

Abstract
The work presented below is related to our companion paper in this issue, entitled: Substance P in solution: trans-to-cis configurational changes of penultimate prolines initiate non-enzymatic peptide bond cleavages. Two-dimensional ion mobility spectrometry (IMS-IMS) and mass spectrometry techniques are used to investigate structural transitions for [M+3H]3+ ions of substance P (subP) upon collisional activation (CA) in the gas phase. In this approach, different conformations of ions having a specified mobility are selected after an initial IMS separation, collisionally activated to produce new conformers, and these product structures are separated again using a second IMS region. In this way it is possible to follow folding and unfolding transitions of different conformations. The analysis shows evidence for five conformations. Unlike other systems, every transition is irreversible. Studies as a function of activation voltage are used to discern pathways of structural changes prior to reaching the energy required for dissociation. Thresholds associated with the onsets of transitions are calibrated to obtain estimates of the energetic barriers between different structures and semi-quantitative potential-energy diagrams are presented. Overall, barriers associated with structural transitions of [subP+3H]3+ in the absence of solvent are on the order of ~40 kJ∙mol−1, substantially lower than the ~90 kJ∙mol−1 required for some similar structural transitions in solutions of ethanol. Comparisons of the transition energies in the gas-phase with thermochemistry for similar transitions in solution provide clues about why reverse transitions are prohibited.
Introduction
It is fair to ask the question: why would anyone want to study the conformations of biomolecules in a vacuum? After all, these molecules rarely find themselves completely stripped of solvent at pressures found in the upper atmosphere. And, such studies are not easy – requiring complex instrumentation that often needs to be designed and constructed in house. Moreover, today, with ~137,000 entries in the protein data bank, much is known about the nearly 1400 unique folds that describe native structures.[1, 2] So much so, that a machine-learning approach developed by the Google subsidiary DeepMind won the 2018 CASP13 protein-folding competition, with the most accurate predictions of 25 of 43 unknown structures; the nearest competitive approach was most accurate for only three sequences.[3]
The ability to predict native structures from primary sequences is a major advance that builds on more than a half century of experimental measurements.[4–8] One might imagine that the protein-folding problem is largely solved. But, native structures are only a part of this problem. Proteins sample many other non-native conformations as they are synthesized, modified, and transported through new environments.[9] Little is known about these states. Non-native conformations may or may not function in the same way, or with the same efficiency, as native structures;[10, 11] but they are critical to living systems. In order to maintain proteostasis, denatured structures must be recognized as such,[12] tagged,[13] and destroyed,[14] in order to prevent deleterious consequences such as aggregation.[15–17] We have previously quoted[18, 19] Lumry’s and Eyring’s now classic 1954 paper,[20] “Conformation Changes of Proteins” which begins by stating, “[t]he term protein denaturation even in its original meaning included all those reactions destroying the solubility of native proteins and has since acquired so many other meanings as to become virtually useless.” In the 65 years since, little has changed. In large part this is because it is extremely difficult to trap, purify, and characterize non-native states.
One exception to this difficulty comes about when solvent is removed as species are transferred into mass spectrometers. Early structural studies of naked biomolecules from Fenselau’s,[21] McLafferty’s,[22] Douglas’,[23] Cooks’,[24] Williams’,[25] Bowers’,[26] and Jarrold’s[27] groups (among others) were perhaps initially driven by curiosity. But, we might now ask: what better place is there to study non-native structures, than in the gas phase? In the absence of a lubricating solvent, some non-native structures are stable for long times,[28] allowing them to be probed with an arsenal of fast and powerful mass spectrometric techniques developed during the last century.[29] Moreover, the evaporative cooling process associated with creating macromolecular ions by electrospray ionization (ESI)[30] rapidly freezes-out specific structures as they dry.[18, 31, 32] These “freeze-dried biomolecules,” as Beauchamp’s group called them[33] are now more than a curiosity; they provide access to non-native states where few options exist. And, studies of naked proteins provide the chance to examine intramolecular interactions without complications due to solvent.[34] As more information becomes available, computational methods will undoubtedly provide a more detailed understanding of how such structures are formed, and what functional or dysfunctional roles they play.
Perhaps it is not all that surprising that structures are stabilized upon removal of solvent. After all, removal of solvent is how protein crystals are stabilized.[35] And, while some were suspicious that early crystal structures may lack key aspects relevant to solution structure,[36] they appear to have caught on and are now widely accepted. Below, we describe the use of ESI with hybrid ion mobility spectrometry-mass spectrometry and collisional-activation techniques (IMS-CA-IMS-MS) to probe structural transitions of the simple, model, undecapeptide substance P (subP) in the gas phase. This peptide, a well-studied member of the tachykinin family,[37] has the sequence Arg1-Pro2-Lys3-Pro4-Gln5-Gln6-Phe7-Phe8-Gly9-Leu10-Met11-NH2. Recent studies, using the cryogenic-IMS techniques pioneered by Russell’s group,[31] found evidence for two types of conformers: a kinetically-trapped structure that emerges in the gas phase upon evaporation of solvent (conformer A); and, an extended gas-phase structure that forms upon annealing desolvated subP ions (conformer B). In another paper in this issue, Conant et al. describe kinetics studies of structural changes of subP that ultimately result in non-enzymatic cleavage of specific bonds, when subP is incubated in ethanol solutions.[38] In ethanol, a trans-Pro2→cis-Pro2 configurational change regulates cleavage of the Pro2-Lys3 peptide bond. After this occurs, the subP(3–11) fragment that is formed undergoes a similar trans-Pro4→cis-Pro4 isomerization before the Pro4-Gln5 bond spontaneously cleaves. In both dissociation events product peptides are accompanied by formation of a cyclic diketopiperazine (DKP) dipeptide. This spontaneous processing is very different than enzymatic dipeptidyl peptidase IV cleavage of penultimate-proline peptide bonds,[39] which occurs only from the trans-configuration and forms dipeptide products rather than DKPs. The presence of these solution intermediates and preservation of kinetically-trapped [subP+3H]3+ ions in the gas phase provides an interesting opportunity to also compare structural changes and bond cleavages in solution with those induced upon collisional activation in the gas phase, which we do below.
Experimental
IMS-CA-IMS-MS measurements.
The instrument used for the studies described here was designed and constructed by Koeniger et al. and a schematic diagram is shown in Figure 1.[40] This instrument uses long drift regions and low pressures. Each drift region is ~300 times longer than the excitation region, such that differences in drift times associated with the higher electric fields in the CA region are smaller than the 60 µs bin sizes used to record drift time distributions. At low pressures, collisional cooling of activated ions occurs more slowly than at high pressures. This makes it possible to activate ions inside of the drift tube using relatively low voltages. Our approach is very similar to the now widely used collision-induced unfolding method (CIU, where ions are injected into a drift tube at different voltages) that was pioneered and perfected by Jarrold’s, Bower’s, and Ruotolo’s groups and is now commercially available.[41, 42] The CIU approach is remarkably sensitive to very subtle differences in structures and stabilities, even for large ions.[43]
Figure 1.
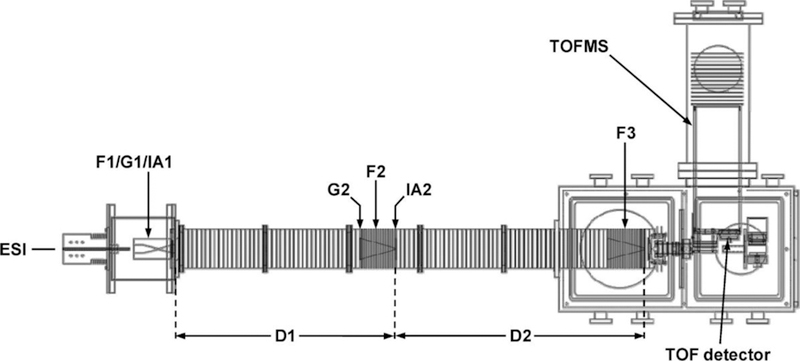
Schematic diagram of the IMS–IMS–MS instrument employed in these studies.
Experimental IMS-CA-IMS-MS measurements are carried out using a method pioneered by Pierson et al.[44] Briefly, ions were produced and introduced to the IMS drift tube using a TriVersa NanoMate (Advion, Ithica, NY) autosampler and nanospray ionization source. The drift tube[40] consists of a source region that periodically releases packets of ions from a gridded electrostatic gate (G1) into the first drift region (D1) where, under the influence of a uniform electric field along the axis of the instrument, ions migrate through a 0.9 m drift tube containing ~3.0 Torr of He buffer gas before entering a collisional activation region where they are activated with an applied voltage. Upon exiting this region the ions (which may have changed conformation or undergone fragmentation) enter a second 1.0 m drift region where they undergo a second separation prior to detection in a time-of-flight mass analyzer. The first and second drift regions are separated by an ion funnel (F2) that serves to radially focus ions and contains an electrostatic gate (G2) that may be raised and lowered periodically to allow ions of a specific mobility to pass. The funnel also contains an activation region (IA2), operated for this experiment at voltages ranging from 6 V to 200 V, that may rapidly accelerate ions for collisional activation. After exiting IA2, ions are rapidly thermalized to the buffer gas temperature and are separated again in D2. An ion funnel (F3) then focuses the ions before they exit into the mass spectrometer.[45]
Determination of experimental collision cross sections from ion mobility distributions.
Collision cross sections (Ω) were determined from ion drift times (tD) using Equation 1.[46]
| (1) |
Included in this equation are terms for Boltzmann’s constant (kb), temperature (T), charge of the ion (z), elementary charge (e), masses of the ion (mI) and buffer gas (mB), and the neutral number density of the buffer gas at standard temperature and pressure (N). The electric field (E), the length of the drift tube (L), and pressure (P) are defined experimentally.
The IMS-CA-IMS-MS instrument (Figure 1) is designed so that cross sections can be measured in several ways. The most accurate measurement is obtained by scanning the delay time associated with release of ions from the G1 and selection of ions at G2 across a peak. In this region the electric field is uniform, the length of the drift region is nearly exactly defined as the difference between the grids of G1 and G2 and the drift time avoids inclusion of any time that ions spend outside of the drift region (e.g., time associated with transfer of ions into the source of the MS) as well as flight times of ions in the MS. This approach can be used to check mobilities that travel through the entire instrument and create a calibration curve for cross section distributions recorded using the entire D1 and D2 regions. Finally, the drift time can be measured with respect to the selection gate G2, allowing cross sections to be determined for activated ions.
Peptide synthesis and sample preparation.
Substance P was obtained from Sigma Aldrich (≥95% purity, St. Louis, MO). Several subP analogues involving a range of Pro→Ala substitutions were synthesized using standard FMOC solid-phase peptide synthesis carried out on an Applied Biosystems 433A Peptide Synthesizer (Applied Biosystems, Foster City, CA).[47] Peptide solutions (10 µM in 1-propanol) were electrosprayed using a TriVersa NanoMate autosampler. We focused these studies on ions produced from 1-propanol because this solvent produces four structures that appear to be trapped during the electrospray process. Thus, this system allows us to study transitions of different structures of the same peptide.
Method for investigating cis/trans configurations of Pro2 and Pro4 peptide bonds.
Proline is unique among the naturally occurring amino acids because the pyrrolidine side-chain restricts formation of the more commonly adopted and energetically favorable trans-configured peptide bond.[48–52] As a result, proline has an increased tendency to occupy the cis-form, which frequently leads to additional structural features.[53–55] Substitution of an alanine residue for a proline residue prevents formation of a cis-configured peptide bond that can be formed by proline. Thus, comparisons of the cross section distributions for subP (containing proline) with distributions recorded for Pro→Ala-substituted analogues allows us to obtain insight about the configuration of the proline peptide bond configuration. To make this comparison it is useful to account for differences in the cross sections that arise from differences in the sizes of proline and alanine residues. This difference is known from values of intrinsic size parameters which were initially determined for all amino acids by Counterman and Valentine.[56–60] With these corrections, Pierson et al. assigned the cis- and trans-configurations of each proline residue in the conformations of the bradykinin peptide backbone configuration.[61] Similarly, Fort et al. utilized alanine-substitution of various subP residues (Pro, Gln, Phe) to characterize the key residues in stabilizing conformer A of [subP+3H]3+.[62] Below, we analyze three Pro→Ala substituted sequences: RAKPQQFFGLM-NH2 [subP(P2A)], RPKAQQFFGLM-NH2 [subP(P4A)], and RAKAQQFFGLM-NH2 [subP(P2,4A)]. Studies of Ala-substituted analogues as a function of activation energy allow us to identify the origin of specific structural changes.
Calibration of threshold voltages to obtain activation energies.
Activation voltages are calibrated to obtain activation energies, as described previously.[44] Briefly, collisional fragmentation threshold voltages from measurements in the drift tube are calibrated to reported thermochemistry. Most of the thermochemistry used to calibrate our method was determined by Armentrout who has pioneered the most rigorous statistical analyses associated with determining fragmentation thresholds from single-collision events that lead to new ions (either fragments or products of ion-molecule reactions).[63–65] The calibration also uses thermochemistry for bradykinin ions from an Arrhenius analysis of dissociation rates measured in a temperature-controlled ion trap by McLuckey and his coworkers,[66] and an average of several reports of thermochemistry for leucine enkephalin.[67–70] Each experimental threshold, defined as the voltage at which a product state abundance reaches 1% normalized abundance, is multiplied by charge, divided by number of vibrational degrees of freedom (d.o.f.) of the activated species, and calibrated to literature values for the dissociation energies as shown in Equation 2.[44]
| (2) |
The calibrated thresholds determined from Equation 2 are in units of eV and these values are reported in kJ∙mol−1. The use of 1% relative intensity as threshold voltages was not based on statistical theory, but was chosen as the point of a signal-to-noise ratio sufficient for confident detection, described previously.[44] Other definitions of the threshold (e.g., 2% or 5%) could be calibrated and used for determination of thresholds. Once calibrated other definitions yield similar values.[71]
One critical caveat of this approach is that it does not capture the effects of entropy. Specifically, it assumes that transition states have similar entropies of activation when approached from the forward or reverse directions. That is, they are both either similarly loose, or tight. This assumption appears to be valid for activation of the main peaks observed in the quasi-equilibrium distribution for bradykinin. But, this is clearly not the case for substance P. The studies described below reveal that four conformers originate from solution and if provided enough activation energy, each of these will form the conformer B – the gas phase structure. But, none of these processes is reversible. This strongly suggests that solvent is required to reach the transition states necessary to form these conformers. This finding introduces an important caveat. Below, we report threshold energies and treat them as transition state energies for the forward direction that results in formation of B; however, strictly speaking these values are upper limits to the transition state energies, and may also be subject to kinetic shifts larger than the reactions with loose transition states that were used to calibrate this method, and so these reactions may give an activation energy that is slightly too large. Interestingly, our reported values for these transitions are relatively low, ~28 to 54 kJ∙mol−1, especially when compared with the solution thermochemistry for similar transitions. Therefore, it seems likely that kinetic shifts in the forward direction are small.
Results & discussion
IMS cross section distributions for [subP+3H]3+.
Figure 2 shows a typical cross section distribution for [subP+3H]3+ ions obtained upon electrospraying from a solution of propanol. The most abundant peak in the spectrum at Ω = 300 Å2 corresponds to a conformation that was observed previously by Russell and coworkers and is called conformer A.[31] One of the smaller peaks, centered at Ω = 354 Å2 was also observed previously and is called conformer B.[31] Additionally, we find evidence for two new very low-intensity peaks centered at Ω = 333 Å2 and 339 Å2, that we have labeled as C1 and C2, respectively, that we discuss in more detail below. The observation of multiple conformations for this peptide is similar to results for other peptides with proline residues, which often show evidence for multiple structures associated with proline residues sampling both cis- and trans-peptide-bond configurations.[53–55]
Figure 2.
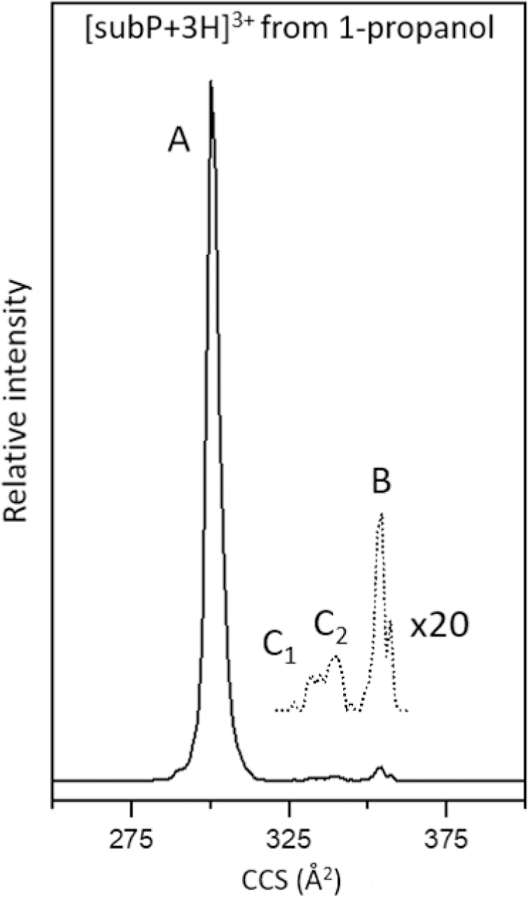
Mobility-separated, cross section distribution for [subP+3H]3+ measured upon electrospraying subP from ethanol. The major peak corresponds to conformer A, as assigned previously [31]. Three very low-abundance peaks are also observed, corresponding to conformers C1, C2, B. The region associated with low-abundance structures is multiplied by a factor of 20 (dashed line).
Selection & activation of individual conformers.
To explore structural changes in this system, each peak was selected based on its mobility in the first drift tube and subjected to CA at different activation voltages. The results for selection and activation for each conformer are shown in Figure 3. We begin by discussing the most abundant species, conformer A. Upon selection and activation, this peak remains the only feature in the distribution below ~50 V. At an applied activation voltage of 56 V we observe in Figure 3 that a small fraction of the conformer A ions unfolds, forming conformer B, which has a larger cross section. As the CA voltage is increased, the distribution shifts to favor B and by 68 V, B dominates the distribution, becoming the only observable feature above CA ~70 V. This result is consistent with results reported by Russell’s group, where ions were activated in the source region.[31]
Figure 3.

IMS-CA-IMS cross section distributions for mobility-selected [subP+3H]3+ conformers upon activation in the IA2 region (see text for details). Each of the four conformers (A, B, C1, C2) shown in Figure 2 was selected and collisionally activated using the voltages that are indicated in the Figure.
Previously we reported that at high activation voltages (prior to fragmentation), bradykinin ions favor a ‘quasi-equilibrium distribution’ (QED).[72] That is, when the activation energy exceeds all of the barriers between different structures, increasing activation voltage no longer results in changes to the populations of different states that are present.[44, 72] Additionally, the QED distribution of bradykinin (which involved three main structures) can be reached upon activating any of the six resolved structures that were produced directly by ESI for this ion. In the case of subP only a single peak is observed at high energies. This peak (conformer B) may be comprised of multiple structures with similar cross sections that are not resolved and reflect the QED of gas phase sub P ions that appears to have been reached at ~70 V. Conformer A is not observed in the QED. We interpret this as an indication that conformer A results from a population of states that are kinetically trapped as ions emerge from solution. This is consistent with the cryogenic-IMS measurements.[31]
Analogous selection and activation experiments were carried out for the smaller peaks (B, C1, and C2) as shown in Figure 3. While conformer B dominates the distribution when formed at high activation energies from conformer A, only a small population is formed directly from the source. Integration of the ion signals in Figure 2 indicates that conformer B comprises only ~1.5% of the total distribution. Selection and activation of the Ω = 354 Å2 conformer B peak results in an interesting set of distributions. Most of these ions (>98%) do not appear to change structure upon activation. This is consistent with the idea that in the gas phase B ions are more stable than A ions. In this case, we suspect that the conformer B ions observed directly from our source are formed by activation of a conformer A, after their emergence into the gas-phase as ions (presumably this slight activation occurs in the ion funnel region of our source, in analogy to Russell’s activation results).[31] Activating the Ω = 354 Å2 peak at 60 V shows evidence for a very small population of conformer A ions. It appears that ~2% of the selected Ω = 354 Å2 ions (which initially comprised only ~1.5% of the distribution of ions from the source) can form conformer A. However, as the activation energy is increased beyond ~80 V this population vanishes and only B is observed. This result requires that an additional, very-low abundance conformer must be present. Unlike other B ions, upon activation, this small population of species (which we call conformer B*) must be kinetically trapped (similarly to A) and upon activation these ions are capable of forming conformer A. The A state that is produced from B* presumably exists over a narrow range of energies (as shown below) because at higher energies, A can convert to B. The B* conformer trapped during the ESI process represents only ~0.03% (2% × 1.5%) of the initial distribution of ions. This analysis not only illustrates the value of selecting and activating ions by IMS-IMS as a means of revealing differences in structures that have identical cross sections, but also highlights the high sensitivity of these methods.
Figure 3 also shows cross section distributions that are recorded upon selection and activation of C1 and C2 ions, which each comprise ~0.5% of the initial source distribution. Both of these ions form conformer A at intermediate voltages. As observed for B*, at higher voltages A disappears and only the final B product remains. The formation of A from C1 and C2 suggests that these are also kinetically-trapped structures from solution. No other structures form either of these species in the gas phase, consistent with this idea.
Careful examination of Figure 3 reveals a very small difference between the C1 and C2 conformers. At intermediate voltages, e.g., 38 V in Figure 3, the C2 peak shows a small shoulder at a slightly larger cross sections (Ω = 354 Å2) consistent with formation of B or B*. As the collision voltage is increased this shoulder decreases in abundance, disappearing entirely by ~48 V. This behavior is consistent with formation of B*. At higher CA voltages the peak at Ω = 354 Å2 returns and dominates the distribution. This is the B conformer, and it becomes the only product observed above ~70 V.
It is important to note that Russell’s cryogenic-IMS results show definitively that conformer A emerges directly from solution, upon evaporation of the last remaining solvent molecules from the ion. But, here, we have shown that selection and activation of C1, C2, and also B* in the gas phase can form a state with the same cross section as conformer A. The only structure that does not form A upon activation is conformer B. This requires that either conformer A can be formed in the gas phase from activation of other structures, or that gas-phase activation of other conformers produces a different species with the same cross section as the conformation that emerges directly from solution. The same is true of B*. Activation of C2 shows that B* can be formed the gas-phase. However, the observation that B* forms A indicates that B* is a kinetically-trapped conformer and could also emerge directly from solution.
Changes in conformer abundances as a function of activation voltage.
A summary of the abundances that are obtained for different structures upon selection and activation of each of the conformers at all of the CA voltages used in these studies is shown in Figure 4. These data are consistent with the changes in the peaks discussed above. Figure 4 also shows simple reaction mechanisms that are consistent with the discussion given above upon activating each ion. One important finding is that the transitions described above show no evidence of being reversible. As can be observed from Figure 4, as the collision voltage is increased, from ~50 to 80 V, the population of conformer A ions decreases as B increases and no further changes are observed until fragmentation is observed at ~90 V. When the Ω = 354 Å2 peak (dominated by B) is selected and activated we observe the small population of B* that forms A. At higher energies A forms B. Again, the large population of B ions does not change over a wide distribution of energies (here from 0 to 90 V) until the threshold for fragmentation is reached. Similarly, the lowest energy product observed upon activation of C1 is A and at higher energies B is observed prior to fragmentation. Activation of C2 is somewhat different. This conformer forms B* and A. At higher energies B* also forms A and at even higher energies conformer B dominates before dissociation.
Figure 4.

(Left) Relative abundance plots of [subP+3H]3+ conformers A, B (and B*), C1, and C2. Observed transition pathways are shown for each conformer. Amplified abundances of low-intensity ions (i.e., B* formed by C2, and A formed by B*) are included for clarity. (Right) Expansion of the 0 – 5% abundance range from activation of the C1 ions, showing abundances associated with formation of each product (A, B*, and fragments). The dashed lines indicate the 1% abundance thresholds for each transition and the approximate threshold voltage.
Extraction of thresholds and determination of activation energies for structural transitions.
Figure 4 also shows a detailed plot of C2 activation showing the threshold regions for each transition. Similar analysis of 1% thresholds for each activated conformer yield activation energies for each transition. These values are summarized in Table 1, and a simple representation of the reaction coordinate associated with these conformational changes is shown in Scheme I. This analysis reveals that barriers for these gas-phase transitions are in the range of ~28 to 54 kJ∙mol−1.
Table 1.
Threshold voltages and calculated activation energies for [subP+3H]3+ transitions.
| conformer | |||
|---|---|---|---|
| Selecteda | Formedb | Voltagec | Threshold Ead, kJ·mol−1 |
| A | B | 48 ± 1 | 42 ± 1 |
| C1 | A | 32 ± 2 | 29 ± 2 |
| B | 48 ± 2 | 42 ± 2 | |
| C2 | B* | 32 ± 1 | 29 ± 1 |
| A | 31 ± 6 | 28 ± 5 | |
| B | 43 ± 4 | 38 ± 4 | |
| B* | A | 51 ± 4 | 44 ± 3 |
| B | 63 ± 2 | 54 ± 2 | |
| A | [subP-NH3+3H]3+ | 85 ± 2 | 71 ± 2 |
| C1 | [subP-NH3+3H]3+ | 92 ± 5 | 76 ± 4 |
| C2 | [subP-NH3+3H]3+ | 90 ± 4 | 75 ± 4 |
| B | [subP-NH3+3H]3+ | 96 ± 5 | 79 ± 4 |
| A | [b10+2H]2+ | 92 ± 4 | 76 ± 4 |
| C1 | [b10+2H]2+ | 102 ± 6 | 85 ± 5 |
| C2 | [b10+2H]2+ | 105 ± 1 | 87 ± 2 |
| B | [b10+2H]2+ | 105 ± 5 | 87 ± 4 |
Mobility peak of [subP+3H]3+ selected for activation.
Structure formed during activation of the selected peak.
Collisional activation voltage applied in the activation region IA2. The indicated uncertainties in voltage represent the standard deviation from triplicate measurements.
Activation energy threshold of the indicated transition, calibrated with equation 2. Activation energy uncertainty was determined through propagation of voltage error and the uncertainty of the energy calibration from ref 44.
Scheme 1.

Simple reaction coordinate showing the conformational changes in substance P sampled by collisional activation.
Fragmentation thresholds and dissociation energies.
Figure 4 also shows that at very high energies (above ~90 V), the B conformers fragment. The fragmentation products that are observed are identical regardless of which conformer is selected for activation. This is not surprising. As ions enter the activation region IA2 they undergo a rapid heating and cooling process. This is a relatively slow cycle (as compared to isomerization); we anticipate that every conformer will convert to B prior to dissociation.
Figure 5 shows fragmentation mass spectra at several energies. The lowest energy process involves loss of ammonia to form a small population of [subP-NH3+3H]3+ ions. While we cannot unambiguously assign where these products are formed (there are several amide groups associated with this peptide) loss of ammonia involves a very specific transition state. Regardless of its origin this process is entropically disfavored, consistent with the relative inefficiency, observed experimentally as a small peak in Figure 5. At higher energies, direct bond cleavage leads to formation of the b102+ ion. As soon as this becomes energetically accessible, this process dominates the mass spectrum. As the activation voltages is increased beyond ~130 V, we observe a third fragment, corresponding to the b92+. This fragment competes directly with b102+ indicating that b92+ is formed from b102+ in a sequential process.
Figure 5.

(Left) Mass spectra corresponding to, from bottom to top, the selection of conformer B from [subP+3H]3+ ions, and resulting fragment ions from CA at 105 V, 135 V, and 156 V. (Right) Normalized abundances of conformer B and product ions as a function of voltage.
Calibrated thresholds are used to obtain dissociation energies associated with two fragmentation pathways: process 1) [subP-NH3+3H]3+ + NH3, which likely requires a significant intramolecular rearrangement in formation of a very specific transition state that eliminates ammonia without cleaving any peptide bond; and, process 2) a peptide bond is cleaved to produce the b102+ + y1+ product ions. A mechanism for process 2 has been proposed in which the backbone carbonyl carbon of Leu10 is subjected to nucleophilic attack from the carbonyl oxygen of Gly9, a relatively localized rearrangement leading to peptide bond cleavage and an oxazolone intermediate fragment that rearranges to the final b102+ product.[73] We do not analyze the thresholds associated with additional products formed at higher energies from fragmentation of b102+ (e.g., the b92+, and other smaller fragments, not shown) because these appear to be formed in sequential processes.
The bond energies that we obtain from this analysis are interesting. While the products of dissociation are independent of the initial selected conformer that is activated (indicating that fragments are formed after formation of the distribution of B ions) the energy required for dissociation differs. That is, this analysis is sensitive to subtle differences in stabilities of different precursor structures. The activation energies necessary for dissociation via pathways 1 and 2 measured from selection and activation of each precursor conformer are tabulated in Table 1. The energy required to eliminate ammonia via process 1 ranges from a lowest value of 71 ± 2 kJ∙mol−1 for conformer A to a highest value of 79 ± 4 kJ∙mol−1 for conformer B. It is interesting to note that these values are similar to the value of 76 ± 3 kJ∙mol−1 measured by McLuckey and coworkers for elimination of water from bradykinin, a process that we expect to be energetically similar to elimination of ammonia.
Table 1 also lists values associated with process 2, which results in formation of b102+. The energetics associated with this fragmentation are also dependent upon which precursor ion has been activated. Our threshold analysis yield, dissociation energies of 76 ± 4 kJ∙mol−1 for conformer A, 85 ± 5 kJ∙mol−1 for conformer C1, 87 ± 2 kJ∙mol−1 for conformer C2, and 87 ± 4 kJ∙mol−1 for conformer B, as listed in Table 1. Comparison of these values with those reported above for process 1 reveals that the former process (elimination of ammonia) is energetically favorable by ~5 to 12 kJ∙mol−1. Interestingly, while elimination of ammonia is favored energetically, as soon as fragmentation pathway 2 is accessible, formation of b102+ dominates the product distribution, suggesting that process 2 is more favored entropically.
Assignment of proline configurations for different conformers.
As mentioned above, substitutions of Ala residues for Pro residues allowed Pierson et al. to assign the cis/trans configurations of different bradykinin conformers. We have carried out analogous substitutions of the Pro2 and Pro4 peptide bonds for different conformations of subP (cross section distributions shown in Figure 6) and find that assignments based on comparisons of cross sections alone are somewhat ambiguous. Still, it is instructive to go through this analysis as some insight is gained by analyzing the subP(P2A), subP(P4A), and subP(P2,4A) Ala-analogues.
Figure 6.
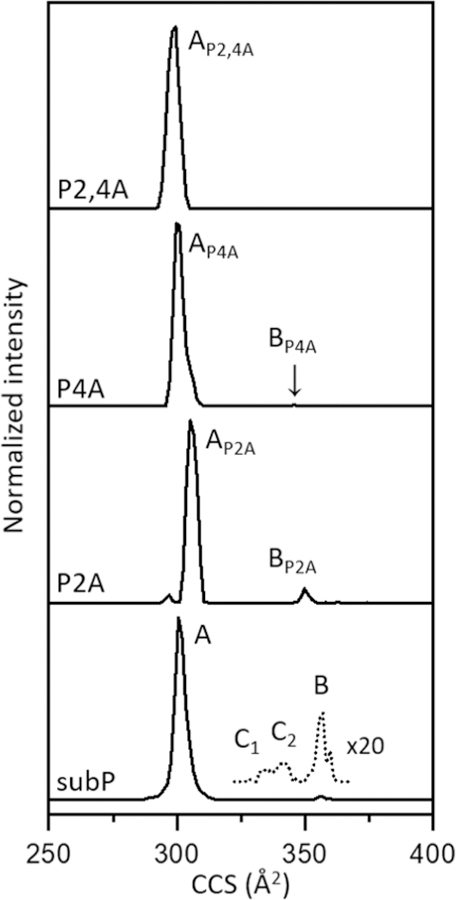
Cross section distributions of [M+3H]3+ corresponding to subP and Pro→Ala substituted analogues. The cross sections of the analogues were shifted according to values of intrinsic size parameters, as described in the experimental section.
We begin by considering the proline configurations for C1 and C2, because these assignments are relatively straightforward. None of the Ala-analogues form the C1 or C2 conformers, within our detection limits (S/N ~ 104 in these experiments). This indicates that each of these conformers has a cis-Pro2 and cis-Pro4 configuration and the trans-peptide bonds associated with the Pro→Ala substitutions prohibit their formation.
Cross section measurements for the Ala-analogues of conformer A (when corrected for the differences in size of the Ala and Pro residues, ~2.5 Å2)[60] yield values of Ω[subP(P2A)+3H]3+ = 305 Å2, Ω[subP(P4A)+3H]3+ = 300 Å2, and Ω[subP(P2,4A)+3H]3+ = 299 Å2, or an average size-corrected value of Ω(A) = 301 ± 2 Å2 (Figure 6). A trans-Pro2 and trans-Pro4 configurational assignment for conformer A is consistent with the idea that cross sections for the Ala-analogues and Ω(A) = 302 ± 2 Å2 for [subP+3H]3+ with no Ala substitutions are identical within the experimental uncertainties (when conformer A is produced from a 1-propanol solution). It is important to note that when electrosprayed from ethanol[38] the peak associated with conformer A becomes broader. It is likely that this broadening is associated with a population of ions having a cis-Pro2 configuration, as this is required for DKP formation in solution.[74] Thus, the trans-Pro2 and trans-Pro4 configurational assignment of peak A from 1-propanol based on cross sections alone is not very satisfying.
This assignment is strengthened upon examining the CA data (see supporting information). When activated each of the three Ala-analogues convert entirely into conformer B; moreover, the thresholds for each of these transitions are ~6 kJ∙mol−1 lower than for activation of [subP+3H]3+ with no substitution. Substitution of the Ala residue imposes a trans-configured peptide bond because it raises the barrier for forming the cis-configuration.[48, 49] Thus, the lower thresholds observed for the Ala-substituted peptides corroborate the trans-Pro2 and trans-Pro4 configurational assignment of A.
Conformer B is produced in the gas phase. For some of the Ala-analogues we must activate A in order to produce B. An average of the size-parameter corrected cross section for all three Ala-analogues is Ω(B) = 349 ± 1 Å2, a value which is 1.7% smaller than Ω(B) = 354 ± 3 Å2 measured for subP with no Ala substitutions (Figure 6). This is slightly outside of the ± 1% relative uncertainty that we expect for identical structures, such that assignment based on comparisons of cross sections is a little ambiguous. The similar values suggest that conformer B has a trans-Pro2 and trans-Pro4 configuration. We note that the Ala and Pro size parameters vary with peptide size (as well as peptide structures)[59] and other size parameter values would yield slightly different corrected cross sections for the Ala-analogues. Moreover, this analysis assumes that the only change in size arises from the differences in these residues, and clearly this substitution could alter the overall structure of this conformer within this range. As mentioned above, the lower thresholds for forming B from A for the Ala-substituted peptides indicates that B (for subP) has a trans-Pro2 and trans-Pro4 configuration.
Finally, activation of the Ω(B) = 349 Å2 peak for each of the Ala-analogues provides a means of assigning the proline configurations for B*. The B* conformer is observed only for the subP(P2A) analogue, with an energy dependence and population that is very similar to subP having no Ala substitutions. Thus, we assign the proline configurations of the B* subP conformer as trans-Pro2 and cis-Pro4 configurations. Table 2 provides a summary of the proline configurations for each of the subP conformations.
Table 2.
Proline peptide bond configurations in each conformer of [subP+3H]3+.
| [subP+3H]3+ conformera | Pro2 | Pro4 |
|---|---|---|
| A | trans | trans |
| C1 | cis | cis |
| C2 | cis | cis |
| B* | trans | cis |
| B | trans | trans |
Conformations A, C1, C2, B*, and B correspond to populations of ions from the ion mobility distribution of [subP+3H]3+ ions electrosprayed from a solution of 1-propanol.
Semi-quantitative potential energy diagram for [subP+3H]3+ conformers.
The activation energies in Table 1 for transitions between structures and fragmentation of each selected structure (with the exception of B*) can be used to construct the semi-quantitative potential energy diagram shown in Figure 7. The energy level for each conformer (except for B*) could be positioned by using the fragmentation patterns or thresholds for structural transitions between states. Thus, there are several ways to produce this diagram. We show only one, which arises as follows. In Figure 7 we define the energy of each state with respect to conformer B, because it is most stable. The difference between fragmentation thresholds for A and B (Table 1) places A at 8 ± 5 kJ∙mol−1. The threshold for the A→B transition is 42 ± 1 kJ∙mol−1 or, 50 ± 5 kJ∙mol−1 higher than B. Similarly, from the experimental thresholds required to form conformer B, we determine the relative energies of C1, C2, and B* to be 8 ± 5 kJ∙mol−1, 12 ± 6 kJ∙mol−1, and −3 ± 5 kJ∙mol−1, respectively. The barriers associated with the remaining transitions are taken from the thresholds for each transition that are given in Table 1: 38 ± 6 kJ∙mol−1 for the C1→A transition; 41 ± 8 kJ∙mol−1 for C2→A; and, 41 ± 6 kJ∙mol−1 for C2→B* as well as B*→A. The threshold for fragmenting conformer B, 79 ± 4 kJ∙mol−1, is also shown.
Figure 7.

Energy diagram of [subP+3H]3+ derived from experimental threshold Ea barriers for structural transitions and the low energy fragmentation products, [subP-NH3+3H]3+ and b102+ ions. Irreversible transitions are shown by a black arrow. A proposed transition pathway is shown at the top of the diagram, which includes experimentally determined cis- and trans-configurational assignments of each Pro residue for each structure.
A cross check of the energies associated with the C1 and C2 conformers can be made by comparing the fragmentation thresholds that are predicted from this energy diagram (that are referenced to the barrier for making B) to the thresholds that we measure experimentally. The diagram predicts fragmentation thresholds of 71 ± 6 kJ∙mol−1 and 67 ± 7 kJ∙mol−1 for C1 and C2, respectively. Experimentally we find a fragmentation threshold of 76 ± 4 kJ∙mol−1 for C1, in agreement with the values calculated from barriers for structural transitions. The experimental threshold for fragmentation of C2 is 75 ± 4 kJ∙mol−1. While the threshold for C2 calculated from Figure 7 is 8 kJ∙mol−1 lower than measured experimentally, we note that within the combined uncertainties they are in agreement. This agreement provides a cross check of the barrier heights for structural transitions. If there was a significant kinetic shift associated with forming B, then the calculated fragmentation threshold for C2 would be significantly higher than was measured experimentally.
Comparisons of structures and stabilities, structural transitions, and fragments for gas-phase, solution-phase, and enzyme-bound subP.
The data presented above provide an opportunity to compare structural transitions across a range of environments. Proline-containing peptides have been studied extensively because cis/trans isomers introduce a significant structural heterogeneity.[53–55] A number of endo- and exo-peptidases are proline specific.[75] Dipeptidyl peptidase IV (DPP IV) is especially relevant to the work presented here as it targets peptides containing trans-configured penultimate proline motifs and catalyzes the elimination Xxx-Pro dipeptides.[39] Without enzymes, in solution (ethanol), only the cis-configured Pro2 can eliminate DKP.[38, 74] The rate-limiting trans→cis isomerization for subP reported by Conant et al. has a free-energy barrier of 88 ± 6 kJ∙mol−1. Based on their findings, Conant speculated that one biological role of DPP IV may be to favor formation of dipeptides rather than DKP products, which avoids the bioactivity of DKP.[76] In the gas phase, the Pro2 trans-configuration of the B state of subP is energetically favored. This configuration cannot eliminate DKP, and unlike enzymatic processing to form dipeptides, upon activation we observe elimination of ammonia and fragmentation at the C-terminal end of the peptide to produce the b102+. Thus, activation of the gas-phase ions leads to fragments that are not observed in solution or upon enzymatic digestion. And, the solution and enzymatic fragments are not accessible in the gas phase.
One final note involves the irreversible nature of the trans-Pro → cis-Pro transitions in the gas phase. Our results indicate that a large entropic barrier prohibits this process. And, we know that from our solution studies that trans-Pro → cis-Pro transitions are observed prior to DKP formation. This implies that addition of ethanol increases the accessibility of the trans-Pro2→cis-Pro2 transition state. While this might be so, we note that in ethanol Conant reports a value of ΔH‡ = 41 ± 5 kJ∙mol−1 and ΔS‡ = −157 ± 12 J∙mol−1∙K−1 for this transition. That is, this transition is extremely difficult to reach in solution as well. It is perhaps no surprise that in most biological systems, the trans-configuration of proline is heavily favored.
Conclusions
IMS-CA-IMS-MS techniques were used to characterize five conformations of [subP+3H]3+. There is evidence that all five structures are produced during the electrospray droplet-drying process. One is the lowest-energy gas-phase structure B, and the other four are kinetically trapped conformations that can be converted to B by collisional activation in the gas phase. Each transition of the kinetically trapped structures was found to be irreversible. This indicates that solvent is required to approach key transition states in reverse. A semi-quantitative potential energy diagram is derived from threshold activation voltages that are calibrated as described in the text. The cis/trans isomerization of Pro2 and Pro4 residues in subP has a significant influence on [subP+3H]3+ conformations and plays a key role in many of the transitions observed. Comparisons of structural changes and dissociation patterns in the gas phase to those from solution show that solvent plays a key role in regulating conformations in solution.
Supplementary Material
Acknowledgements
This work is supported in part by funds from the National Institute of Health, R01 GM117207–04, and the Indiana University Robert and Marjorie Mann fellowships (CRC, DRF). The work at TAMU (DHR) was funded by NSF (CHE-1707675) and NIH (P41GM121751–01A1).
References
- 1.Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P: The Protein Data Bank Nucleic Acids Research, 28: 235–242. URL: www.rcsb.org Citation (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dill KA, MacCallum JL: The protein-folding problem, 50 years on. science. 338, 1042–1046 (2012) [DOI] [PubMed] [Google Scholar]
- 3.Service RF: Google’s Deep Mindaces protein folding. http://www.sciencemag.org/news/2018/12/google-s-deepmind-aces-protein-folding Accessed 13 Jan 2019
- 4.Kendrew JC, Bodo G, Dintzis HM, Parrish R, Wyckoff H, Phillips DC: A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature. 181, 662–666 (1958) [DOI] [PubMed] [Google Scholar]
- 5.Epstein CJ, Goldberger RF, Anfinsen CB: The genetic control of tertiary protein structure: studies with model systems In: Cold Spring Harbor symposia on quantitative biology. Cold Spring Harbor Laboratory Press; 28, 439–449 (1963) [Google Scholar]
- 6.Williamson MP, Havel TF, Wüthrich K: Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1 H nuclear magnetic resonance and distance geometry. World Scientific, (1995) [DOI] [PubMed]
- 7.Fersht AR: From the first protein structures to our current knowledge of protein folding: delights and scepticisms. Nature Reviews Molecular Cell Biology. 9, 650 (2008) [DOI] [PubMed] [Google Scholar]
- 8.Boutet S, Lomb L, Williams GJ, Barends TR, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL: High-resolution protein structure determination by serial femtosecond crystallography. Science. 1217737 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter P, Ron D: The unfolded protein response: from stress pathway to homeostatic regulation. Science. 334, 1081–1086 (2011) [DOI] [PubMed] [Google Scholar]
- 10.Desai UR, Osterhout JJ, Klibanov AM: Protein structure in the lyophilized state: A hydrogen isotope exchange/NMR study with bovine pancreatic trypsin inhibitor. Journal of the American Chemical Society. 116, 9420–9422 (1994) [Google Scholar]
- 11.Zaks A, Klibanov AM: Enzymatic catalysis in nonaqueous solvents. Journal of Biological Chemistry. 263, 3194–3201 (1988) [PubMed] [Google Scholar]
- 12.Freeman BC, Morimoto RI: The human cytosolic molecular chaperones hsp90, hsp70 (hsc70) and hdj-1 have distinct roles in recognition of a non-native protein and protein refolding. The EMBO journal. 15, 2969–2979 (1996) [PMC free article] [PubMed] [Google Scholar]
- 13.Hershko A, Ciechanover A: The ubiquitin system. Annu. Rev. Biochem 67, 425–479 (1998) [DOI] [PubMed] [Google Scholar]
- 14.Ciechanover A, Schwartz AL: The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proceedings of the National Academy of Sciences. 95, 2727–2730 (1998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguzzi A, O’connor T: Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature reviews Drug discovery. 9, 237 (2010) [DOI] [PubMed] [Google Scholar]
- 16.Ross CA, Poirier MA: Protein aggregation and neurodegenerative disease. Nature medicine. 10, S10 (2004) [DOI] [PubMed] [Google Scholar]
- 17.Goldberg AL: Protein degradation and protection against misfolded or damaged proteins. Nature. 426, 895 (2003) [DOI] [PubMed] [Google Scholar]
- 18.El-Baba TJ, Fuller DR, Woodall DW, Raab SA, Conant CR, Dilger JM, Toker Y, Williams ER, Russell DH, Clemmer DE: Melting proteins confined in nanodroplets with 10.6 μm light provides clues about early steps of denaturation. Chemical Communications. 54, 3270–3273 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE: Biomolecule analysis by ion mobility spectrometry. Annu. Rev. Anal. Chem 1, 293–327 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumry R, Eyring H: Conformation Changes of Proteins. The Journal of Physical Chemistry. 58, 110–120 (1954) [Google Scholar]
- 21.Cheng X, Wu Z, Fenselau C: Collision energy dependence of proton-bound dimer dissociation: entropy effects, proton affinities, and intramolecular hydrogen-bonding in protonated peptides. Journal of the American Chemical Society. 115, 4844–4848 (1993) [Google Scholar]
- 22.Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler F, McLafferty FW: Coexisting stable conformations of gaseous protein ions. Proceedings of the National Academy of Sciences. 90, 790–793 (1993) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Covey T, Douglas D: Collision cross sections for protein ions. Journal of the American Society for Mass Spectrometry. 4, 616–623 (1993) [DOI] [PubMed] [Google Scholar]
- 24.Cox K, Julian R, Cooks R, Kaiser R: Conformer selection of protein ions by ion mobility in a triple quadrupole mass spectrometer. Journal of the American Society for Mass Spectrometry. 5, 127–136 (1994) [DOI] [PubMed] [Google Scholar]
- 25.Gross DS, Schnier PD, Rodriguez-Cruz SE, Fagerquist CK, Williams ER: Conformations and folding of lysozyme ions in vacuo. Proceedings of the National Academy of Sciences. 93, 3143–3148 (1996) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Von Helden G, Wyttenbach T, Bowers MT: Conformation of macromolecules in the gas phase: use of matrix-assisted laser desorption methods in ion chromatography. Science. 267, 1483–1485 (1995) [DOI] [PubMed] [Google Scholar]
- 27.Clemmer DE, Hudgins RR, Jarrold MF: Naked protein conformations: cytochrome c in the gas phase. Journal of the American Chemical Society. 117, 10141–10142 (1995) [Google Scholar]
- 28.Freitas MA, Hendrickson CL, Emmett MR, Marshall AG: High-field Fourier transform ion cyclotron resonance mass spectrometry for simultaneous trapping and gas-phase hydrogen/deuterium exchange of peptide ions. Journal of the American Society for Mass Spectrometry. 9, 1012–1019 (1998) [Google Scholar]
- 29.Grayson MA. Chemical Heritage Foundation, (2002)
- 30.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM: Electrospray ionization for mass spectrometry of large biomolecules. Science. 246, 64–71 (1989) [DOI] [PubMed] [Google Scholar]
- 31.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH: From solution to the gas phase: stepwise dehydration and kinetic trapping of Substance P reveals the origin of peptide conformations. Journal of the American Chemical Society. 135, 19147–19153 (2013) [DOI] [PubMed] [Google Scholar]
- 32.Pierson NA, Chen L, Valentine SJ, Russell DH, Clemmer DE: Number of solution states of bradykinin from ion mobility and mass spectrometry measurements. Journal of the American Chemical Society. 133, 13810–13813 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S-W, Freivogel P, Schindler T, Beauchamp J: Freeze-dried biomolecules: FT-ICR studies of the specific solvation of functional groups and clathrate formation observed by the slow evaporation of water from hydrated peptides and model compounds in the gas phase. Journal of the American Chemical Society. 120, 11758–11765 (1998) [Google Scholar]
- 34.Liu Y, Valentine SJ, Counterman AE, Hoaglund CS, Clemmer DE: Peer Reviewed: Injected-Ion Mobility Analysis of Biomolecules. Analytical Chemistry. 69, 728A–735A (1997) [Google Scholar]
- 35.McPherson A, Gavira JA: Introduction to protein crystallization. Acta Crystallographica Section F: Structural Biology Communications. 70, 2–20 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Billeter M: Comparison of protein structures determined by NMR in solution and by X-ray diffraction in single crystals. Quarterly reviews of biophysics. 25, 325–377 (1992) [DOI] [PubMed] [Google Scholar]
- 37.Severini C, Improta G, Falconieri-Erspamer G, Salvadori S, Erspamer V: The tachykinin peptide family. Pharmacological reviews. 54, 285–322 (2002) [DOI] [PubMed] [Google Scholar]
- 38.Conant CR, Fuller DR, El-Baba TJ, Zhang Z, Russell DH, Clemmer DE: Spontaneous non-enzymatic bond cleavage of Substance P in ethanol. submitted.
- 39.Fischer G, Heins J, Barth A: The conformation around the peptide bond between the P1-and P2-positions is important for catalytic activity of some proline-specific proteases. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology. 742, 452–462 (1983) [DOI] [PubMed] [Google Scholar]
- 40.Koeniger SL, Merenbloom SI, Valentine SJ, Jarrold MF, Udseth HR, Smith RD, Clemmer DE: An IMS− IMS Analogue of MS− MS. Analytical chemistry. 78, 4161–4174 (2006) [DOI] [PubMed] [Google Scholar]
- 41.Shelimov KB, Clemmer DE, Hudgins RR, Jarrold MF: Protein structure in vacuo: gas-phase conformations of BPTI and cytochrome c. Journal of the American Chemical Society. 119, 2240–2248 (1997) [Google Scholar]
- 42.Dixit SM, Polasky DA, Ruotolo BT: Collision induced unfolding of isolated proteins in the gas phase: past, present, and future. Current opinion in chemical biology. 42, 93–100 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian Y, Han L, Buckner AC, Ruotolo BT: Collision induced unfolding of intact antibodies: rapid characterization of disulfide bonding patterns, glycosylation, and structures. Analytical chemistry. 87, 11509–11515 (2015) [DOI] [PubMed] [Google Scholar]
- 44.Pierson NA, Clemmer DE: An IMS–IMS threshold method for semi-quantitative determination of activation barriers: Interconversion of proline cis↔ trans forms in triply protonated bradykinin. International journal of mass spectrometry. 377, 646–654 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoaglund CS, Valentine SJ, Sporleder CR, Reilly JP, Clemmer DE: Three-dimensional ion mobility/TOFMS analysis of electrosprayed biomolecules. Analytical chemistry. 70, 2236–2242 (1998) [DOI] [PubMed] [Google Scholar]
- 46.Mason EA, McDaniel EW. Wiley Online Library, (1988)
- 47.Coin I, Beyermann M, Bienert M: Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nature protocols. 2, 3247–3256 (2007) [DOI] [PubMed] [Google Scholar]
- 48.Stewart DE, Sarkar A, Wampler JE: Occurrence and role of cis peptide bonds in protein structures. Journal of molecular biology. 214, 253–260 (1990) [DOI] [PubMed] [Google Scholar]
- 49.Ramachandran GT, Sasisekharan V: Conformation of polypeptides and proteins. Elsevier, (1968) [DOI] [PubMed] [Google Scholar]
- 50.Jorgensen WL, Gao J: Cis-trans energy difference for the peptide bond in the gas phase and in aqueous solution. Journal of the American Chemical Society. 110, 4212–4216 (1988) [Google Scholar]
- 51.Zimmerman SS, Scheraga HA: Stability of cis, trans, and nonplanar peptide groups. Macromolecules. 9, 408–416 (1976) [DOI] [PubMed] [Google Scholar]
- 52.Zhong H, Carlson HA: Conformational studies of polyprolines. Journal of chemical theory and computation. 2, 342–353 (2006) [DOI] [PubMed] [Google Scholar]
- 53.Glover MS, Bellinger EP, Radivojac P, Clemmer DE: Penultimate proline in neuropeptides. Analytical chemistry. 87, 8466–8472 (2015) [DOI] [PubMed] [Google Scholar]
- 54.Glover MS, Shi L, Fuller DR, Arnold RJ, Radivojac P, Clemmer DE: On the split personality of penultimate proline. Journal of the American Society for Mass Spectrometry. 26, 444–452 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Counterman AE, Clemmer DE: Cis− Trans Signatures of Proline-Containing Tryptic Peptides in the Gas Phase. Analytical chemistry. 74, 1946–1951 (2002) [DOI] [PubMed] [Google Scholar]
- 56.Henderson SC, Li J, Counterman AE, Clemmer DE: Intrinsic size parameters for Val, Ile, Leu, Gln, Thr, Phe, and Trp residues from ion mobility measurements of polyamino acid ions. The Journal of Physical Chemistry B. 103, 8780–8785 (1999) [Google Scholar]
- 57.Valentine SJ, Counterman AE, Hoaglund-Hyzer CS, Clemmer DE: Intrinsic amino acid size parameters from a series of 113 lysine-terminated tryptic digest peptide ions. The Journal of Physical Chemistry B. 103, 1203–1207 (1999) [Google Scholar]
- 58.Counterman AE, Clemmer DE: Volumes of individual amino acid residues in gas-phase peptide ions. Journal of the American Chemical Society. 121, 4031–4039 (1999) [Google Scholar]
- 59.Valentine SJ, Counterman AE, Clemmer DE: A database of 660 peptide ion cross sections: use of intrinsic size parameters for bona fide predictions of cross sections. Journal of the American Society for Mass Spectrometry. 10, 1188–1211 (1999) [DOI] [PubMed] [Google Scholar]
- 60.Srebalus Barnes CA, Clemmer DE: Assessing intrinsic side chain interactions between i and i+ 4 residues in solvent-free peptides: a combinatorial gas-phase approach. The Journal of Physical Chemistry A. 107, 10566–10579 (2003) [Google Scholar]
- 61.Pierson NA, Chen L, Russell DH, Clemmer DE: Cis–trans isomerizations of proline residues are key to bradykinin conformations. Journal of the American Chemical Society. 135, 3186–3192 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fort KL, Silveira JA, Pierson NA, Servage KA, Clemmer DE, Russell DH: From solution to the gas phase: factors that influence kinetic trapping of Substance P in the gas phase. The Journal of Physical Chemistry B. 118, 14336–14344 (2014) [DOI] [PubMed] [Google Scholar]
- 63.Moision R, Armentrout P: The special five-membered ring of proline: an experimental and theoretical investigation of alkali metal cation interactions with proline and its four- and six-membered ring analogues. The Journal of Physical Chemistry A. 110, 3933–3946 (2006) [DOI] [PubMed] [Google Scholar]
- 64.Heaton A, Moision R, Armentrout P: Experimental and theoretical studies of sodium cation interactions with the acidic amino acids and their amide derivatives. The Journal of Physical Chemistry A. 112, 3319–3327 (2008) [DOI] [PubMed] [Google Scholar]
- 65.Armentrout P, Gabriel A, Moision R: An experimental and theoretical study of alkali metal cation/methionine interactions. International Journal of Mass Spectrometry. 283, 56–68 (2009) [Google Scholar]
- 66.Butcher DJ, Asano KG, Goeringer DE, McLuckey SA: Thermal dissociation of gaseous bradykinin ions. The Journal of Physical Chemistry A. 103, 8664–8671 (1999) [Google Scholar]
- 67.Sztáray J, Memboeuf A, Drahos L, Vékey K: Leucine enkephalin—a mass spectrometry standard. Mass spectrometry reviews. 30, 298–320 (2011) [DOI] [PubMed] [Google Scholar]
- 68.Meot-Ner M, Dongré AR, Somogyi A, Wysocki VH: Thermal decomposition kinetics of protonated peptides and peptide dimers, and comparison with surface-induced dissociation. Rapid communications in mass spectrometry. 9, 829–836 (1995) [DOI] [PubMed] [Google Scholar]
- 69.Asano KG, Goeringer DE, McLuckey SA: Thermal dissociation in the quadrupole ion trap: ions derived from leucine enkephalin1. International journal of mass spectrometry. 185, 207–219 (1999) [Google Scholar]
- 70.Paech K, Jockusch RA, Williams ER: Slow infrared laser dissociation of molecules in the rapid energy exchange limit. The Journal of Physical Chemistry A. 106, 9761–9766 (2002) [Google Scholar]
- 71.Rodgers MT, Armentrout PB: A Thermodynamic “Vocabulary” for Metal Ion Interactions in Biological Systems. Accounts of Chemical Research. 37, 989–998 (2004) [DOI] [PubMed] [Google Scholar]
- 72.Pierson NA, Valentine SJ, Clemmer DE: Evidence for a quasi-equilibrium distribution of states for bradykinin [M+ 3H] 3+ ions in the gas phase. The journal of physical chemistry B. 114, 7777–7783 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen X, Turecček F: Simple b ions have cyclic oxazolone structures. A neutralization-reionization mass spectrometric and computational study of oxazolone radicals. Journal of the American Society for Mass Spectrometry. 16, 1941–1956 (2005) [DOI] [PubMed] [Google Scholar]
- 74.Capasso S, Vergara A, Mazzarella L: Mechanism of 2, 5-dioxopiperazine formation. Journal of the American Chemical Society. 120, 1990–1995 (1998) [Google Scholar]
- 75.Walter R, Simmons WH, Yoshimoto T: Proline specific endo-and exopeptidases. Molecular and cellular biochemistry. 30, 111–127 (1980) [DOI] [PubMed] [Google Scholar]
- 76.Borthwick AD: 2, 5-Diketopiperazines: synthesis, reactions, medicinal chemistry, and bioactive natural products. Chemical reviews. 112, 3641–3716 (2012) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.