitment, an established bioluminescence resonance energy transfer (BRET) assay in FlpInCHO cells transiently expressing GLP-1R-Rluc and β-arrestin-Venus was used.32 In the case of β-Arrestin-1, G1, G2, and G3 of the O-GlcNAc modified peptides displayed comparable potencies to GLP-1; however, G4 potency was significantly reduced (Figure 2e and Table S1). In addition, there was a selective attenuation of the Emax for the G4 analogue, while for the G1 analogue, there was a small but statistically significant increase in maximal response (Table S1). In contrast, we observed a statistically significant shift in EC50 for the recruitment of β-Arrestin-2 by each of the peptides G1–G4 (Figure 2f and Table S1). For β-Arrestin-2 recruitment, there was again an attenuated response for G4 that manifests as both a reduction in Emax and potency (Figure 2f and Table S1). Similar to the observation for β-Arrestin-1, the G1 peptide resulted in higher maximal recruitment of β-Arrestin-2 compared to GLP-1 (Figure 2f and Table S1). We previously showed that AIB substitution at both residues 8 and 16 can cause a modest bias toward arrestin recruitment relative to cAMP when compared to GLP-1.20 However, here, using operational modeling to calculate bias factors, biased agonism was observed in the opposite direction (Figure S5 and Table S2), biasing the peptide responses away from arrestin recruitment relative to cAMP for G4, indicating that the O-GlcNAc modification could contribute to the direction and extent of bias that is manifested. Interestingly, a similar pattern of bias relative to cAMP was observed for G2 for β-Arrestin-2 but not β-Arrestin-1.
O-GlcNAc Modification Stabilizes GLP-1 from Degradation in Serum and Improves Its Function in Vivo.
To complement our cell-based characterization of the GLP-1 analogs G1–G4, we next tested our original hypothesis that O-GlcNAcylation would improve the stability of GLP-1 in the presence of serum proteases ex vivo. Accordingly, unmodified GLP-1, G1, or G4 were incubated in human serum for 24 h (n = 3 for each peptide). After different lengths of time, aliquots were removed and analyzed by RP-HPLC to measure the remaining full-length peptide (Figure 3a and Figure S6).33 These data were then fit to an exponential decay curve to calculate the peptide half-lives (Figure 3a). Consistent with our underlying hypothesis, the half-life of peptide G1 was twice that of unmodified GLP-1, while the incorporation of AIB subsitutions in G4 further increased this stability increasing the half-life by 7-fold relative to GLP-1. Interestingly, we could identify the major DPP-4 cleavage product of GLP-1 and G1 (residue 8 in both peptides) by mass spectrometry (see marked HPLC peak in Figure S6) and found that O-GlcNAcylation appears to inhibit this proteolysis event despite the fact that this modification is 10 amino acids away from the DPP-4 cleavage site in the primary sequence. Given this promising in vitro data, we next tested our peptides in vivo using a glucose clearance assay or glucose tolerance test (GTT).34 More specifically, after fasting, mice were injected intraperitoneally (IP) with 1 mg/kg of glucose and either vehicle (n = 11), GLP-1 (n = 14), or one of our O-GlcNAcylated analogs (n = 5 per group). After different lengths of time, tail blood-glucose levels were measured (Figure 3b,c). In this acute GTT, all of the modified peptides (G1–G4) displayed significantly improved glucose disposal efficiency compared to unmodified GLP-1. These data demonstrate that O-GlcNAc modification results in improved in vivo activity in a mouse model of glucose disposal.
Figure 3.
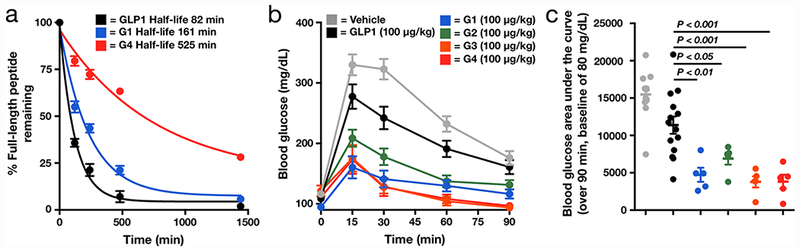
O-GlcNAcylation stabilizes GLP-1 from proteolysis and improves glucose clearance in vivo. (a) O-GlcNAc modification improves the stability of GLP-1. The indicated peptides (n = 3) were incubated with human serum for 24 h. The stability of each peptide was measured using RP-HPLC after the indicated lengths of time. (b) O-GlcNAcylation of GLP-1 improves glucose clearance in a glucose tolerance test. Lean mice were subjected to a single intraperitoneal coinjection of vehicle (n = 11) or the indicated concentrations of peptide (GLP1, n = 14; G1–G4, n = 5) with glucose challenge. Blood glucose levels were then measured after the indicated lengths of time. (c) Blood glucose area under the curve (AUC) calculated from the data in b over 90 min with a baseline of 80 mg/dL. Error bars represent ± s.e.m., and statistical significance of differences in AUC from GLP-1 was calculated using one-way ANOVA followed by Dunnett’s post-test.
O-GlcNAcylation Analogs of PTH Maintain Canonical cAMP Signaling.
Much like GLP-1R, activation of PTHR1 can result in the recruitment of Gαs that promotes cAMP production and associated downstream signaling events in bone and the kidneys to regulate blood calcium levels. To test the ability of our PTH analogs (n = 4) to stimulate cAMP production, we used a HEK-293 cell line, GP-2.3, which stably overexpresses the human PTHR1 and a cAMP sensor (Figure 4a and Table S3), as well as a human osteoblastic cell line (SGS-72) that endogenously expresses PTHR1 (Figure 4b and Table S3). Overall, there was limited impact of the O-GlcNAcylation modification on PTH-mediated cAMP responses, with only small reductions in potency observed, in a peptide- and cell-specific manner. However, similar to what we observed with GLP-1 modifications, the most robust effect was an enhancement of Emax, particularly in the case of the GP-2.3 model.
Figure 4.
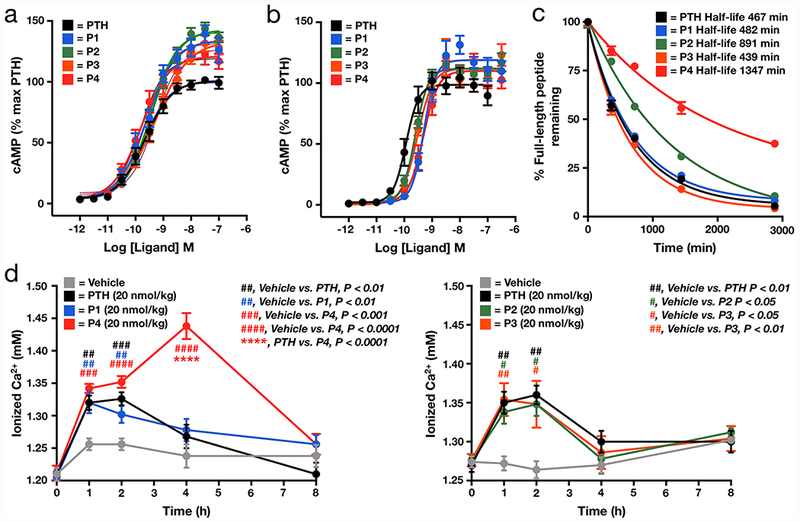
Analysis of O-GlcNAcylated PTH-analogs. (a,b) O-GlcNAcylation cAMP production when compared to unmodified PTH in cells that either overexpress (a, GP-2.3) or endogenously express (b, SGS-72) PTHR1 (n = 4 for each peptide). (c) O-GlcNAc modification improves the stability of PTH. Unmodified PTH or the O-GlcNAcylated analogs (n = 3) were incubated with human serum for 48 h. The stability of each peptide was measured using RP-HPLC after the indicated lengths of time. (d) O-GlcNAcylation of PTH can site-selectively increase serum Ca2+ levels compared to PTH in vivo. Mice (n = 5) were subjected to a single subcutaneous injection of vehicle or the indicated concentrations of PTH or the indicated O-GlcNacylated analog. Blood Ca2+ levels were then measured after the indicated lengths of time. Error bars represent ± s.e.m., and statistical significance was calculated using one-way ANOVA with a Dunnett post-test.
O-GlcNAc Modification Stabilizes PTH from Degradation in Serum and Improves Its Function in Vivo.
To determine if the O-GlcNAc modified PTH analogs could also enhance serum stability, we separately incubated either PTH or the O-GlcNAcylated analogs with human serum for 48 h (n = 3 for each peptide) ex vivo. As with the GLP-1 peptides, aliquots of the reaction mixture were removed after different lengths of time and analyzed by RP-HPLC (Figure S7) and peptide half-lives were calculated (Figure 4c). We found that P1 and P3 had essentially the same stability as PTH in serum. However, P2 and P4 were approximately 2- and 3-times, respectively, as stable as unmodified PTH. We next tested the peptides in vivo measuring blood Ca2+ levels in mice, which increase upon PTH administration through release of calcium from bone. Specifically, either PTH or O-GlcNAcylated peptides were injected subcutaneously into the tail vein at a dose of 20 nmol/kg (n = 5 per group). In this case, we split the peptides into two sets with independent controls due to experimental limitations with the number of mice that can be tested simultaneously. We then analyzed the ionizable Ca2+ levels from blood that was withdrawn immediately before injection and at various time points after injection (Figure 4d). As expected, we observed an increase in blood Ca2+ levels upon administration of PTH. This increase was indistinguishable from the amounts of Ca2+ mobilized by peptides P1, P2, or P3. In contrast, we found enhanced/prolonged blood Ca2+ levels following P4 administration, consistent with its improved stability compared to all of the other peptides in serum. These results suggest that the stability and improved in vivo activity conferred by O-GlcNAc is context and position dependent but also confirm that our strategy is transferable to multiple therapeutically relevant peptides.
Structure-Based Modeling and Receptor Mutagenesis Experiments Indicate the Molecular Determinants of O-GlcNAcylated Peptide Binding.
In order to gain molecular insight into how O-GlcNAcylation might affect the binding of GLP-1 and PTH, we performed conformational modeling of the corresponding ligand–receptor complexes, as described in the experimental methods. The GLP-1R peptide binding models were based on Cryo-EM structure of GLP-1R in complex with unmodified GLP-1 peptide.22 The generated models of G1 and G4 in their complexes with GLP-1R suggest that the O-GlcNAc moiety introduced at S18 is accommodated in the shallow cleft located between the extracellular loops ECL2 and ECL3 of the 7 transmembrane (7TM) domain and ECD of GLP-1R (Figures 5a and 6a). The energy optimized conformation of the sugar shows a potential hydrophobic interaction between O-GlcNAc and L142, as well as a potential hydrogen bond between N300 of the receptor ECL2. Additionally, O-GlcNAc can potentially form an internal hydrogen bond with the side chain of D15, which is further stabilized by a salt bridge with R376 and R380. In the native GLP-1R-GLP-1 bound cryo-EM structure the Asp15 conformation is stabilized by R380 of ECL3, and this salt bridge is potentially maintained in O-GlcNAc peptide model (Figure 5b).
Figure 5.
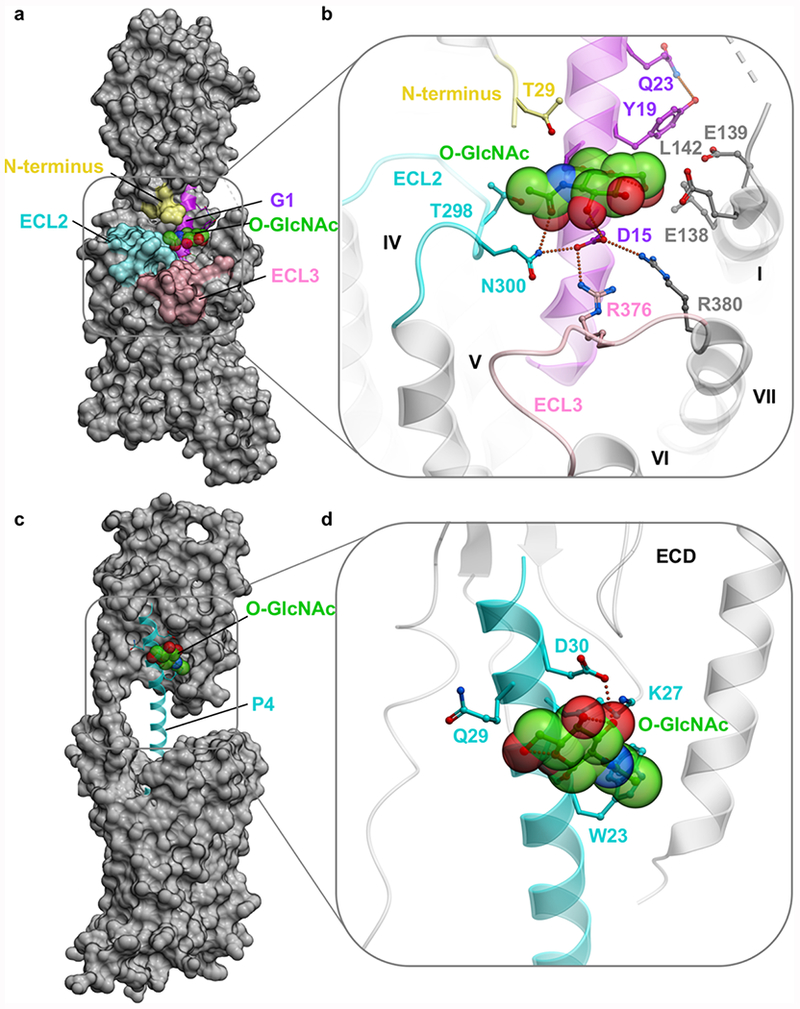
Models of full length GLP-1R-G1 and PTH1R-P2 complexes. (a) Overview of the G1-GLP-1R represented as a molecular surface. (b) Close up showing predicted stabilizing interactions of the O-GlcNAc moiety. In both panels, GLP-1R is shown as gray, except for ECL2 (cyan), ECL3 (pink), and truncated N-terminus (yellow). G1 peptide is represented as a ribbon (magenta), where the O-GlcNAc moiety is highlighted as transparent spheres with carbon atoms colored green. Predicted intermolecular hydrogen bonds and salt bridges are shown by green dotted lines. (c) Overview of the P4-PTH1R represented as a molecular surface. (d) Close up showing predicted stabilizing interactions of the O-GlcNAc moiety. In both panels, PTH1R is shown as gray. P4 peptide is represented as ribbon (cyan), where the O-GlcNAc moiety is highlighted as transparent spheres with carbon atoms colored green.
Figure 6.
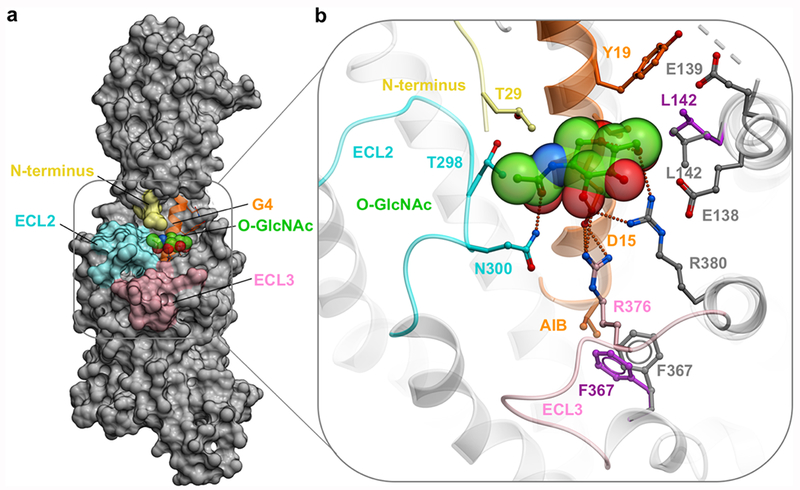
Overview of the GLP-1R-G4 complex. (a) Overview of the GLP-1R-G4, molecular surface of GLP-1R (gray), ECL2 (cyan), ECL3 (pink), ECD far N-terminus (yellow), G4 is represented as a ribbon (orange), with the O-GlcNAc moiety represented as spheres (green). (b) Mutation of V16 into AIB16 alters the side chain position of L142 residue (gray); initial conformation of L142side chain (magenta). Mutation of A8 into AIB8 alters the side chain position of F367 (gray); initial conformation of F367 side chain (magenta).
This model is consistent with our experimental data, where the O-GlcNAcylation of S18 is accommodated by GLP-1R and does not inhibit potency of our GLP-1 peptide analogs in the cell-based signaling assays (Figure 2 and Table S1). Moreover, mutation of key residues within this pocket alter the potency and/or maximum response of GLP-1, G1, and G4 peptides when assessed in both cAMP and pERK1/2 assays, albeit there are distinctions in the extent of these effects when comparing between cAMP and pERK1/2 and between ligands (Figure S8). For example, L142A reduces potency for all ligands in cAMP but has more limited effect in pERK1/2, whereas R376AECL3 had a greater effect of pERK1/2 potency relative to cAMP (Figure S8 and Table S4). Ala mutation of N300ECL2 heavily impaired cAMP signaling mediated by all three peptides, but G4 was more sensitive to this mutation with a maximal response about half of GLP-1. Ala mutation of N300ECL2 also reduced pERK1/2 signaling by all three peptides; however, G1 and G4 were most affected by this mutation with maximal responses again about half of GLP-1. Similarly, Ala mutation of R380ECL3 heavily impaired cAMP signaling mediated by all three peptides; however, both G1 and G4 show reduced maximal responses, with G4 being having the most pronounced affect. For pERK1/2, potency was impacted for all three peptides, but maximal response was only slightly reduced for G1 (Figure S8 and Table S4). These results are consistent with the additional interactions predicted by the modeling between the sugar of the O-GlcNAc peptides and receptor residues N300, R376, and R380 (Figures 5b and 6b).
In the case of PTH, we based our model on the high resolution crystal structure of the full length PTH1R in complex with ePTH, a peptide agonist with 79% sequence homology to PTH (2.5 Å).35 We generated a model of P4 in complex with PTH1R by introducing an O-GlcNAcylated serine at Lys26 of PTH and found that this moiety is likely accommodated in the proximity of the ECD, but without making any contacts with the receptor, and is mostly solvent exposed (Figure 5c,d). The O-GlcNAc may form a hydrogen bond interaction with the D30 side chain residue of P4, which could potentially increase its rigidity when bound to the receptor and explain the observed boost in maximal signaling (Figure 4 and Table S3).
Modeling of Peptide GLP-1R Peptide G4 Suggests a Potential Mechanism of Biased Signaling.
As reported above, the introduction of both O-GlcNAcylation on Ser18 and an AIB residue at position 8 of GLP-1 resulted in a biased agonist (G4 in Figure S5 and Table S2). To investigate molecular mechanism of this effect, we analyzed the corresponding structure-based model of the GLP-1R-G4 complex. In this energy-optimized model, introduction of the additional methyl moiety of AIB8 impinges on the receptor F367 phenyl ring, resulting in a dramatic shift of this side-chain (Figure 6b). We suggest that this shift could result in a rotation of the helix VI backbone, where conformational changes can result in signaling bias. Consistent with this hypothesis, our recent Cryo-EM structure with another biased agonist, Exendin-P5,33 shows a similar outward shift and rotation of residue F367 and the tip of helix VI (PDB ID 6B3J) and Exendin-P5 has a bulky residue (V8) in the same position as AIB8 in G4. This analysis suggests that interactions of the N-terminal residues of ligands with helix VI might be a factor in G-protein bias through the GLP-1R. As noted above, AIB modifications without the O-GlcNAcylation induce distinct and more subtle bias.19,20 This could be explained by some flexibility in the GLP-1 peptide, which can absorb the introduced strain of the ligand, helix VIinteraction. In contrast, the snug fit of the O-GlcNAc of G4 into the binding pocket on the interface of 7TM and ECD domains of GLP-1R is likely to increase the ligand–receptor complex rigidity. In other words, AIB8 may create a lever for inducing the conformational change in helix VI, while the O-GlcNAc and AIB16 could enhance the foothold for this lever and make it more rigid. As with the G1 and P4 peptides described above, this model accommodates our cellular data, although direct structural or biochemical information will be required to validate the mechanism of G4 biased agonism relative to native GLP-1. This is a goal we are also currently exploring.
DISCUSSION
Inspired by our experiments showing that O-GlcNAcylation can inhibit caspase-8 activation and α-synuclein proteolysis,17,18 we find here that this property is portable to increase the serum stability of at least two peptide hormones (GLP-1 and PTH) and may be generalizable. More specifically, we first find that modification of GLP-1 at S18 (peptide G1) has no detrimental effects on the ability of GLP-1 to engage its receptor in living cells and initiate physiologically relevant signaling pathways (Figure 2 and Table S1). In fact, the O-GlcNAc appears capable of generating higher cAMP signaling in certain contexts (G1 or G3, Figure 2 and Table S1). However, this monosaccharide does indeed slow the degradation of GLP-1 in human serum in vitro by 2-fold compared to the unmodified peptide (Figure 3a) and significantly improves glucose disposal in vivo (Figure 3b). Remarkably, the increased stability of GLP-1 in vitro extends to the major cleavage of the peptide at residue 8 by the protease DDP-4 (Figure S6) that is 10 amino acids away from the O-GlcNAc modification. In addition to this single modification of GLP-1 by O-GlcNAc, we also explored the potential for additional effects when glycosylation was combined with backbone modifications. Given the dramatic stabilization of GLP-1 by AIB substitution,19–21 we do not believe that O-GlcNAc would significantly increase the peptide stability further, but two AIB substitutions at residues 8 and 16 of G1, yielding G4, resulted in biased agonism toward cAMP accumulation and away from arrestin recruitment (Figures 2 and S5 and Table S2). There was also a trend toward bias away from calcium mobilization. This further supports previous data showing altered signaling when AIB at positions 8 and 16 was combined with other backbone modifications at residue 18. Next, we tested whether an unrelated peptide hormone, PTH, could also be stabilized by O-GlcNAcylation. Gratifyingly, we found that modification of PTH can increase the serum half-life of PTH in vitro, while not inhibiting the ability of the peptide to agonize the corresponding receptor in cells (Figure 4). Additionally, the peptide most stabilized by O-GlcNAc (P4) gave an increased in vivo response measured by increased blood Ca2+ levels compared to unmodified PTH (Figure 4d).
Molecular modeling was also used to gain insight into how O-GlcNAcylation could be accommodated by the GLP-1 and PTH receptors. In the case of both G1 and P4, our models (Figure 5) illustrate that the O-GlcNAc modifications could be easily accommodated in the receptor-peptide complexes, supporting our data reporting minimal impact on canonical receptor signaling in cell lines (Figures 2 and 4). While in GLP-1 receptor, O-GlcNAc moiety is partially solvent exposed, and additional potential steric and polar contacts form with the modification indicating that O-GlcNAcylation may directly participate in receptor interactions. In the PTH receptor model, however, the O-GlcNAc moiety is fully exposed to the solvent without making contacts with the receptor itself. Additional potential polar interactions can be formed with the peptide itself, suggesting that rigidifying the peptide with O-GlcNAc modification may be sufficient to improve the potency for PTH. Importantly, our analysis of GLP-1R mutants of the residues that participate in these predicted interactions experimentally support this model (Figure S8). Additionally, we believe that O-GlcNAc-receptor interactions could be exploited to discover new types of ligands. Our cellular data (Figure 2) and model (Figure 6) of the biased peptide G4 in complex with GLP-1R support this hypothesis. Specifically, we believe that the positioning of the O-GlcNAc in the cleft of GLP-1R may rigidify the peptide with respect to the receptor and allow the AIB substitution at position 8 to alter interactions with helix VI, contributing to biased agonism. These models need to be confirmed through further structural and/or biochemical experiments but together with our cellular data suggest that O-GlcNAc may participate in direct interactions with receptors and affect changes to the downstream signaling pathways.
Like all methods employed to stabilize peptide hormones (artificial amino acids, polyethylene glycol, etc.), we do not expect that O-GlcNAcylation will be a panacea for every peptide or protein of interest. We also do not yet understand the exact mechanism of stabilization. For example, our previous data show that O-GlcNAc can block calpain mediated cleavage at positions remote from the site of modification.18 However, a wide range of proteomics experiments aimed at identifying endogenous O-GlcNAcylation sites suggest that the sugar does not block the activity of trypsin or Lys-C. Additionally, we have shown that O-GlcNAcylation of α-synuclein does not block proteinase K, a broad-spectrum enzyme.16 Furthermore, the stability and increased in vivo activity conferred to PTH by O-GlcNAc is site-selective (Figure 4c,d) and does not correlate with the degree of structural perturbation in solution (Figure S4). Taken together, these data show that O-GlcNAc mediated stabilization will be context-dependent on both the modified peptide and the principal pathways/enzymes of degradation. We are currently investigating these parameters in more detail and the potential that our observed stability in serum could be due to increased interactions with proteins rather than direct inhibition of protease recognition.
Despite these areas of future investigation, we believe that our data strongly suggests that this modification strategy should be a part of the therapeutic peptide toolkit. In particular, we predict that the “remote stabilization” property of O-GlcNAc will make it significantly more flexible in its application than other strategies that target the specific site of proteolysis. For example, the N-terminus of GLP-1 is essential for signaling, and previous strategies that directly stabilize the amide bond at residue 810,36 or use larger hydrophilic groups37 can result in reduced signaling potency. O-GlcNAcylation could be employed in other such peptides or proteins at sites away from the proteolytically labile residues that are also key for receptor binding, since the modification might be placed in a solvent-exposed position distant to such sites. Additionally, our discovery of biased GLP-1R agonists indicates that O-GlcNAc might be readily combined with other strategies, including other natural and unnatural amino acids, to further improve the clinical properties of peptides. Importantly, because O-GlcNAc is a native modification of intracellular proteins and certain extracellular receptors, we speculate that it will have a low potential to elicit immune responses. In summary, we anticipate that O-GlcNAcylation will be a generalizable strategy to slow the proteolysis and degradation of known and potentially undiscovered signaling peptides for both biological studies and therapeutic applications.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (R01GM114537 to M.R.P and P01-DK11794 and P30-AR066261 to T.J.G.), the University of Southern California, The Scripps Research Institute, and the National Health and Medical Research Council of Australia (NHMRC) (1157539 and 1126857 to D.W. and 1150083 to P.M.S.). Circular dichroism was performed at the USC Nano Biophysics Core Facility. P.M.S. is a NHMRC Senior Principal Research Fellow, and D.W. is a NHMRC Senior Research Fellow.
Footnotes
Supporting Information
Materials and methods, supporting figures, and data tables. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b05365.
Characterization of unmodified and O-GlcNAcylated GLP-1 and PTH analogs, EC50 and maximal activity data, web of bias illustrating distinctions in the pattern of signaling of peptides, bias agonism, and effect of alanine mutations on cAMP and pERK1/2 mediated signaling from GLP-1Rs (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Henninot A; Collins JC; Nuss JM The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem 2018, 61 (4), 1382–1414. [DOI] [PubMed] [Google Scholar]
- (2).Trujillo JM; Nuffer W; Ellis SL GLP-1 Receptor Agonists: a Review of Head-to-Head Clinical Studies. Ther. Adv. Endocrinol. Metab 2015, 6 (1), 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zaykov AN; Mayer JP; DiMarchi RD Pursuit of a Perfect Insulin. Nat. Rev. Drug Discovery 2016, 15 (6), 425–439. [DOI] [PubMed] [Google Scholar]
- (4).Cheloha RW; Gellman SH; Vilardaga J-P; Gardella TJ PTH Receptor-1 Signalling-Mechanistic Insights and Therapeutic Prospects. Nat. Rev. Endocrinol 2015, 11 (12), 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lovshin JA; Drucker DJ Incretin-Based Therapies for Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol 2009, 5 (5), 262–269. [DOI] [PubMed] [Google Scholar]
- (6).Fraher LJ; Klein K; Marier R; Freeman D; Hendy GN; Goltzman D; Hodsman AB Comparison of the Pharmacokinetics of Parenteral Parathyroid Hormone-(1–34) [PTH-(1–34)] and PTH-Related Peptide-(1–34) in Healthy Young Humans. J. Clin. Endocrinol. Metab 1995, 80 (1), 60–64. [DOI] [PubMed] [Google Scholar]
- (7).Sato AK; Viswanathan M; Kent RB; Wood CR Therapeutic Peptides: Technological Advances Driving Peptides Into Development. Curr. Opin. Biotechnol 2006, 17 (6), 638–642. [DOI] [PubMed] [Google Scholar]
- (8).Lau JL; Dunn MK Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem 2018, 26 (10), 2700–2707. [DOI] [PubMed] [Google Scholar]
- (9).Green BD; Mooney MH; Gault VA; Irwin N; Bailey CJ; Harriott P; Greer B; O’Harte FPM; Flatt P R N-Terminal His(7)-Modification of Glucagon-Like Peptide-1(7–36) Amide Generates Dipeptidyl Peptidase IV-Stable Analogues with Potent Antihyperglycaemic Activity. J. Endocrinol 2004, 180 (3), 379–388. [DOI] [PubMed] [Google Scholar]
- (10).Chen X; Mietlicki-Baase EG; Barrett TM; McGrath LE; Koch-Laskowski K; Ferrie JJ; Hayes MR; Petersson EJ Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones. J. Am. Chem. Soc 2017, 139 (46), 16688–16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bond MR; Hanover JA A Little Sugar Goes a Long Way: the Cell Biology of O-GlcNAc. J. Cell Biol 2015, 208 (7), 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yang X; Qian K Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol 2017, 18 (7), 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hart GW Nutrient Regulation of Signaling and Transcription. J. Biol. Chem 2019, 294 (7), 2211–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rexach JE; Clark PM; Mason DE; Neve RL; Peters EC; Hsieh-Wilson LC Dynamic O-GlcNAc Modification Regulates CREB-Mediated Gene Expression and Memory Formation. Nat. Chem. Biol 2012, 8 (3), 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marotta NP; Lin YH; Lewis YE; Ambroso MR; Zaro BW; Roth MT; Arnold DB; Langen R; Pratt MR O-GlcNAc Modification Blocks the Aggregation and Toxicity of the Protein A-Synuclein Associated with Parkinson’s Disease. Nat. Chem 2015, 7 (11), 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Levine PM; Galesic A; Balana AT; Mahul-Mellier A-L; Navarro MX; De Leon CA; Lashuel HA; Pratt MR A-Synuclein O-GlcNAcylation Alters Aggregation and Toxicity, Revealing Certain Residues as Potential Inhibitors of Parkinson’s Disease. Proc. Natl. Acad. Sci U. S. A 2019, 116 (5), 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chuh KN; Batt AR; Zaro BW; Darabedian N; Marotta NP; Brennan CK; Amirhekmat A; Pratt MR The New Chemical Reporter 6-Alkynyl-6-Deoxy-GlcNAc Reveals O-GlcNAc Modification of the Apoptotic Caspases That Can Block the Cleavage/Activation of Caspase-8. J. Am. Chem. Soc 2017, 139 (23), 7872–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Levine PM; De Leon CA; Galesic A; Balana A; Marotta NP; Lewis YE; Pratt MR O-GlcNAc Modification Inhibits the Calpain-Mediated Cleavage of A-Synuclein. Bioorg. Med. Chem 2017, 25 (18), 4977–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hager MV; Johnson LM; Wootten D; Sexton PM; Gellman SH B-Arrestin-Biased Agonists of the GLP-1 Receptor From B-Amino Acid Residue Incorporation Into GLP-1 Analogues. J. Am. Chem. Soc 2016, 138 (45), 14970–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hager MV; Clydesdale L; Gellman SH; Sexton PM; Wootten D Characterization of Signal Bias at the GLP-1 Receptor Induced by Backbone Modification of GLP-1. Biochem. Pharmacol 2017, 136, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Deacon CF; Knudsen LB; Madsen K; Wiberg FC; Jacobsen O; Holst JJ Dipeptidyl Peptidase IV Resistant Analogues of Glucagon-Like Peptide-1 Which Have Extended Metabolic Stability and Improved Biological Activity. Diabetologia 1998, 41 (3), 271–278. [DOI] [PubMed] [Google Scholar]
- (22).Zhang Y; Sun B; Feng D; Hu H; Chu M; Qu Q; Tarrasch JT; Li S; Sun Kobilka T; Kobilka BK; Skiniotis G Cryo-EM Structure of the Activated GLP-1 Receptor in Complex with a G Protein. Nature 2017, 546 (7657), 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liang Y-L; Khoshouei M; Glukhova A; Furness SGB; Zhao P; Clydesdale L; Koole C; Truong TT; Thal DM; Lei S; Radjainia M; Danev R; Baumeister W; Wang M-W; Miller LJ; Christopoulos A; Sexton PM; Wootten D Phase-Plate Cryo-EM Structure of a Biased Agonist-Bound Human GLP-1 Receptor-Gs Complex. Nature 2018, 555 (7694), 121–125. [DOI] [PubMed] [Google Scholar]
- (24).Marotta NP; Cherwien CA; Abeywardana T; Pratt MR O-GlcNAc Modification Prevents Peptide-Dependent Acceleration of A-Synuclein Aggregation. ChemBioChem 2012, 13 (18), 2665–2670. [DOI] [PubMed] [Google Scholar]
- (25).De Leon CA; Lang G; Saavedra MI; Pratt MR Simple and Efficient Preparation of O- and S-GlcNAcylated Amino Acids Through InBr3-Catalyzed Synthesis of B-N-Acetylglycosides From Commercially Available Reagents. Org. Lett 2018, 20 (16), 5032–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pioszak AA; Xu HE Molecular Recognition of Parathyroid Hormone by Its G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (13), 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang R; Xie X Tools for GPCR Drug Discovery. Acta Pharmacol. Sin 2012, 33 (3), 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Werry TD; Gregory KJ; Sexton PM; Christopoulos A Characterization of Serotonin 5-HT2C Receptor Signaling to Extracellular Signal-Regulated Kinases 1 and 2. J. Neurochem 2005, 93 (6), 1603–1615. [DOI] [PubMed] [Google Scholar]
- (29).Sonoda N; Imamura T; Yoshizaki T; Babendure JL; Lu J-C; Olefsky JM Beta-Arrestin-1 Mediates Glucagon-Like Peptide-1 Signaling to Insulin Secretion in Cultured Pancreatic Beta Cells. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (18), 6614–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Quoyer J; Longuet C; Broca C; Linck N; Costes S; Varin E; Bockaert J; Bertrand G; Dalle S GLP-1 Mediates Antiapoptotic Effect by Phosphorylating Bad Through a Beta-Arrestin 1-Mediated ERK1/2 Activation in Pancreatic Beta-Cells. J. Biol. Chem 2010, 285 (3), 1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lefkowitz RJ; Shenoy SK Transduction of Receptor Signals by Beta-Arrestins. Science 2005, 308 (5721), 512–517. [DOI] [PubMed] [Google Scholar]
- (32).Wootten D; Savage EE; Willard FS; Bueno AB; Sloop KW; Christopoulos A; Sexton PM Differential Activation and Modulation of the Glucagon-Like Peptide-1 Receptor by Small Molecule Ligands. Mol. Pharmacol 2013, 83 (4), 822–834. [DOI] [PubMed] [Google Scholar]
- (33).Jenssen H; Aspmo SI Serum Stability of Peptides. Methods Mol. Biol 2008, 494, 177–186. [DOI] [PubMed] [Google Scholar]
- (34).Zhang H; Sturchler E; Zhu J; Nieto A; Cistrone PA; Xie J; He L; Yea K; Jones T; Turn R; Di Stefano PS; Griffin PR; Dawson PE; McDonald PH; Lerner RA Autocrine Selection of a GLP-1R G-Protein Biased Agonist with Potent Antidiabetic Effects. Nat. Commun 2015, 6, 8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ehrenmann J; Schöppe J; Klenk C; Rappas M; Kummer L ; Doré AS; Plückthun A High-Resolution Crystal Structure of Parathyroid Hormone 1 Receptor in Complex with a Peptide Agonist. Nat. Struct. Mol. Biol 2018, 25 (12), 1086–1092. [DOI] [PubMed] [Google Scholar]
- (36).Meng H; Krishnaji ST; Beinborn M; Kumar K Influence of Selective Fluorination on the Biological Activity and Proteolytic Stability of Glucagon-Like Peptide-1. J. Med. Chem 2008, 51 (22), 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Selis F; Schrepfer R; Sanna R; Scaramuzza S; Tonon G; Dedoni S; Onali P; Orsini G; Genovese S Enzymatic Mono-Pegylation of Glucagon-Like Peptide 1 Towards Long Lasting Treatment of Type 2 Diabetes. Results Pharma Sci. 2012, 2, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.