

Abstract
There is currently no accepted classification of autosomal recessive cerebellar ataxias, a group of disorders characterized by important genetic heterogeneity and complex phenotypes. The objective of this task force was to build a consensus on the classification of autosomal recessive ataxias in order to develop a general approach to a patient presenting with ataxia, organize disorders according to clinical presentation, and define this field of research by identifying common pathogenic molecular mechanisms in these disorders. The work of this task force was based on a previously published systematic scoping review of the literature that identified autosomal recessive disorders characterized primarily by cerebellar motor dysfunction and cerebellar degeneration. The task force regrouped 12 international ataxia experts who decided on general orientation and specific issues. We identified 59 disorders that are classified as primary autosomal recessive cerebellar ataxias. For each of these disorders, we present geographical and ethnical specificities along with distinctive clinical and imagery features. These primary recessive ataxias were organized in a clinical and a pathophysiological classification, and we present a general clinical approach to the patient presenting with ataxia. We also identified a list of 48 complex multisystem disorders that are associated with ataxia and should be included in the differential diagnosis of autosomal recessive ataxias. This classification is the result of a consensus among a panel of international experts, and it promotes a unified understanding of autosomal recessive cerebellar disorders for clinicians and researchers.
Keywords: Spinocerebellar degenerations, Cerebellar ataxia, Friedreich ataxia, Ataxia telangiectasia, Genetics, Classification
Introduction
The classification of hereditary ataxias represents a significant challenge due to the large number of neurological and metabolic diseases that present with cerebellar dysfunction and the phenotypic heterogeneity in known genetically defined disorders. Indeed, ataxia is a presenting feature in degenerative disorders that target mainly the cerebellum, but it may be present in hereditary spastic paraplegias, inborn errors of metabolism, and various encephalopathies. Proper classification and phenotypic understanding is of primary importance in this field where the high prevalence of repeat expansion disorders, which are not adequately covered by the next-generation sequencing (NGS) techniques [1, 2], precludes NGS as a first diagnostic step and requires phenotypic evaluation to perform custom gene testing when applicable. Nevertheless, autosomal recessive cerebellar ataxias have remained an ill-defined and disorganized group of disorders for two main reasons. First, unlike the dominant ataxias that have been organized with a numerical naming system, recessive disorders presenting with ataxia have been named in a highly heterogeneous manner according to clinical features, physicians’ surname, or regions of high prevalence. Second, several recessive multisystemic or complex metabolic disorders present with ataxia, such that it is difficult to properly circumscribe this group of disorders and classify it in a meaningful way for both clinicians and researchers. Hence, the Society for Research on the Cerebellum and Ataxias (SRCA) Task Force on the Classification of Recessive Cerebellar Ataxias was created in 2016 to regroup a panel of international ataxia experts in order to propose a classification relevant to clinical practice and researchers. As a first step, we undertook a systematic scoping review of the literature to identify all recessive disorders presenting with ataxia, select those in which cerebellar degeneration was a core feature, and propose a first classification. This systematic scoping review has been previously published [3] and served as the basis for the current work.
Recently, the Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders proposed a revised naming system based on the gene name associated with a phenotypical prefix. They presented a list of 92 gene-defined recessive disorders associated with ataxia for which this naming system would be applied and an exhaustive list of disorders that may occasionally present with ataxia [4]. This represents a useful reference for interpretation of NGS results. However, in a significant number of listed disorders, the cerebellum is only one of the many affected organs in multisystemic and metabolic disorders. For example, maple syrup urine disease, caused by BCKDHB mutations, and congenital disorders of glycosylation 1a, 1c, and 1q have been included. These disorders are inborn errors of metabolism characterized by developmental delay, hypotonia, and metabolic defects, and ataxia is only mild, found in a minority of patients, or present solely during episodes of metabolic decompensation. Hence, there remains a need for a classification system that focuses on disorders affecting primarily the cerebellum and organizes clinical and paraclinical information to promote an understanding of cerebellar disorders useful not only to ataxia experts but also to general neurologists, learners, patients, and researchers.
The objective of this task force was to build a consensus on the classification of autosomal recessive ataxias in order to develop a general approach to a patient presenting with ataxia, organize disorders according to clinical presentation, and define this field of research by identifying common pathophysiological mechanisms in recessive disorders presenting with ataxia. This aims at bringing together clinicians and researchers to promote a common understanding of recessive cerebellar disorders in order to advance research and improve patient care.
Materials and Methods
The first step was to identify all recessive disorders presenting with ataxia. Recessive cerebellar ataxias were defined as disorders with autosomal recessive inheritance characterized by a cerebellar motor syndrome of gait ataxia, dysmetria, adiadochokinesia, nystagmus, and dysarthria associated with cerebellar degeneration as demonstrated by imagery or pathology. A pathogenic mutation had to be identified in at least two independent families for a specific gene to be included. Purely malformative disorders were excluded, and disorders with complex phenotypes where ataxia is a secondary or late feature were also excluded. We conducted a systematic scoping review of the literature to identify relevant reports. The methodology and results of this systematic review have been published previously [3]. In the first publication, this review process had allowed the identification of 2354 records and was current as of September 2016. The literature search was updated and is current as of October 2018.
The second step was to regroup a panel of 12 international ataxia experts to create a logical classification system and build a consensus. Ataxia experts were identified from various geographical regions and areas of expertise within the field of ataxias, ensuring proper representation of regional differences in prevalence and clinical approach to ataxias. Discussions spanned over 2 years, included meetings at two SRCA international conferences, and concerned general orientation, clinical approach, specific disorders, classification issues, and regional specificities. The first author (MB) reviewed identified records for inclusion, extracted clinical, epidemiological, and molecular data to build the classifications and wrote the text integrating all authors’ input and comments. All authors approved the final manuscript and list of included disorders.
Results
The final list of included autosomal recessive cerebellar ataxias is presented in Table 1 and includes 59 primary recessive ataxias, which regroup 15 disorders that are more prevalent and widely distributed and 44 disorders that are less frequent and reported only in certain populations or few families. Because ethnic and regional specificities are an essential element to consider in the appraisal of a patient with a recessive ataxia, areas where the disorder has been reported to date are listed. Metabolic or mitochondrial disorders where ataxia is only a secondary nonspecific finding in a multisystemic phenotype were excluded, as cerebellar pathology is not central in these disorders. However, clinicians must bear in mind that some of these disorders may present with a milder juvenile or adult onset phenotype where cerebellar ataxia may predominate, for example, in Niemann-Pick disease type C, Tay-Sachs disease, sialic acid storage disorders, congenital disorders of glycosylation, and Zellweger spectrum disorders. As some of these metabolic disorders may benefit from early treatment, clinicians must keep a high index of suspicion to test for these disorders, and they should be included in large NGS gene panels for ataxia. These and other complex disorders that may occasionally present with ataxia are presented in Table 2. This second list is not exhaustive and presents only the main or most frequent disorders occasionally associated with ataxia. Disorders in which the cerebellar phenotype is not clearly established have been excluded.
Table 1.
Primary autosomal recessive cerebellar ataxias
| MDS nomenclature1 or gene name | Alternate nomenclature2 | OMIM | Geographic specificities | Additional clinical clues and neuroimaging findings | References |
|---|---|---|---|---|---|
| Most prevalent ataxias | |||||
| ATX-FXN | FRDA | 229300 | Most prevalent in populations of European descent, Middle East, and North Africa; absent in Far East populations | Bilateral Babinski sign, square-wave jerks, scoliosis, hypertrophic cardiomyopathy, sensory involvement, teenage onset, spinal cord atrophy, absence of cerebellar atrophy | [5, 6] |
| ATX-ATM | AT | 208900 | Second most common cause of recessive ataxia worldwide, especially in regions with low inbreeding | Telangiectasias, oculomotor apraxia, photosensitivity, immunodeficiency, predisposition for cancer, dystonia, myoclonus, choreoathetosis, tremor, elevation of α-fetoprotein, infantile onset, cerebellar atrophy | [7–9] |
| ATX-APTX | AOA1/EAOH | 208920 | Most prevalent in Japan; second most prevalent ataxia in Portugal | Oculomotor apraxia, cognitive impairment, axonal motor polyneuropathy, late onset of hypoalbuminemia, elevated α-fetoprotein and hypercholesterolemia, childhood onset, cerebellar atrophy | [10–12] |
| ATX-SETX | AOA2 | 606002 | Worldwide, second most prevalent in Eastern France | Axonal sensorimotor polyneuropathy, pyramidal signs, oculomotor apraxia, head tremor, chorea, dystonia, elevation of α-fetoprotein, teenage onset, cerebellar atrophy | [13–15] |
| ATX/HSP-SACS | ARSACS | 270550 | Worldwide | Spastic paraparesis, retinal striation with thickened retinal nerve fibers, sensorimotor neuropathy, pes cavus, infantile or childhood onset, anterior superior cerebellar atrophy, occasional T2-weighted linear hypointensities in pons | [16, 17] |
| POLG | MIRAS, SANDO, SCAE | 607459 | Prevalent in populations of European descent, especially Scandinavia, UK, and Belgium | Cerebellar and sensory ataxia, dysarthria, progressive external ophthalmoplegia, myoclonus, epilepsy, myopathy, migraine, variable age at onset, signal abnormalities in the cerebellum and thalamus | [18–20] |
| ATX-SYNE1 | ARCA1 | 610743 | Worldwide | Pure cerebellar ataxia with occasional upper and/or lower motor neuron involvement, cognitive impairment, late onset, cerebellar atrophy | [21–23] |
| HSP/ATX-SPG7 | SPG7 | 607259 | Described worldwide, frequent in Europe | Spasticity, pyramidal signs, optic neuropathy, ptosis, ophthalmoparesis, bladder dysfunction, adult onset, cerebellar atrophy | [24, 25] |
| COQ8A (ATX-ADCK3) | ARCA2 | 612016 | European descent, Algeria, Middle East | Exercise intolerance, epilepsy, myoclonus, developmental delay, intellectual disability, childhood onset, cerebellar atrophy, occasional stroke-like cerebral lesions | [26, 27] |
| ATX-ANO10 | ARCA3 | 613728 | European descent, Middle East, West Indies, Japan | Pure cerebellar ataxia with occasional upper motor neuron signs, cognitive impairment, epilepsy, nystagmus, teenage or adult onset, cerebellar atrophy | [28–30] |
| ATX-TTPA | AVED | 277460 | Worldwide, high prevalence around Mediterranean sea | Dorsal column involvement, areflexia, retinitis pigmentosa, head titubation, low serum vitamin E, skeletal deformities, teenage onset, spinal cord atrophy, occasional cerebellar atrophy | [31–33] |
| ATX-CYP27A1 | CTX | 213700 | Worldwide | Dementia, pyramidal signs, epilepsy, tendon xanthomas, atherosclerosis, cataracts, diarrhea, elevated serum cholestanol, polyneuropathy, childhood to adult onset, variable cerebellar atrophy, cerebellar or cerebral white matter anomalies | [34–36] |
| ATX-SIL1 | MSS | 248800 | Worldwide | Cataracts, intellectual disability, myopathy, short stature, childhood onset, cerebellar atrophy | [37, 38] |
| TWNK (ATX-C10orf2) | IOSCA/MTDPS7 | 271245 | Described worldwide, highly prevalent in Finland | Athetosis, sensory axonal neuropathy, hypotonia, optic atrophy, ophthalmoplegia, sensorineural deafness, epilepsy, hypogonadism, liver involvement, infantile onset, atrophy of the brainstem and cerebellum | [39, 40] |
| Rare ataxias or described only in few families | |||||
| ATX-ABHD12 | PHARC | 612674 | Europe, USA, Middle East, Algeria | Demyelinating sensorimotor neuropathy, pes cavus, cataracts, hearing loss, retinitis pigmentosa, teenage onset, cerebellar atrophy | [41, 42] |
| ATX/HSP-AFG3L2 | SPAX5 | 614487 | Colombia, Saudi Arabia | Ataxia, spasticity, oculomotor apraxia, myoclonic epilepsy, neuropathy, extrapyramidal involvement, optic atrophy, severe cases with developmental regression, microcephaly, hypsarrhythmia and intractable epilepsy, infantile to childhood onset, cerebellar atrophy | [43, 44] |
| ATCAY | Cayman ataxia | 601238 | Grand Cayman Islands, Pakistan | Psychomotor retardation, hypotonia, strabismus, bradykinesia, occasional dystonia, neonatal or infantile onset, cerebellar hypoplasia | [45, 46] |
| ATX-CA8 | CAMRQ3 | 613227 | Iran, Saudi Arabia, Syria | Mild intellectual disability, occasional quadrupedal gait, tremor, hyperreflexia, congenital onset, cerebellar atrophy, periventricular white matter anomalies | [47, 48] |
| HSP/ATX-CAPN1 | SPG76 | 616907 | Europe, Middle East, Brazil, Japan, Punjab | Pyramidal signs, pes cavus, dysarthria, ataxia, slow saccades, cognitive impairment, teenage to adult onset, cerebellar vermian atrophy | [49, 50] |
| HSP/ATX-CLCN2 | Leucoencephalopathy with ataxia | 615651 | Europe, North Africa, Turkey, Japan | Chorioretinopathy, optic neuropathy, learning disability, headaches, occasional mild spasticity, childhood to adult onset, T2 hypersignal in cerebellar and cerebral peduncles with internal capsule, myelin microvacuolation | [51, 52] |
| COA7 | MC4D, SCAN3 | 220110 | Italy, Japan | Sensorimotor neuropathy, hyporeflexia, mild cognitive impairment, elevated serum creatine kinase, elevated lactate and pyruvate, ragged red fibers, infantile to childhood onset, cerebellar atrophy, supratentorial leucopathy, spinal cord atrophy | [53, 54] |
| ATX-COX20 | Mitochondrial complex IV deficiency | 220110 | Turkey | Growth retardation, pyramidal signs, sensory neuropathy, extrapyramidal features, elevation of blood lactate, childhood or teenage onset, cerebellar atrophy | [55, 56] |
| ATX-CWF19L1 | SCAR17 | 616127 | Turkey, Netherlands | Intellectual disability, congenital to infantile onset, cerebellar atrophy | [57, 58] |
| HSP/ATX-CYP7B1 | SPG5A | 270800 | Worldwide, prevalent in Europe | Pyramidal signs, dorsal column sensory deficits, urge incontinence or voiding, childhood or teenage onset, white matter lesions | [59, 60] |
| ATX/HSP-DARS2 | LBSL | 611105 | Worldwide, high carrier rate in Finland | Pyramidal signs, dorsal column dysfunction, axonal neuropathy, tremor, cerebral lactic acidosis, seizures, infantile to adult onset, signal abnormalities in cerebral white matter and specific brainstem and spinal cord tracts | [61, 62] |
| ATX-DNAJC19 | DCMA/MGCA5 | 610198 | Canadian Hutterite population, Finland, Turkey | Dilated cardiomyopathy, nonprogressive cerebellar ataxia, intellectual disability, testicular dysgenesis, anemia, increased urinary 3-methylglutaconic acid, infantile onset, progressive cerebellar atrophy | [63–65] |
| HSP/ATX-GBA2 | SPG46 | 614409 | Tunisia, Cyprus, Italy, Norway | Pyramidal signs, spastic dysarthria, cognitive impairment, hearing loss, cataracts, urge incontinence, axonal sensorimotor neuropathy, childhood onset, cerebellar and cerebral atrophy, thin corpus callosum | [66, 67] |
| GDAP2 | – | – | Belgium, Dutchland, Egypt | Pyramidal signs, cognitive impairment, adult onset, cerebellar atrophy | [68] |
| ATX/HSP-GJC2 | HLD2 or Pelizaeus-Merzbacher-like disease | 608804 | Worldwide | Nystagmus, hypotonia progressing to spastic tetraparesis, developmental delay, dystonia, chorea, neonatal to infantile onset, diffuse hypomyelination | [69, 70] |
| MYC/ATX-GOSR2 | Progressive myoclonic epilepsy 6 | 614018 | North Sea region | Areflexia, myoclonic seizures, scoliosis, late cognitive impairment, axonal sensory neuropathy and anterior horn cell involvement, raised creatine kinase, infantile onset, occasional cerebellar atrophy | [71, 72] |
| ATX-GRID2 | SCAR18 | 616204 | Middle East, Mexico, Morocco | Tonic upgaze, vertical nystagmus, oculomotor apraxia, intellectual disability, developmental delay, hypotonia, infantile onset, cerebellar atrophy; possible autosomal dominant transmission | [73, 74] |
| GRM1 | SCAR13 | 614831 | Roma ethnic group in Bulgaria | Developmental delay, intellectual disability, occasional pyramidal signs, short stature, seizures, congenital onset, cerebellar atrophy; allelic with SCA44 | [75, 76] |
| ATX-GRN | CLN11 | 614706 | Italy, Portugal, Brazil | Myoclonic epilepsy, retinopathy, dementia, adult onset, cerebellar atrophy | [77, 78] |
| ATX-ITPR1 | Gillespie syndrome | 206700 | Brazil, Europe, North Africa, Middle East, Asia, Caribbean Islands | Autosomal recessive and dominant transmission. Nonprogressive cerebellar ataxia, iris hypoplasia, hypotonia, intellectual disability, facial dysmorphism, neonatal onset, progressive cerebellar atrophy; allelic with SCA15 and SCA29 | [79, 80] |
| HSP/ATX-KIF1C | SPAX2/SPG58 | 611302 | Palestine, Morocco, Turkey, Germany | Spastic paraparesis with pyramidal signs, tremor, childhood or teenage onset, T2 hyperintensity in internal capsules, parietal and occipital white matter, cerebellar peduncles, and pyramidal tracts | [81, 82] |
| ATX-KCNJ10 | EAST/SeSAME syndrome | 612780 | Africa, Middle East, India, Caucasian, Afro-Caribbean population | Epilepsy, sensorineural deafness, intellectual disability, tubulopathy and electrolyte imbalance, hypotonia progressing to spasticity, infantile onset, cerebellar hypoplasia, signal anomaly in dentate nuclei | [83–85] |
| ATX-L2HGDH | L-2-hydroxyglutaric aciduria | 236792 | Worldwide | Developmental delay, macrocephaly, hypotonia, elevated levels of L-2-hydroxyglutaric acid, infantile to adult onset, subcortical white matter, dentate nucleus and basal ganglia signal anomalies, cerebellar atrophy | [86, 87] |
| ATX-MRE11A | ATLD | 604391 | Described in Europe, Saudi Arabia, Canada, Pakistan and Japan | Oculomotor apraxia, extrapyramidal movement disorders, occasional myoclonus, childhood onset, cerebellar atrophy | [88, 89] |
| MTPAP | SPAX4 | 613672 | Amish families | Pyramidal signs, optic atrophy, sensibility to ionizing radiations, developmental delay, cognitive impairment, growth failure, infantile onset | [90, 91] |
| ATX-PEX10 | PBD 6B or ZSD | 614871 | Caucasians, Japan | Axonal motor or sensorimotor neuropathy, variable cognitive impairment, nystagmus, hypo or hyperreflexia, childhood to adolescent onset, cerebellar atrophy | [92, 93] |
| ATX-PMPCA | SCAR2 | 213200 | Lebanon, France, French Canadians | Intellectual disability, hypotonia, short stature, severe phenotype with lactic academia and ophthalmoplegia, congenital or infantile onset, cerebellar atrophy | [94–96] |
| PNKP | AOA4 | 616267 | European descent | Dystonia, oculomotor apraxia, sensorimotor polyneuropathy, cognitive impairment, childhood onset, cerebellar atrophy | [97–99] |
| ATX/HSP-PNPLA6 | BNS/GHS/OMCS | 215470, 275400 | Worldwide | Hypogonadotropic hypogonadism, chorioretinal dystrophy, pyramidal signs, childhood onset, atrophy of the cerebellum and pons; allelic to HSP39 | [100, 101] |
| ATX/HSP-POLR3A | HLD7, 4H syndrome | 607694 | Worldwide | Tremor, variable cognitive impairment, spasticity, hyperreflexia, variable hypodontia and dysmorphism, hypogonadotropic hypogonadism, myopia, short stature, infantile to childhood onset, diffuse cerebral hypomyelination, cerebellar atrophy, thin corpus callosum, T2 hypointense thalamus | [102, 103] |
| ATX-POLR3B | HLD8 | 614381 | Japan, Caucasians, Syria, African American, Mediterranean | Intellectual disability, vertical gaze limitation, hypodontia, hypogonadotropic hypogonadism, myopia, mild hyperreflexia, short stature, infantile to childhood onset, diffuse cerebral hypomyelination with partly myelinated internal capsule, cerebellar atrophy, thin corpus callosum, T2 hypointense thalamus | [104, 105] |
| ATX-RNF216 | Ataxia and hypogonadotropism/GHS | 212840 | Middle East, Caucasians | Hypogonadotropic hypogonadism, dementia, occasional chorea, childhood to young adult onset, cerebellar atrophy, cerebral white matter anomalies | [106, 107] |
| SCYL1 | SCAR21 | 616719 | European, Middle East, Cuba, Ashkenazi Jews | Transient episodes of liver failure, intention tremor, peripheral sensorimotor neuropathy, mild cognitive impairment, occasional short stature, infantile to childhood onset, cerebellar vermis atrophy | [108, 109] |
| ATX-SNX14 | SCAR20 | 616354 | Portugal, Middle East, North Africa, Central Asia | Intellectual disability, developmental delay, macrocephaly, dysmorphism, hypotonia, skeletal anomalies, occasional sensorineural hearing loss, infantile onset, cerebellar atrophy | [110, 111] |
| SLC9A1 | LIKNS/SCAR19 | 616291 | Turkey, Han Chinese | Occasional sensorineural hearing loss, mild psychomotor delay, infantile to childhood onset, progressive cerebellar atrophy | [112, 113] |
| ATX-SPTBN2 | SCAR14/SPARCA1 | 615386 | Middle East, Egypt, North America | Cognitive impairment, developmental delay, nystagmus, hypotonia, occasional tremor, infantile to childhood onset, cerebellar atrophy; allelic to SCA5 | [114, 115] |
| ATX-STUB1 | SCAR16 | 615768 | China, Middle East, Caucasians | Pyramidal signs, variable cognitive impairment, occasional hypogonadism, variable age at onset, cerebellar atrophy; allelic to SCA48 | [116, 117] |
| TDP2 | SCAR23 | 616949 | Ireland, USA | Seizures, developmental delay, dysmorphism, hypotonia, hypersomnia, failure to thrive, infantile to childhood onset, absence of cerebellar atrophy | [118, 119] |
| ATX-TPP1 | SCAR7 | 609270 | The Netherlands, African American population | Occasional pyramidal signs, posterior column involvement, tremor, square-wave jerks, nystagmus, childhood to adolescent onset, pontocerebellar atrophy; allelic to CLN2 | [120, 121] |
| HSP/ATX-UCHL1 | SPG79 | 615491 | Norway, Turkey | Optic atrophy, nystagmus, intention tremor, pyramidal signs, dorsal column involvement, mild cognitive impairment, childhood onset, cerebellar atrophy | [122, 123] |
| ATX-VLDLR | CAMRQ1/DES | 224050 | North American Hutterite population, Middle East, Europe | Nonprogressive cerebellar ataxia, moderate to severe intellectual disability, hypotonia, strabismus, delayed ambulation with occasional quadripedal gait, seizures, congenital to infantile onset, inferior cerebellar hypoplasia, pontine hypoplasia, cortical gyral simplification | [124, 125] |
| VPS13D | SCAR4 | 607317 | Europe, USA, French Canadian, Egyptian, Javanese | Pyramidal signs, axial hypotonia, oculomotor abnormalities, chorea or dystonia, cognitive impairment, infantile to adult onset, cerebellar atrophy, basal ganglia T2/F hyperintensity | [126, 127] |
| ATX-WDR81 | CAMRQ2/DES2 | 610185 | Turkey, Yemen | Occasional quadrupedal gait, intellectual disability, congenital onset, cerebellar hypoplasia; allelic with Congenital hydrocephalus type 3 with brain anomalies | [128, 129] |
| XRCC1 | SCAR26 | 617633 | India, Pakistan | Oculomotor apraxia with nystagmus, peripheral sensorimotor axonal neuropathy, cognitive impairment, childhood to adult onset, progressive cerebellar atrophy | [130, 131] |
In part inspired from [3]
1MDS nomenclature: nomenclature proposed by the Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders [4] with a phenotypical prefix followed by the gene name. ATX ataxia, HSP hereditary spastic paraplegia, MYC myoclonus
2AOA ataxia with oculomotor apraxia, ARCA autosomal recessive cerebellar ataxia, ARSACS autosomal recessive spastic ataxia of Charlevoix-Saguenay, AT ataxia telangiectasia, ATLD ataxia telangiectasia-like disorder, AVED ataxia with vitamin E deficiency, BNS Boucher-Neuhäuser syndrome, CA Cayman ataxia, CAMRQ cerebellar ataxia mental retardation with or without quadrupedal locomotion, DCMA dilated cardiomyopathy with ataxia, DES disequilibrium syndrome, EAOH early-onset ataxia with oculomotor apraxia and hypoalbuminemia, FRDA Friedreich ataxia, GHS Gordon Holmes syndrome, HLD hypomyelinating leukodystrophy, IOSCA infantile onset spinocerebellar ataxia, LIKNS Lichtenstein-Knorr syndrome, MGCA5 3-methyglutaconic aciduria type 5, MIRAS mitochondrial recessive ataxia syndrome, MC4D mitochondrial complex 4 deficiency, MSS Marinesco-Sjogren syndrome, MTDPS7 mitochondrial DNA depletion syndrome 7, NBIA neurodegeneration with brain iron accumulation, OMCS Oliver McFarlane syndrome, PBD peroxisome biogenesis disorder, PEOA3 progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant 3, PHARC polyneuropathy hearing loss ataxia retinitis pigmentosa and cataract, SANDO sensory ataxic neuropathy with dysarthria and ophthalmoparesis, SCAE spinocerebellar ataxia with epilepsy, SCAN1 spinocerebellar ataxia with axonal neuropathy 1, SCAR spinocerebellar ataxia autosomal recessive, SeSAME seizures sensorineural deafness ataxia mental retardation and electrolyte imbalance, SPAX spastic ataxia, SPG spastic paraplegia, UMN upper motor neuron, ZSD Zellweger spectrum disorder
Table 2.
Other metabolic or complex autosomal recessive disorders that have ataxia as an associated feature
| MDS nomenclature1 or gene name | Alternate nomenclature2 | OMIM | Additional clinical clues | References |
|---|---|---|---|---|
|
ATX-AHI1 ATX-ARL13B ATX-CEP290 ATX-CC2D2A ATX-OFD1 ATX-TMEM231 ATX-TMEM67 ATX-RPGRIP1L Others |
Joubert syndrome (including COACH syndrome) | Many, see 213300 | Developmental delay, ataxia, hypotonia, neonatal breathing abnormalities, intellectual disability, nephronophthisis, congenital onset, agenesis of the cerebellar vermis with molar tooth sign; in COACH syndrome, associated with ocular colobomas and hepatic fibrosis | [132, 133] |
| ATX-ALDH5A1 | Succinic semialdehyde dehydrogenase deficiency | 603147 | Developmental delay, intellectual disability, language dysfunction, hypotonia, hyporeflexia, autistic behavior and hallucinations, infantile to childhood onset, T2 hypersignal in globi pallidi | [134, 135] |
| ATX-ALG6 | CDG1c | 603147 | Developmental delay, hypotonia, seizures, protein-losing enteropathy, psychiatric manifestations, nystagmus, strabismus, failure to thrive, dysmorphic features, neonatal to infantile onset, occasional brain atrophy | [136, 137] |
| DYT/ATX-ATP7B | Wilson disease | 277900 | Tremor, dystonia, parkinsonism, choreoathetosis, liver disease, psychiatric involvement, Kayser-Fleischer rings, childhood to adult onset, T2 hypersignal in basal ganglia or brainstem | [138] |
| ATP8A2 | CAMRQ4 | 615268 | Global development delay, cognitive impairment, microcephaly, ataxia or quadrupedal gait, choreoathetoid movement, congenital onset, cerebellar and cerebral atrophy or delay in myelination | [139, 140] |
| HSP/ATX-B4GALNT1 | SPG26 | 609195 | Pyramidal signs, amyotrophy, progressive hyporeflexia, cognitive impairment, axonal peripheral neuropathy, occasional cerebellar ataxia and extrapyramidal signs, scoliosis, childhood to teenage onset, cerebral cortical atrophy, T2/F white matter hyperintensity | [141] |
| ATX-BTD | Biotinidase deficiency | 253260 | Seizures, hypotonia, developmental delay, optic atrophy, sensorineural hearing loss, skin rash, alopecia, hepatosplenomegaly, optic atrophy, exacerbation during infections, infantile to childhood onset, white matter anomalies including delayed demyelination | [142, 143] |
| MYC-CLN5 | CLN | 256731 | Myoclonic epilepsy, psychomotor retardation or regression, ataxia, visual loss, ataxia, infantile to adult onset, cerebellar and cortical atrophy | [144] |
| NBIA/DYT/PARK-CP | Aceruloplasminemia | 604290 | Diabetes, dementia, parkinsonism, dystonia, cerebellar ataxia, retinal degeneration, involuntary movements, anemia, low serum and urinary copper, adult onset, decreased signal intensity in thalamus, basal ganglia and dentate nucleus | [145] |
| MYC/ATX-CSTB1 | Unverricht and Lundborg disease/EPM1 | 254800 | Stimulus-sensitive and action-sensitive myoclonus, tonic-clonic generalized seizures, mild cerebellar ataxia, cognitive impairment, emotional lability, childhood to adolescent onset, normal brain MRI | [146] |
|
EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5 |
Vanishing white matter disease | 603896 | Cerebellar ataxia with spasticity, clinical deterioration following head trauma, febrile illness or surgery, infantile to adult onset, symmetric and diffusely abnormal cerebral white matter that appears isointense to CSF | [147, 148] |
|
MYC/ATX-EPM2A MYC/ATX-NHLRC1 |
Lafora disease | 607566 | Myoclonus, generalized tonic-clonic seizures, occipital seizures, headaches, behavioral deterioration, rapidly progressive dementia, cerebellar ataxia, spasticity, adolescent onset, normal initial brain MRI with progressive diffuse atrophy | [149, 150] |
| ERCC4 | Xeroderma pigmentosum/Cockayne syndrome | 278760 | Photosensitivity, solar lentigine growth retardation, microcephaly, ataxia, chorea, cognitive impairment, adolescent to adult onset, cerebellar and brainstem atrophy | [151, 152] |
| HSP/ATX/NBIA-FA2H | SPG35/FAHN | 612319 | Spastic paraparesis, pyramidal signs, dystonia, ataxia, dysarthria, optic atrophy, seizures, cognitive impairment, childhood to adolescent onset, T2 subcortical and periventricular white matter hyperintensity, atrophy of the cerebellum and brainstem | [153] |
| ATX/HSP-FOLR1 | Neurodegeneration due to cerebral folate transport deficiency | 613068 | Developmental regression, hypotonia, myoclonic, tonic or astatic seizures, cerebellar ataxia, chorea, tremor, autism spectrum disorder, occasional pyramidal signs, infantile onset, delayed myelination in cerebral white matter, cerebellar atrophy | [154, 155] |
| HSP/ATX-GAN1 | Giant axonal neuropathy 1 | 256850 | Peripheral sensorimotor neuropathy, weakness, amyotrophy, areflexia, pes cavus, typical frizzly hair, ataxia, nystagmus, pyramidal signs, seizures, cognitive impairment, childhood onset, cerebellar or cerebral white matter T2 hypersignal | [156, 157] |
| DYT/PARK-GLB1 | GM1 gangliosidosis type II | 230600 | Developmental regression in childhood with gait disorder and cognitive impairment, dystonia, hepatosplenomegaly, ataxia, skeletal dysplasia, cardiomyopathy, infantile to childhood onset, progressive diffuse brain atrophy | [158, 159] |
| ATX/HSP-HEXA | Tay-Sachs disease | 272800 | Infantile form with weakness, motor regression, startle reaction, myoclonic jerks, decreased attentiveness, cherry red spots, dementia, blindness. Juvenile form with ataxia, dysarthria, incoordination; adult form with ALS-like symptomatology | [160, 161] |
| ATX/HSP-HEXB | Sandhoff disease | 268800 | Similar to Tay-Sachs with organomegaly | [162] |
| HSD17B4 | Perreault syndrome1, D-bifunctional protein deficiency | 233400 | Sensorineural hearing loss, ovarian dysfunction, ataxia, dysarthria, dysmetria, hyperreflexia, cognitive impairment, sensory neuropathy, childhood onset, cerebellar atrophy | [163, 164] |
| HSP-KIAA1840 | SPG11 | 604360 | Spasticity, ataxia, cognitive impairment, sensorimotor neuropathy, childhood or teenage onset, thin corpus callosum, signal abnormalities in cervical cord | [165, 166] |
| MYC/ATX-KCTD7 | EPM3/CLN14 | 611726 | Multifocal myoclonic seizures, status myoclonus, motor and language regression, intellectual disability, cerebellar ataxia, infantile onset, diffuse cerebral and cerebellar atrophy, T2 periventricular white matter hyperintensity | [167, 168] |
| ATX-MAN2B1 | Alpha-mannosidosis | 248500 | Dysmorphism, skeletal abnormalities, visceromegaly, sensorineural hearing loss, immunodeficiency, cognitive impairment, psychosis, ataxia, prenatal to adult onset, cerebellar atrophy, partially empty sella turcica, white matter abnormalities | [169] |
| HSP/ATX-MLC1 | Megalencephalic leukoencephalopathy with subcortical cysts | 604004 | Macrocephaly, initial radiological-clinical discrepancy, eventual motor regression, ataxia, spasticity, epilepsy, cognitive decline, infantile onset, diffuse supratentorial white matter signal anomalies | [170] |
| ATX-MSTO1 | MMYAT | 617619 | Myalgia, proximal muscle weakness, psychiatric manifestations, developmental delay, tremor, dysmetria, pigmentary retinopathy, growth retardation, neonatal to childhood onset, cerebellar atrophy | [171, 172] |
| MTTP | Abetalipoproteinemia | 200100 | Fat malabsorption symptoms, hypocholesterolemia, hypotriglyceridemia, acanthocytosis, sensory loss, hyporeflexia, ataxia, neonatal onset, absence of cerebellar atrophy | [173] |
| MYC/ATX-NEU1 | Neuraminidase deficiency or sialidosis type I and II | 256550 | Myoclonic epilepsy, visual impairment, cherry red spots, ataxia, hyperreflexia, severe phenotype with dysmorphic features, dysostosis multiplex, hepatomegaly, developmental delay, increased urinary bound sialic acid, variable age at onset, diffuse cerebellar and cerebral atrophy | [174, 175] |
| NKX6-2 | SPAX8 with hypomyelinating leukodystrophy | 617560 | Nystagmus, developmental delay, hypotonia followed by rapidly progressive spasticity, weakness, dystonia, dysphagia, ataxia, visual impairment, infantile to childhood onset, brain hypomyelination, occasional cerebellar atrophy | [176, 177] |
|
ATX-NPC1 ATX-NPC2 |
Niemann-Pick type C |
257220 607625 |
Vertical supranuclear ophthalmoplegia, gelastic cataplexy, premature cognitive decline, dystonia, hepatosplenomegaly, respiratory failure, seizures, psychiatric features, neonatal to adult onset, variable cerebellar or cerebral atrophy | [178–180] |
| OPA1 | Behr syndrome | 210000 | Optic atrophy, pyramidal signs, sensorimotor peripheral neuropathy, cerebellar ataxia, developmental delay, gastrointestinal symptoms, infantile or childhood onset, cerebellar atrophy; allelic to dominant optic atrophy 1 | [181, 182] |
| PEX2 | PBD5B/Zellweger spectrum disorder | 614867 | Hypotonia, seizures, inability to feed, ataxia, hyporeflexia, slow saccades, sensorimotor neuropathy, childhood to adult onset, cerebellar atrophy | [183, 184] |
| ATX-PEX7 | PBD9B | 614879 | Retinitis pigmentosa, polyneuropathy, ataxia, anosmia, pes cavus, skeletal abnormalities, ichthyosis, hearing loss, cataracts, cardiomyopathy, elevated phytanic acid, childhood or teenage onset, absence of cerebellar atrophy | [185] |
| ATX-PHYH | Refsum disease | 266500 | Retinitis pigmentosa, polyneuropathy, increased CSF protein, anosmia, sensorineural hearing loss, ichthyosis, ataxia, teenage onset, elevated serum phytanic acid, absence of cerebellar atrophy | [186] |
| NBIA/DYT/PARK-PLA2G6 | NBIA 2A | 256600 | Psychomotor retardation or regression, hypotonia followed by spastic quadriparesis, ataxia, strabismus, nystagmus, infantile to teenage onset, cerebellar atrophy and variable iron accumulation in globi pallidi with associated T2 hypointensity | [187, 188] |
| ATX-PMM2 | CDG 1a | 212065 | Intellectual disability, axial hypotonia, visceral involvement with feeding difficulties and cardiac involvement, dysmorphic features, cerebellar ataxia, strabismus, peripheral neuropathy, retinitis pigmentosa, skeletal abnormalities, infantile to adult onset, cerebellar hypoplasia or atrophy | [189, 190] |
| PxMD/DYT/ATX-PRRT2 | Episodic kinesigenic dyskinesia 1 | 614386 | Seizures, paroxysmal nonkinesigenic dyskinesia, paroxysmal vertigo, episodic ataxia, hemiplegic migraine, rare progressive ataxia, infantile to childhood onset, occasional cerebellar atrophy | [191, 192] |
| ATX-PTRH2 | IMNEPD | 616263 | Developmental delay, intellectual disability, hypotonia, muscular weakness, demyelinating sensorimotor neuropathy, dysmorphism, ataxia, microcephaly, growth retardation, sensorineural deafness, pancreatic insufficiency, infantile onset, variable cerebellar atrophy | [193, 194] |
| SEPSECS | PCH 2D | 613811 | Developmental delay, intellectual disability, hypotonia, nystagmus, microcephaly, seizure, ataxia, spasticity, chorea, congenital to infantile onset, cerebellar and cerebral atrophy, thinning of corpus callosum | [195, 196] |
| ATX-SLC17A5 | Sialic acid storage diseases |
604369 269920 |
Severe neonatal phenotype with ascites, failure to thrive and early death. Milder infantile phenotype with hypotonia, cerebellar ataxia and intellectual disability, infantile to adult onset, hypomyelination, cerebellar atrophy | [197–199] |
| SLC2A1 | GLUT1 deficiency | 606777 | Epileptic encephalopathy, psychomotor retardation, hypotonia, dystonia, microcephaly, ataxia, spasticity, seizures, infantile onset, absence of cerebellar atrophy | [200, 201] |
| ATX-SLC52A2 | SCAR3/BVVLS2 |
271250 614707 |
Sensorimotor neuropathy, optic atrophy, blindness, sensorineural hearing loss, respiratory insufficiency, bulbar involvement, childhood onset, absence of cerebellar atrophy; ataxia is on a spectrum between Brown-Vialetto-Van Laere syndrome type 2 and SCAR3 | [202–204] |
| SLC6A19 | Hartnup disorder | 234500 | Transient manifestations of pellagra, cerebellar ataxia, psychosis, nystagmus and ophthalmoparesis, cognitive impairment, amino aciduria, early onset | [205] |
| SLC25A46 | CMT6B | 616505 | Optic atrophy, blindness, severe sensorimotor neuropathy, hyporeflexia, amyotrophy, pes cavus, sensory loss in lower limbs, sensitive and cerebellar ataxia, nystagmus, divergent strabismus, neonatal to childhood onset, cerebellar and brain atrophy, T2 hyperintensity in cerebellar white matter | [206, 207] |
| ATX-SRD5A3 | CDG 1q | 612379 | Hypotonia, intellectual disability, optic nerve atrophy, nystagmus, ocular colobomas, ichthyosis, palmoplantar keratodermia, mild ataxia, congenital to childhood onset, cerebellar vermis hypoplasia | [208, 209] |
| ATX-TTC19 | MC3DN2 | 615157 | Muscular hypotonia progressing to spasticity, developmental delay, neurological regression with loss of language and ambulation, cognitive regression, rapid evolution, axonal motor neuropathy, psychiatric features, infantile to adult onset, cerebral and cerebellar atrophy, T2 hypersignal in basal ganglia, bilateral inferior olive involvement | [210–212] |
| ATX-WDR73 | GMS/SCAR5 | 251300 | Intellectual disability, nephrotic syndrome, microcephaly, hypotonia, epilepsy, optic atrophy, skin abnormalities, infantile to childhood onset, cerebellar and cerebral atrophy | [213, 214] |
| WFS1 | Wolfram syndrome | 222300 | Diabetes mellitus, optic atrophy, diabetes insipidus, deafness, renal abnormalities, ataxia, intellectual disability, psychiatric features, childhood to adolescent onset, generalized brain and cerebellar atrophy | [215] |
| WWOX | SCAR12 | 614322 | Tonic-clonic epilepsy, intellectual disability, spasticity, neonatal to childhood onset, variable cerebellar or cerebral atrophy, phenotypic spectrum with infantile epileptic encephalopathy associated with psychomotor retardation and growth retardation | [216, 217] |
1MDS nomenclature: nomenclature proposed by the Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders [4] with a phenotypical prefix followed by the gene name. ATX ataxia, DYT dystonia, HSP hereditary spastic paraplegia, MYC myoclonus, NBIA neurodegeneration with brain iron accumulation, PARK Parkinsonism
2ALS amyotrophic lateral sclerosis, BVVLS2 Brown-Vialetto-Van Laere syndrome type 2, CAMRQ cerebellar ataxia mental retardation with or without quadrupedal locomotion, CDG congenital disorder of glycosylation, CLN neuronal ceroid lipofuscinosis, CMT Charcot-Marie-Tooth, COACH cerebellar vermis hypoplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis, EPM progressive myoclonic epilepsy, FAHN fatty acid hydroxylase-associated neurodegeneration, GMS Galloway-Mowat syndrome, IMNEPD infantile-onset multisystem neurologic, endocrine, and pancreatic disease, MC3DN2 mitochondrial complex III deficiency, nuclear type 2, MMYAT mitochondrial myopathy and ataxia, NBIA neurodegeneration with brain iron accumulation, PBD peroxisome biogenesis disorder, PCH pontocerebellar hypoplasia, SCAR spinocerebellar ataxia autosomal recessive, SPAX spastic ataxia, SPG spastic paraplegia
Clinical Approach to a Patient Presenting with Ataxia
The first step in evaluating a patient with ataxia is to perform a detailed clinical evaluation that includes a clinical history, a family history, a targeted neurological and systemic physical evaluation, and relevant paraclinical tests. The temporal course is a central element in determining the underlying etiology. Indeed, a chronic progressive evolution over months to years, without trauma or toxin exposure, is suggestive of a hereditary disorder, whereas acute or subacute onset points towards an acquired etiology. A clinical history and physical examination are essential to assess the severity of the cerebellar syndrome and the presence of associated neurological features or systemic involvement. Headache, fever, or an associated autoimmune disorder should prompt the consideration of acquired etiologies. A detailed family history should be obtained to search for relatives with similar symptomatology. Laboratory tests may be useful to rule out acquired causes or as biomarkers for certain disorders. Neuroimaging, preferably with magnetic resonance imaging, is an essential tool to evaluate the presence of cerebellar atrophy or signal anomalies, to search for associated pontine atrophy, and to rule out space-occupying lesions. Electromyography and nerve conduction studies can prove the presence of clinically suspected or subclinical neuropathy and provide evidence of associated myopathy.
-
2.
Following the clinical assessment, one should verify that acquired and treatable causes for ataxia have been excluded. These include vascular disease, trauma, infection, primary or metastatic tumor, excess alcohol consumption, vitamin deficiency, Creutzfeldt-Jakob disease, and immune-mediated cerebellar ataxias such as multiple sclerosis, gluten ataxia, anti-GAD (glutamic acid decarboxylase) ataxia, and paraneoplastic cerebellar degenerations. Clinical evaluation should reveal previous exposure to toxins or traumatic injuries, along with specific signs and symptoms suggestive of infectious, vascular, or metastatic disease. Laboratory tests are useful to identify vitamin deficiencies or autoimmune conditions. Specifically, testing for antibodies involved in paraneoplastic or autoimmune cerebellar degeneration may be particularly useful for patients with a subacute progression, older age at onset, and absence of family history. The paraneoplastic antibodies most associated with cerebellar degeneration are anti-Yo, anti-Hu, anti-Tr, and anti-mGluR1 antibodies; the tumors most often involved are breast and gynecological tumors, Hodgkin lymphoma, and small-cell lung carcinoma [218]. Large paraneoplastic autoantibody panels are now available and may reduce the delay associated with serial testing.
-
3.
Once acquired causes have been ruled out, a genetic etiology may be considered, especially in the presence of a positive family history, early onset, chronic progressive course, or with a set of clinical signs and symptoms that is reminiscent of a well-described genetic disorder. One should bear in mind that a negative family history does not rule out a genetic cause, and sporadic cases may be due to recessive or mitochondrial inheritance, de novo mutations, genetic anticipation, incomplete penetrance, variability in disease expression, paternity error, gonadic mosaicism, or incomplete phenotyping of family members. Indeed, recessive disorders may appear as sporadic in small kindred or with incomplete family history. In other cases, a complete family history should allow identification of the mode of transmission.
-
4.
If autosomal recessive inheritance is suspected, the next step in clinical evaluation is to consider age at onset and clinical signs and symptoms to evaluate if the clinical picture is reminiscent of a well-described disorder. Presentation in infancy suggests ataxia telangiectasia or autosomal recessive ataxia of Charlevoix-Saguenay. Childhood or teenage onset should raise the suspicion for Friedreich ataxia, ataxia with oculomotor apraxia 1 and 2, and POLG-related disorders. Finally, recessive ataxia with onset in adulthood is evocative of autosomal recessive cerebellar ataxia 1 and 3 and spastic paraplegia 7. However, there are large variations in the age at onset of most of the presented disorders, and Friedreich ataxia is one of the best examples with some patients presenting with late-onset (> 25 years of age) or very-late-onset Friedreich ataxia (> 40 years of age). Clinical signs and symptoms may provide clues to identify the mutated gene. Indeed, certain discriminating clinical features or combinations of neurological symptoms may be helpful to guide the clinician towards specific genes (Fig. 1 and Table 1). As one may observe in Fig. 1, none of the autosomal recessive ataxias reported up to now presents with a pure cerebellar phenotype. Even SYNE1-related autosomal recessive cerebellar ataxia 1, which used to be the prototype of a pure cerebellar phenotype [21], has recently been reported to be associated with upper and/or lower motor neuron involvement in 58% of cases, with some rare patients presenting with a very severe early-onset neuromuscular phenotype [22]. The presence of motor neuron involvement, polyneuropathy, extrapyramidal movement disorders, eye movement abnormalities such as oculomotor apraxia, intellectual impairment, and associated multisystemic involvement may guide the clinician towards a particular diagnosis. Some clinical syndromes are particularly evocative of specific disorders. Multisystemic involvement with sensory loss, muscle weakness, cardiomyopathy, diabetes, optic atrophy, and sensorineuronal hearing loss is characteristic of Friedreich ataxia, which is the prototype of a disorder associated with mitochondrial dysfunction. Other associated disorders present with similar features and occasionally epilepsy, retinal involvement, or ophthalmoplegia, such as POLG-related disorders, autosomal recessive cerebellar ataxia 2, and Marinesco-Sjogren syndrome. Extrapyramidal involvement with oculomotor apraxia, elevated α-fetoprotein, and occasional polyneuropathy are typical findings of ataxia telangiectasia, ataxia telangiectasia-like disorder, spinocerebellar ataxia recessive 26, and ataxia with oculomotor apraxia types 1, 2, and 4. Nevertheless, autosomal recessive ataxias are characterized by important phenotypic variability and significant clinical overlap between different pathologies, such that predicting the mutated gene according to the clinical phenotype is prone to errors even for ataxia experts [219]. Some laboratory tests may serve as useful biomarkers for recessive ataxias. Altered levels of vitamin E, α-fetoprotein, albumin, coenzyme Q10, cholesterol, cholestanol, lactate, sex hormones, and gonadotropins have been associated with specific disorders (see Table 1). Dosing of immunoglobulins, very long chain fatty acids, and hexosaminidase may be relevant according to clinical suspicion.
Fig. 1.

Clinical classification of autosomal recessive ataxias. The gene associated with each primary recessive ataxia is classified according to the most frequent clinical syndrome described for this disorder. Note that some disorders have more complex or variable phenotypes and are placed in the overlapping areas between two categories. Genes presented in larger font represent the most prevalent ataxias
-
5.
Once the clinical assessment is complete, genetic testing is indicated to confirm the mutated gene or allow a more specific diagnosis if the clinical picture is nonspecific. Initial testing should include searching for the Friedreich ataxia-associated trinucleotide repeat expansion in the FXN gene considering the high prevalence of this mutation, its incomplete coverage through the next-generation sequencing methods [1], and the heterogeneous clinical phenotype. Searching for a FXN repeat expansion can be done with frataxin protein analysis or gene analysis with Southern blot or PCR. Moreover, clinicians may consider testing for another specific gene through Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA) if the clinical and paraclinical data are highly evocative of a particular disorder, if there is a confirmed mutation in a relative or in isolated populations where selected disorders are highly prevalent. Finally, a panel for the dominantly inherited CAG-repeat expansion spinocerebellar ataxias may also be considered as part of the initial assessment if family history is inconclusive regarding the mode of inheritance and considering the high prevalence of these mutations and their incomplete coverage through the next-generation sequencing methods [1].
-
6.
If single gene testing does not provide a molecular diagnosis, one should consider the high-throughput NGS methods either with a multigene panel, whole exome sequencing, or whole genome sequencing. Several studies have demonstrated the efficacy and cost efficiency of multigene panels [220], targeted exome sequencing [219, 221], or whole exome sequencing [222, 223], with a diagnostic yield varying between 18 and 80%. The highest yield is obtained for patients with early-onset ataxia and positive family history and consanguinity among parents. NGS panels allow for better coverage of included genes and reduce the volume of genetic variants that are unrelated to the clinical phenotype, while exome sequencing may reveal mutations in genes that were not previously known to be associated with ataxia [1]. Whole genome sequencing may be considered in selected cases with appropriate genetic counseling, but its diagnostic yield is uncertain [224]. Once genetic testing is completed and a pathogenic mutation has been identified, it is of primary importance to provide specialized genetic counseling for the patient and his or her relatives along with symptom management and disease treatment when available. Figure 2 presents a graphical summary of the proposed clinical approach.
Fig. 2.
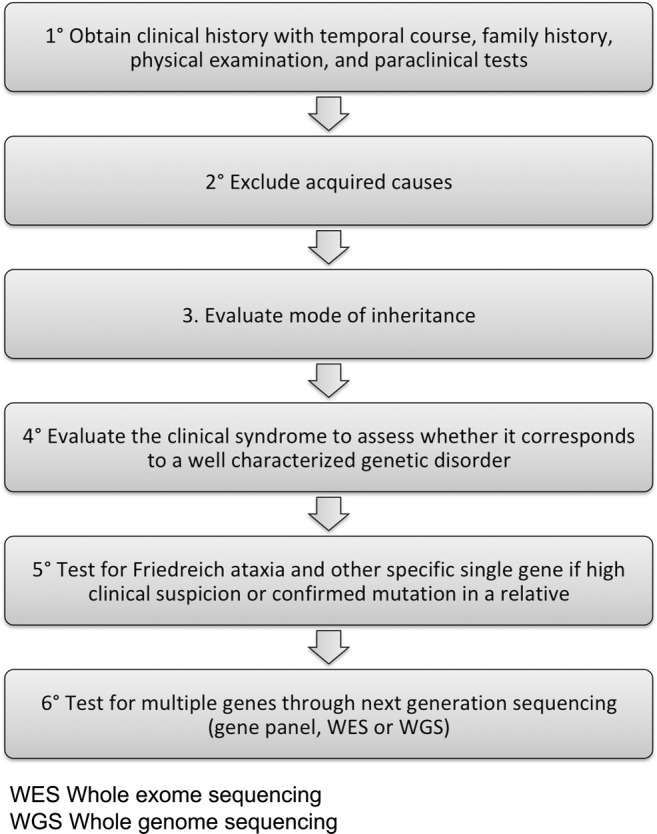
Graphical summary of the clinical approach to a patient presenting with ataxia
Pathophysiological Mechanisms Underlying Autosomal Recessive Cerebellar Ataxias
The importance of a proper recessive ataxia classification goes beyond the clinical diagnosis perspective. Autosomal recessive ataxias can be regrouped according to the deficient cellular and metabolic pathways involved, which provide a better understanding of cerebellar physiology and of its selective vulnerability to certain metabolic defects. This is also essential from a therapeutic perspective, as disorders that belong to the same metabolic pathway may respond to the same treatment options, indicating potential for drug repurposing. Figure 3 presents a pathophysiological classification of autosomal recessive ataxias. Certain genes are presented more than once since some proteins are involved in several metabolic pathways or may interfere with other cellular processes as they accumulate in neurons or glial cells. Table 3 presents a more detailed listing of the pathogenic pathways involved along with relevant references.
Fig. 3.

Pathophysiological classification of autosomal recessive ataxias. A Purkinje cell is depicted along with a granule cell and parallel fibers. Subcellular organelles and structures are represented graphically. Each gene is classified at one or more subcellular localizations according to the different metabolic pathways involved
Table 3.
Detailed pathogenic mechanisms involved in autosomal recessive cerebellar ataxias
| Pathophysiological mechanism | Genes involved | References | |
|---|---|---|---|
| Mitochondrial defect | Mitochondrial DNA maintenance | PNKP, POLG, TWNK | [225–227] |
| Mitochondrial protein synthesis or quality control | AFG3L2, PMPCA, SPG7 | [94, 228, 229] | |
| Increased reactive oxygen species and oxidative stress | ATM, FXN, TTPA | [230–233] | |
| Coenzyme Q10 metabolism | APTX, COQ8A | [234, 235] | |
| Altered mitochondrial dynamics | SACS, VPS13D | [127, 236–238] | |
| Mitochondrial respiratory chain assembly | COA7, COX20 | [54, 55] | |
| Mitochondrial RNA maturation and processing | DARS2, MTPAP | [91, 239, 240] | |
| Others | DNAJC19, L2HGDH, STUB1 | [241–243] | |
| DNA break repair dysfunction | Double-strand break repair | ATM, MRE11A, SETX, TDP2 | [118, 230, 244, 245] |
| Single-strand break repair | APTX, PNKP, XRCC1 | [130, 225] | |
| RNA transcription or processing defect | CWF19L1, POLR3A, POLR3B, SETX | [57, 104, 246] | |
| Synaptic dysfunction | Aberrant morphology at the PC/parallel fibers synapse | CA8, CAPN1, GRID2, ITPR1 | [50, 247–249] |
| Impaired dendritic architecture | SPTBN2, SYNE1 (MF/CGN synapse) | [250, 251] | |
| Dysregulation of glutamate transmission | ATCAY, GRM1 | [252, 253] | |
| Others | GOSR2, RNF216, SLC9A1, UCHL1 | [254–257] | |
| Abnormal cytoskeleton architecture | SACS, SYNE1 | [251, 258] | |
| Abnormal protein folding or quality control | SACS, SIL1, STUB1 | [259–261] | |
| Golgi apparatus dysfunction | COQ8A, GOSR2, SCYL1 | [235, 255, 262] | |
| Calcium homeostasis dysregulation | AFG3L2, ANO10, CA8, ITPR1, SPG7 | [79, 247, 263–266] | |
| Lysosomal dysfunction | GRN, TPP1 | [267, 268] | |
| Disrupted autophagy | SNX14, VPS13D | [238, 269] | |
| Defective ubiquitin-proteasome pathway | RNF216, STUB1, UCHL1 | [256, 257, 261] | |
| Altered intracellular trafficking | GOSR2, SCYL1, SPTBN2, SYNE1, TTPA | [250, 251, 255, 262, 270] | |
| Altered lipid metabolism | ABHD12, CYP27A1, CYP7B1, DNAJC19, GBA2, PNPLA6, SNX14, TTPA | [60, 233, 241, 269, 271–274] | |
| Axonal dysfunction | DARS2, GJC2, KCNJ10, PNPLA6, SLC9A1 | [239, 274–277] | |
| Abnormal myelin structure or composition | ABHD12, CLCN2, GJC2, KCNJ10, KIF1C, L2HGDH, POLR3A, POLR3B | [242, 276–281] | |
| Disrupted intrinsic Purkinje cell firing | SACS, SPTBN2 | [250, 282] | |
| Abnormal cellular stress response | GDAP2 | [68] | |
| Peroxisome dysfunction | PEX10 | [92] | |
| Impaired mitosis | WDR81 | [283] | |
| Abnormal neuronal migration | VLDLR | [284] | |
CGN cerebellar granule neuron, MF mossy fiber, PC Purkinje cell
Certain pathways are predominantly involved, notably mitochondrial dysfunction, which may result from abnormal mitochondrial DNA maintenance with progressive mutagenesis, defective mitochondrial protein synthesis and quality control, increased levels of reactive oxygen species and oxidative stress, deficient coenzyme Q10 metabolism, altered mitochondrial dynamics, defective mitochondrial chain assembly, or abnormal mitochondrial RNA maturation and processing (Table 3). Interestingly, many of the disorders caused by mitochondrial dysfunction also present with a mitochondrial clinical syndrome as shown in Fig. 1. Disorders of DNA repair mechanisms are also common, with double-strand break repair pathway or single-strand break repair complexes predominantly involved. Pathogenic mutations in these genes are also associated with a susceptibility to ionizing radiations and predisposition for cancers, but the neurological syndrome is characterized by cerebellar involvement and extrapyramidal movement disorders. It remains debated whether defective DNA repair is the main pathogenic mechanism causing the neurological phenotype [230], but the fact that several interacting genes in this pathway are involved in degenerative cerebellar ataxias suggests that the cerebellum has a peculiar susceptibility to DNA damage for which the underlying mechanism is not understood. Finally, altered synaptic morphology or synaptic dysfunction of Purkinje cells (PC) is frequently involved in recessive ataxias and is associated with aberrant morphology at the PC/parallel fiber synapse, impaired dendritic architecture, or dysregulation of glutamate transmission. Other disorders have been implicated in synaptic dysfunction through indirect evidence, for example, SLC9A1, which localizes in presynaptic terminals and is involved in the modulation of synaptic activity [254, 275]. Of interest, many of these disorders are characterized by significant cognitive impairment that goes beyond what is expected in the cerebellar cognitive-affective syndrome and cause intellectual disability, developmental delay, or dementia, highlighting the importance of synaptogenesis in cognitive development.
Discussion
We present a new clinical classification of autosomal recessive ataxias in parallel with a pathophysiological classification. The objective of this classification is to provide a tool for clinicians and researchers that facilitates the understanding of this complex group of disorders and defines this field of research. This work is based on the results of our systematic scoping review of the literature [3]. We updated this literature review and regrouped a panel of 12 international ataxia experts to build a consensus on the definition and classification of cerebellar ataxias. The task force vision is that a classification goes beyond the listing of disorders and must organize diseases in a way that allows better understanding and clinical mastery of this group of disorders. Hence, we proposed a clinical classification along with a pathophysiological classification, which enabled us to observe that there is significant overlap between these two classifications, highlighting how clinical presentation is in some cases a good projection of the underlying biochemical defect. This has potential applications from bench to bedside since treatments that address a specific pathogenic pathway may have therapeutic potential in all disorders in which this pathway is affected. The clinical classification is presented along with a structured clinical approach to a patient presenting with ataxia, which is intended as a clinical tool for expert and nonexpert clinicians. Despite the increasing accessibility of the NGS techniques, there remains a critical place for clinical judgment in the prescription of genetic tests and interpretation of results, taking into account the technical limitations and risk of finding variants of unknown significance. Recently, Renaud and colleagues published the results of a diagnostic algorithm for recessive ataxias that integrates 124 clinical features to propose three potential diagnoses among a list of 67 recessive disorders that may present with ataxia [285]. This is a very promising tool, but its pragmatic impact on molecular testing strategy, final diagnostic rate, patient management, or time efficiency remains to be validated. In the meantime, it is essential for clinicians to be at ease with a general approach to recessive ataxias with the NGS techniques often permitting molecular diagnosis when the clinical picture is nonspecific.
One of the major strengths of this classification proposal is that it is based on a consensus from a panel of international ataxia experts, thereby ensuring a proper representation of regional differences in the prevalence and clinical approach to ataxias. Moreover, the literature search was based on a systematic scoping review of the literature whose methodology has been published before and which permitted an unbiased appraisal of all potentially relevant articles. Nevertheless, there are some limitations to this classification proposal that are inherent to classifying a group of diseases that evolves very rapidly and that is highly heterogeneous. First, as new evidence emerges regarding the identification of novel ataxia-associated genes and as new phenotypes are described for previously described disorders, this classification will need to be updated. This was highlighted by the significant additions to the list of primary recessive ataxias since the original systematic review was conducted in 2016. Indeed, many new genes and new phenotypes of previously described genes have been reported in only 2 years, which suggests that there is a need for periodic updates to the present classification or an online resource. Moreover, several decisions were made in the elaboration of this classification regarding general orientation, purpose of a classification, inclusion of specific disorders, and classification categories. The lists presented here offer in our opinion the best compromise between synthesis and exhaustiveness for the expert and nonexpert clinician.
Compared with a previously published report by the Movement Disorders Society Task Force [4], we decided to exclude disorders in which cerebellar involvement is a minor or late finding in a complex multisystem phenotype or disorders that are already classified on their own, such as genes associated with Joubert syndrome. The objective was to identify the core disorders that are involved in autosomal recessive ataxias in order to define this field of research and build a classification that would be accessible for all clinicians. Indeed, with the progressive advent of affordable NGS diagnostic testing, we believe that it is most important for clinicians to be at ease with one classification and familiar with the most frequent disorders in their unique ethnical and clinical context. Disorders in which ataxia has been reported as a rare or late finding should be included in large NGS testing strategies, but in our opinion should not be categorized as primary ataxias per se. From this perspective, our classification complements the proposal by the Movement Disorders Society Task Force.
There remain some important challenges to be addressed in the field of autosomal recessive ataxias. First, the issue of a proper nomenclature system has been much debated. Recently, the Movement Disorders Society Task Force proposed a revised naming system based on an ataxia prefix associated with the gene name [4]; this was part of a larger effort to revise the nomenclature of all genetic movement disorders. This system overcomes the limitations of the numbered nomenclature, notably unconfirmed genes, and erroneously attributed phenotypes, but its ease of use by nonexperts and patients remains uncertain. Moreover, some disorders were assigned as many as three phenotypic prefixes while some other disorders that are among the most prevalent causes of recessive ataxia, such as POLG, were not assigned an ataxia prefix. Hence, there remains a debate concerning the attribution of prefixes and the integration of this naming system with other fields in neurology and other specialties as many genes involved in ataxia have very complex multisystem phenotypes. Finally, one of the most important challenges in this field of orphan diseases is to develop targeted treatment strategies that address the pathogenic mechanism underlying symptom progression. To this end, we believe that identifying common pathophysiological pathways may provide an opportunity for drug repurposing or enlarge the number of patients that are admissible for drug trials in order to find treatments for these rare but debilitating diseases.
Conclusion or Summary
We present a clinical and a pathophysiological classification of autosomal recessive cerebellar ataxias along with a clinical approach to a patient presenting with ataxia. This classification is the result of a consensus among a panel of international experts, and it promotes a unified understanding of autosomal recessive cerebellar disorders for clinicians and researchers.
Acknowledgments
We thank Miruna Anohim for her contribution to the data collection on geographical specificities. Marie Beaudin is supported by the Canadian Institutes of Health Research.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Klein CJ, Foroud TM. Neurology individualized medicine: when to use next-generation sequencing panels. Mayo Clin Proc. 2017;92(2):292–305. doi: 10.1016/j.mayocp.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Bahlo Melanie, Bennett Mark F, Degorski Peter, Tankard Rick M, Delatycki Martin B, Lockhart Paul J. Recent advances in the detection of repeat expansions with short-read next-generation sequencing. F1000Research. 2018;7:736. doi: 10.12688/f1000research.13980.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaudin M, Klein CJ, Rouleau GA, Dupre N. Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias. 2017;4:3. doi: 10.1186/s40673-017-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi M, Anheim M, Durr A, Klein C, Koenig M, Synofzik M, et al. The genetic nomenclature of recessive cerebellar ataxias. Mov Disord. 2018;33(7):1056–1076. doi: 10.1002/mds.27415. [DOI] [PubMed] [Google Scholar]
- 5.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 6.Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335(16):1169–1175. doi: 10.1056/nejm199610173351601. [DOI] [PubMed] [Google Scholar]
- 7.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 8.Wright J, Teraoka S, Onengut S, Tolun A, Gatti RA, Ochs HD, et al. A high frequency of distinct ATM gene mutations in ataxia-telangiectasia. Am J Hum Genet. 1996;59(4):839–846. [PMC free article] [PubMed] [Google Scholar]
- 9.Levy Ariel, Lang Anthony E. Ataxia-telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Movement Disorders. 2018;33(8):1238–1247. doi: 10.1002/mds.27319. [DOI] [PubMed] [Google Scholar]
- 10.Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- 11.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29(2):189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 12.Renaud M, Moreira MC, Ben Monga B, Rodriguez D, Debs R, Charles P, et al. Clinical, biomarker, and molecular delineations and genotype-phenotype correlations of ataxia with oculomotor apraxia type 1. JAMA Neurol. 2018;75(4):495–502. doi: 10.1001/jamaneurol.2017.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36(3):225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 14.Le Ber I, Bouslam N, Rivaud-Pechoux S, Guimaraes J, Benomar A, Chamayou C, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127(Pt 4):759–767. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- 15.Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11(1):1–12. doi: 10.1007/s10048-009-0196-y. [DOI] [PubMed] [Google Scholar]
- 16.Engert JC, Berube P, Mercier J, Dore C, Lepage P, Ge B, et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000;24(2):120–125. doi: 10.1038/72769. [DOI] [PubMed] [Google Scholar]
- 17.Criscuolo C, Banfi S, Orio M, Gasparini P, Monticelli A, Scarano V, et al. A novel mutation in SACS gene in a family from southern Italy. Neurology. 2004;62(1):100–102. doi: 10.1212/wnl.62.1.100. [DOI] [PubMed] [Google Scholar]
- 18.Van Goethem G, Martin JJ, Dermaut B, Lofgren A, Wibail A, Ververken D, et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord. 2003;13(2):133–142. doi: 10.1016/s0960-8966(02)00216-x. [DOI] [PubMed] [Google Scholar]
- 19.Winterthun S, Ferrari G, He L, Taylor RW, Zeviani M, Turnbull DM, et al. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. 2005;64(7):1204–1208. doi: 10.1212/01.wnl.0000156516.77696.5a. [DOI] [PubMed] [Google Scholar]
- 20.Cohen BH, Chinnery PF, Copeland WC. POLG-related disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K et al., editors. GeneReviews((R)). Seattle (WA) 1993.
- 21.Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39(1):80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- 22.Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmuller C, Baets J, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain. 2016;139(Pt 5):1378–1393. doi: 10.1093/brain/aww079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izumi Y., Miyamoto R., Morino H., Yoshizawa A., Nishinaka K., Udaka F., Kameyama M., Maruyama H., Kawakami H. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology. 2013;80(6):600–601. doi: 10.1212/WNL.0b013e3182815529. [DOI] [PubMed] [Google Scholar]
- 24.Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93(6):973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 25.Pfeffer G, Pyle A, Griffin H, Miller J, Wilson V, Turnbull L, et al. SPG7 mutations are a common cause of undiagnosed ataxia. Neurology. 2015;84(11):1174–1176. doi: 10.1212/WNL.0000000000001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagier-Tourenne C, Tazir M, Lopez LC, Quinzii CM, Assoum M, Drouot N, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82(3):661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82(3):623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vermeer S, Hoischen A, Meijer RP, Gilissen C, Neveling K, Wieskamp N, et al. Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am J Hum Genet. 2010;87(6):813–819. doi: 10.1016/j.ajhg.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chamova T, Florez L, Guergueltcheva V, Raycheva M, Kaneva R, Lochmuller H, et al. ANO10 c.1150_1151del is a founder mutation causing autosomal recessive cerebellar ataxia in Roma/Gypsies. J Neurol. 2012;259(5):906–911. doi: 10.1007/s00415-011-6276-6. [DOI] [PubMed] [Google Scholar]
- 30.Renaud M, Anheim M, Kamsteeg EJ, Mallaret M, Mochel F, Vermeer S, et al. Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: delineation and genotype-phenotype correlation study. JAMA Neurol. 2014;71(10):1305–10. doi: 10.1001/jamaneurol.2014.193. [DOI] [PubMed] [Google Scholar]
- 31.Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, et al. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet. 1995;9(2):141–145. doi: 10.1038/ng0295-141. [DOI] [PubMed] [Google Scholar]
- 32.Yokota T, Shiojiri T, Gotoda T, Arita M, Arai H, Ohga T, et al. Friedreich-like ataxia with retinitis pigmentosa caused by the His101Gln mutation of the alpha-tocopherol transfer protein gene. Ann Neurol. 1997;41(6):826–832. doi: 10.1002/ana.410410621. [DOI] [PubMed] [Google Scholar]
- 33.El Euch-Fayache G, Bouhlal Y, Amouri R, Feki M, Hentati F. Molecular, clinical and peripheral neuropathy study of Tunisian patients with ataxia with vitamin E deficiency. Brain. 2014;137(Pt 2):402–410. doi: 10.1093/brain/awt339. [DOI] [PubMed] [Google Scholar]
- 34.Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266(12):7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 35.Leitersdorf E, Reshef A, Meiner V, Levitzki R, Schwartz SP, Dann EJ, et al. Frameshift and splice-junction mutations in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis in Jews or Moroccan origin. J Clin Invest. 1993;91(6):2488–2496. doi: 10.1172/JCI116484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong JC, Walsh K, Hayden D, Eichler FS. Natural history of neurological abnormalities in cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2018;41(4):647–656. doi: 10.1007/s10545-018-0152-9. [DOI] [PubMed] [Google Scholar]
- 37.Anttonen AK, Mahjneh I, Hamalainen RH, Lagier-Tourenne C, Kopra O, Waris L, et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet. 2005;37(12):1309–1311. doi: 10.1038/ng1677. [DOI] [PubMed] [Google Scholar]
- 38.Senderek J, Krieger M, Stendel C, Bergmann C, Moser M, Breitbach-Faller N, et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet. 2005;37(12):1312–1314. doi: 10.1038/ng1678. [DOI] [PubMed] [Google Scholar]
- 39.Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lonnqvist T, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet. 2005;14(20):2981–2990. doi: 10.1093/hmg/ddi328. [DOI] [PubMed] [Google Scholar]
- 40.Park MH, Woo HM, Hong YB, Park JH, Yoon BR, Park JM, et al. Recessive C10orf2 mutations in a family with infantile-onset spinocerebellar ataxia, sensorimotor polyneuropathy, and myopathy. Neurogenetics. 2014;15(3):171–182. doi: 10.1007/s10048-014-0405-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiskerstrand T, H’Mida-Ben Brahim D, Johansson S, M’Zahem A, Haukanes BI, Drouot N, et al. Mutations in ABHD12 cause the neurodegenerative disease PHARC: an inborn error of endocannabinoid metabolism. Am J Hum Genet. 2010;87(3):410–417. doi: 10.1016/j.ajhg.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisenberger T, Slim R, Mansour A, Nauck M, Nurnberg G, Nurnberg P, et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J Rare Dis. 2012;7:59. doi: 10.1186/1750-1172-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierson TM, Adams D, Bonn F, Cherikuri PF, Teer JK, Hanson NF, et al. Whole exome sequencing identifies AFG3L2 mutation in a novel recessive progressive myoclonic epilepsy-ataxia-neuropathy syndrome. Ann Neurol. 2010;68:S68–SS9. [Google Scholar]
- 44.Eskandrani A, AlHashem A, Ali ES, AlShahwan S, Tlili K, Hundallah K, et al. Recessive AFG3L2 mutation causes progressive microcephaly, early onset seizures, spasticity, and basal ganglia involvement. Pediatr Neurol. 2017;71:24–28. doi: 10.1016/j.pediatrneurol.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 45.Bomar JM, Benke PJ, Slattery EL, Puttagunta R, Taylor LP, Seong E, et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35(3):264–269. doi: 10.1038/ng1255. [DOI] [PubMed] [Google Scholar]
- 46.Manzoor Humera, Brüggemann Norbert, Hussain Hafiz Muhammad Jafar, Bäumer Tobias, Hinrichs Frauke, Wajid Muhammad, Münchau Alexander, Naz Sadaf, Lohmann Katja. Novel homozygous variants in ATCAY , MCOLN1 , and SACS in complex neurological disorders. Parkinsonism & Related Disorders. 2018;51:91–95. doi: 10.1016/j.parkreldis.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Paternoster L, Soblet J, Aeby A, Vilain C, Smits G, Deconinck N. A new mutation of carbonic anhydrase 8 gene expanding the cerebellar ataxia, mental retardation and disequilibrium syndrome (CAMRQ) subtype 3. J Neurol Sci. 2017;381:1136–1137. doi: 10.1016/j.jns.2017.08.3200. [DOI] [PubMed] [Google Scholar]
- 48.Kaya N, Aldhalaan H, Al-Younes B, Colak D, Shuaib T, Al-Mohaileb F, et al. Phenotypical spectrum of cerebellar ataxia associated with a novel mutation in the CA8 gene, encoding carbonic anhydrase (CA) VIII. Am J Med Genet B Neuropsychiatr Genet. 2011;156b(7):826–834. doi: 10.1002/ajmg.b.31227. [DOI] [PubMed] [Google Scholar]
- 49.Gan-Or Z, Bouslam N, Birouk N, Lissouba A, Chambers DB, Veriepe J, et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet. 2016;98(6):1271. doi: 10.1016/j.ajhg.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Hersheson J, Lopez D, Hammer M, Liu Y, Lee KH, et al. Defects in the CAPN1 gene result in alterations in cerebellar development and cerebellar ataxia in mice and humans. Cell Rep. 2016;16(1):79–91. doi: 10.1016/j.celrep.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12(7):659–668. doi: 10.1016/s1474-4422(13)70053-x. [DOI] [PubMed] [Google Scholar]
- 52.Zeydan B, Uygunoglu U, Altintas A, Saip S, Siva A, Abbink TEM, et al. Identification of 3 novel patients with CLCN2-related leukoencephalopathy due to CLCN2 mutations. Eur Neurol. 2017;78(3–4):125–127. doi: 10.1159/000478089. [DOI] [PubMed] [Google Scholar]
- 53.Martinez Lyons A, Ardissone A, Reyes A, Robinson AJ, Moroni I, Ghezzi D, et al. COA7 (C1orf163/RESA1) mutations associated with mitochondrial leukoencephalopathy and cytochrome c oxidase deficiency. J Med Genet. 2016;53(12):846–849. doi: 10.1136/jmedgenet-2016-104194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Higuchi Yujiro, Okunushi Ryuta, Hara Taichi, Hashiguchi Akihiro, Yuan Junhui, Yoshimura Akiko, Murayama Kei, Ohtake Akira, Ando Masahiro, Hiramatsu Yu, Ishihara Satoshi, Tanabe Hajime, Okamoto Yuji, Matsuura Eiji, Ueda Takehiro, Toda Tatsushi, Yamashita Sumimasa, Yamada Kenichiro, Koide Takashi, Yaguchi Hiroaki, Mitsui Jun, Ishiura Hiroyuki, Yoshimura Jun, Doi Koichiro, Morishita Shinichi, Sato Ken, Nakagawa Masanori, Yamaguchi Masamitsu, Tsuji Shoji, Takashima Hiroshi. Mutations in COA7 cause spinocerebellar ataxia with axonal neuropathy. Brain. 2018;141(6):1622–1636. doi: 10.1093/brain/awy104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet. 2013;22(4):656–667. doi: 10.1093/hmg/dds473. [DOI] [PubMed] [Google Scholar]
- 56.Doss S, Lohmann K, Seibler P, Arns B, Klopstock T, Zuhlke C, et al. Recessive dystonia-ataxia syndrome in a Turkish family caused by a COX20 (FAM36A) mutation. J Neurol. 2014;261(1):207–212. doi: 10.1007/s00415-013-7177-7. [DOI] [PubMed] [Google Scholar]
- 57.Burns R, Majczenko K, Xu J, Peng W, Yapici Z, Dowling JJ, et al. Homozygous splice mutation in CWF19L1 in a Turkish family with recessive ataxia syndrome. Neurology. 2014;83(23):2175–2182. doi: 10.1212/wnl.0000000000001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen M, Boesten I, Hellebrekers DM, Vanoevelen J, Kamps R, de Koning B, et al. Pathogenic CWF19L1 variants as a novel cause of autosomal recessive cerebellar ataxia and atrophy. Eur J Hum Genet. 2016;24(4):619–622. doi: 10.1038/ejhg.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsaousidou MK, Ouahchi K, Warner TT, Yang Y, Simpson MA, Laing NG, et al. Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am J Hum Genet. 2008;82(2):510–515. doi: 10.1016/j.ajhg.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schols L, Rattay TW, Martus P, Meisner C, Baets J, Fischer I, et al. Hereditary spastic paraplegia type 5: natural history, biomarkers and a randomized controlled trial. Brain. 2017;140(12):3112–3127. doi: 10.1093/brain/awx273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39(4):534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 62.van Berge L, Hamilton EM, Linnankivi T, Uziel G, Steenweg ME, Isohanni P, et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain. 2014;137(Pt 4):1019–1029. doi: 10.1093/brain/awu026. [DOI] [PubMed] [Google Scholar]
- 63.Al Teneiji A, Siriwardena K, George K, Mital S, Mercimek-Mahmutoglu S. Progressive cerebellar atrophy and a novel homozygous pathogenic DNAJC19 variant as a cause of dilated cardiomyopathy ataxia syndrome. Pediatr Neurol. 2016;62:58–61. doi: 10.1016/j.pediatrneurol.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 64.Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, et al. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43(5):385–393. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ucar SK, Mayr JA, Feichtinger RG, Canda E, Coker M, Wortmann SB. Previously unreported biallelic mutation in DNAJC19: are sensorineural hearing loss and basal ganglia lesions additional features of dilated cardiomyopathy and ataxia (DCMA) syndrome? JIMD Rep. 2017;35:39–45. doi: 10.1007/8904_2016_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hammer MB, Eleuch-Fayache G, Schottlaender LV, Nehdi H, Gibbs JR, Arepalli SK, et al. Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am J Hum Genet. 2013;92(2):245–251. doi: 10.1016/j.ajhg.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Votsi C, Zamba-Papanicolaou E, Middleton LT, Pantzaris M, Christodoulou K. A novel GBA2 gene missense mutation in spastic ataxia. Ann Hum Genet. 2014;78(1):13–22. doi: 10.1111/ahg.12045. [DOI] [PubMed] [Google Scholar]
- 68.Eidhof I, Baets J, Kamsteeg EJ, Deconinck T, van Ninhuijs L, Martin JJ, et al. GDAP2 mutations implicate susceptibility to cellular stress in a new form of cerebellar ataxia. Brain. 2018;141(9):2592–2604. doi: 10.1093/brain/awy198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75(2):251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henneke M, Combes P, Diekmann S, Bertini E, Brockmann K, Burlina AP, et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70(10):748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 71.Corbett MA, Schwake M, Bahlo M, Dibbens LM, Lin M, Gandolfo LC, et al. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet. 2011;88(5):657–663. doi: 10.1016/j.ajhg.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Egmond ME, Verschuuren-Bemelmans CC, Nibbeling EA, Elting JW, Sival DA, Brouwer OF et al. Ramsay Hunt syndrome: clinical characterization of progressive myoclonus ataxia caused by GOSR2 mutation. Mov Disord. 2014;29(1):139–143. doi: 10.1002/mds.25704. [DOI] [PubMed] [Google Scholar]
- 73.Utine GE, Haliloglu G, Salanci B, Cetinkaya A, Kiper PO, Alanay Y, et al. A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J Child Neurol. 2013;28(7):926–932. doi: 10.1177/0883073813484967. [DOI] [PubMed] [Google Scholar]
- 74.Hills LB, Masri A, Konno K, Kakegawa W, Lam AT, Lim-Melia E, et al. Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology. 2013;81(16):1378–1386. doi: 10.1212/WNL.0b013e3182a841a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guergueltcheva V, Azmanov DN, Angelicheva D, Smith KR, Chamova T, Florez L, et al. Autosomal-recessive congenital cerebellar ataxia is caused by mutations in metabotropic glutamate receptor 1. Am J Hum Genet. 2012;91(3):553–564. doi: 10.1016/j.ajhg.2012.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rossi PI, Musante I, Summa M, Pittaluga A, Emionite L, Ikehata M, et al. Compensatory molecular and functional mechanisms in nervous system of the Grm1(crv4) mouse lacking the mGlu1 receptor: a model for motor coordination deficits. Cereb Cortex. 2013;23(9):2179–2189. doi: 10.1093/cercor/bhs200. [DOI] [PubMed] [Google Scholar]
- 77.Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90(6):1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging. 2016;41:200.e1–200.e5. doi: 10.1016/j.neurobiolaging.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 79.Gerber S, Alzayady KJ, Burglen L, Bremond-Gignac D, Marchesin V, Roche O, et al. Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome. Am J Hum Genet. 2016;98(5):971–980. doi: 10.1016/j.ajhg.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paganini Leda, Pesenti Chiara, Milani Donatella, Fontana Laura, Motta Silvia, Sirchia Silvia Maria, Scuvera Giulietta, Marchisio Paola, Esposito Susanna, Cinnante Claudia Maria, Tabano Silvia Maria, Miozzo Monica Rosa. A novel splice site variant in ITPR1 gene underlying recessive Gillespie syndrome. American Journal of Medical Genetics Part A. 2018;176(6):1427–1431. doi: 10.1002/ajmg.a.38704. [DOI] [PubMed] [Google Scholar]
- 81.Dor T, Cinnamon Y, Raymond L, Shaag A, Bouslam N, Bouhouche A, et al. KIF1C mutations in two families with hereditary spastic paraparesis and cerebellar dysfunction. J Med Genet. 2014;51(2):137–142. doi: 10.1136/jmedgenet-2013-102012. [DOI] [PubMed] [Google Scholar]
- 82.Yucel-Yilmaz D, Yucesan E, Yalnizoglu D, Oguz KK, Sagiroglu MS, Ozbek U, et al. Clinical phenotype of hereditary spastic paraplegia due to KIF1C gene mutations across life span. Brain Dev. 2018;40(6):458–464. doi: 10.1016/j.braindev.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 83.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360(19):1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Celmina M., Micule I., Inashkina I., Audere M., Kuske S., Pereca J., Stavusis J., Pelnena D., Strautmanis J. EAST/SeSAME syndrome: Review of the literature and introduction of four new Latvian patients. Clinical Genetics. 2018;95(1):63–78. doi: 10.1111/cge.13374. [DOI] [PubMed] [Google Scholar]
- 86.Topcu M, Jobard F, Halliez S, Coskun T, Yalcinkayal C, Gerceker FO, et al. L-2-Hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum Mol Genet. 2004;13(22):2803–2811. doi: 10.1093/hmg/ddh300. [DOI] [PubMed] [Google Scholar]
- 87.Steenweg ME, Jakobs C, Errami A, van Dooren SJ, Adeva Bartolome MT, Aerssens P, et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype-phenotype study. Hum Mutat. 2010;31(4):380–390. doi: 10.1002/humu.21197. [DOI] [PubMed] [Google Scholar]
- 88.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99(6):577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 89.Pitts SA, Kullar HS, Stankovic T, Stewart GS, Last JI, Bedenham T, et al. hMRE11: genomic structure and a null mutation identified in a transcript protected from nonsense-mediated mRNA decay. Hum Mol Genet. 2001;10(11):1155–1162. doi: 10.1093/hmg/10.11.1155. [DOI] [PubMed] [Google Scholar]
- 90.Crosby AH, Patel H, Chioza BA, Proukakis C, Gurtz K, Patton MA, et al. Defective mitochondrial mRNA maturation is associated with spastic ataxia. Am J Hum Genet. 2010;87(5):655–660. doi: 10.1016/j.ajhg.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin NT, Nakamura K, Paila U, Woo J, Brown C, Wright JA, et al. Homozygous mutation of MTPAP causes cellular radiosensitivity and persistent DNA double-strand breaks. Cell Death Dis. 2014;5:e1130. doi: 10.1038/cddis.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Regal L, Ebberink MS, Goemans N, Wanders RJ, De Meirleir L, Jaeken J, et al. Mutations in PEX10 are a cause of autosomal recessive ataxia. Ann Neurol. 2010;68(2):259–263. doi: 10.1002/ana.22035. [DOI] [PubMed] [Google Scholar]
- 93.Yamashita T, Mitsui J, Shimozawa N, Takashima S, Umemura H, Sato K, et al. Ataxic form of autosomal recessive PEX10-related peroxisome biogenesis disorders with a novel compound heterozygous gene mutation and characteristic clinical phenotype. J Neurol Sci. 2017;375:424–429. doi: 10.1016/j.jns.2017.02.058. [DOI] [PubMed] [Google Scholar]
- 94.Jobling RK, Assoum M, Gakh O, Blaser S, Raiman JA, Mignot C, et al. PMPCA mutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain. 2015;138(Pt 6):1505–1517. doi: 10.1093/brain/awv057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choquet K, Zurita-Rendon O, La Piana R, Yang S, Dicaire MJ, Boycott KM, et al. Autosomal recessive cerebellar ataxia caused by a homozygous mutation in PMPCA. Brain. 2016;139(Pt 3):e19. doi: 10.1093/brain/awv362. [DOI] [PubMed] [Google Scholar]
- 96.Joshi M, Anselm I, Shi J, Bale TA, Towne M, Schmitz-Abe K, et al. Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-alpha protein (PMPCA) cause a severe mitochondrial disease. Cold Spring Harb Mol Case Stud. 2016;2(3):a000786. doi: 10.1101/mcs.a000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T, et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet. 2015;96(3):474–479. doi: 10.1016/j.ajhg.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paucar M, Malmgren H, Taylor M, Reynolds JJ, Svenningsson P, Press R, et al. Expanding the ataxia with oculomotor apraxia type 4 phenotype. Neurol Genet. 2016;2(1):e49. doi: 10.1212/nxg.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schiess N, Zee DS, Siddiqui KA, Szolics M, El-Hattab AW. Novel PNKP mutation in siblings with ataxia-oculomotor apraxia type 4. J Neurogenet. 2017;31(1–2):23–25. doi: 10.1080/01677063.2017.1322079. [DOI] [PubMed] [Google Scholar]
- 100.Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014;137(Pt 1):69–77. doi: 10.1093/brain/awt326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wiethoff Sarah, Bettencourt Conceição, Paudel Reema, Madon Prochi, Liu Yo-Tsen, Hersheson Joshua, Wadia Noshir, Desai Joy, Houlden Henry. Pure Cerebellar Ataxia with Homozygous Mutations in the PNPLA6 Gene. The Cerebellum. 2016;16(1):262–267. doi: 10.1007/s12311-016-0769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bernard G, Chouery E, Putorti ML, Tetreault M, Takanohashi A, Carosso G, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89(3):415–423. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bugiani M, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. 2014;83(21):1898–1905. doi: 10.1212/WNL.0000000000001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saitsu H, Osaka H, Sasaki M, Takanashi J, Hamada K, Yamashita A, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. 2011;89(5):644–651. doi: 10.1016/j.ajhg.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tetreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89(5):652–655. doi: 10.1016/j.ajhg.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368(21):1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alqwaifly M, Bohlega S. Ataxia and hypogonadotropic hypogonadism with intrafamilial variability caused by RNF216 mutation. Neurol Int. 2016;8(2):6444. doi: 10.4081/ni.2016.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schmidt WM, Rutledge SL, Schule R, Mayerhofer B, Zuchner S, Boltshauser E, et al. Disruptive SCYL1 mutations underlie a syndrome characterized by recurrent episodes of liver failure, peripheral neuropathy, cerebellar atrophy, and ataxia. Am J Hum Genet. 2015;97(6):855–861. doi: 10.1016/j.ajhg.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shohet Adi, Cohen Lior, Haguel Danielle, Mozer Yael, Shomron Noam, Tzur Shay, Bazak Lily, Basel Salmon Lina, Krause Irit. Variant in SCYL1 gene causes aberrant splicing in a family with cerebellar ataxia, recurrent episodes of liver failure, and growth retardation. European Journal of Human Genetics. 2018;27(2):263–268. doi: 10.1038/s41431-018-0268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thomas AC, Williams H, Seto-Salvia N, Bacchelli C, Jenkins D, O’Sullivan M, et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet. 2014;95(5):611–621. doi: 10.1016/j.ajhg.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Akizu N, Cantagrel V, Zaki MS, Al-Gazali L, Wang X, Rosti RO, et al. Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat Genet. 2015;47(5):528–534. doi: 10.1038/ng.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guissart C, Li X, Leheup B, Drouot N, Montaut-Verient B, Raffo E, et al. Mutation of SLC9A1, encoding the major Na(+)/H(+) exchanger, causes ataxia-deafness Lichtenstein-Knorr syndrome. Hum Mol Genet. 2015;24(2):463–470. doi: 10.1093/hmg/ddu461. [DOI] [PubMed] [Google Scholar]
- 113.Iwama K, Osaka H, Ikeda T, Mitsuhashi S, Miyatake S, Takata A, et al. A novel SLC9A1 mutation causes cerebellar ataxia. J Hum Genet. 2018;63(10):1049–1054. doi: 10.1038/s10038-018-0488-x. [DOI] [PubMed] [Google Scholar]
- 114.Lise S, Clarkson Y, Perkins E, Kwasniewska A, Sadighi Akha E, Schnekenberg RP, et al. Recessive mutations in SPTBN2 implicate beta-III spectrin in both cognitive and motor development. PLoS Genet. 2012;8(12):e1003074. doi: 10.1371/journal.pgen.1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yildiz Bolukbasi E, Afzal M, Mumtaz S, Ahmad N, Malik S, Tolun A. Progressive SCAR14 with unclear speech, developmental delay, tremor, and behavioral problems caused by a homozygous deletion of the SPTBN2 pleckstrin homology domain. Am J Med Genet A. 2017;173(9):2494–2499. doi: 10.1002/ajmg.a.38332. [DOI] [PubMed] [Google Scholar]
- 116.Shi Y, Wang J, Li JD, Ren H, Guan W, He M, et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One. 2013;8(12):e81884. doi: 10.1371/journal.pone.0081884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Synofzik M, Schule R, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A, et al. Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis. 2014;9:57. doi: 10.1186/1750-1172-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gomez-Herreros F, Schuurs-Hoeijmakers JH, McCormack M, Greally MT, Rulten S, Romero-Granados R, et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat Genet. 2014;46(5):516–521. doi: 10.1038/ng.2929. [DOI] [PubMed] [Google Scholar]
- 119.Zagnoli-Vieira G, Bruni F, Thompson K, He L, Walker S, de Brouwer APM, et al. Confirming TDP2 mutation in spinocerebellar ataxia autosomal recessive 23 (SCAR23) Neurol Genet. 2018;4(4):e262. doi: 10.1212/nxg.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun Y, Almomani R, Breedveld GJ, Santen GW, Aten E, Lefeber DJ, et al. Autosomal recessive spinocerebellar ataxia 7 (SCAR7) is caused by variants in TPP1, the gene involved in classic late-infantile neuronal ceroid lipofuscinosis 2 disease (CLN2 disease) Hum Mutat. 2013;34(5):706–713. doi: 10.1002/humu.22292. [DOI] [PubMed] [Google Scholar]
- 121.Dy ME, Sims KB, Friedman J. TPP1 deficiency: rare cause of isolated childhood-onset progressive ataxia. Neurology. 2015;85(14):1259–1261. doi: 10.1212/wnl.0000000000001876. [DOI] [PubMed] [Google Scholar]
- 122.Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A. 2013;110(9):3489–3494. doi: 10.1073/pnas.1222732110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rydning SL, Backe PH, Sousa MML, Iqbal Z, Oye AM, Sheng Y, et al. Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum Mol Genet. 2017;26(6):1217–1218. doi: 10.1093/hmg/ddx072. [DOI] [PubMed] [Google Scholar]
- 124.Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77(3):477–483. doi: 10.1086/444400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ali BR, Silhavy JL, Gleeson MJ, Gleeson JG, Al-Gazali L. A missense founder mutation in VLDLR is associated with dysequilibrium syndrome without quadrupedal locomotion. BMC Med Genet. 2012;13:80. doi: 10.1186/1471-2350-13-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Recessive mutations in >VPS13D cause childhood onset movement disorders. Ann Neurol. 2018;83(6):1089–1095. doi: 10.1002/ana.25204. [DOI] [PubMed] [Google Scholar]
- 127.Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Bruggemann N, et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol. 2018;83(6):1075–1088. doi: 10.1002/ana.25220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gulsuner S, Tekinay AB, Doerschner K, Boyaci H, Bilguvar K, Unal H, et al. Homozygosity mapping and targeted genomic sequencing reveal the gene responsible for cerebellar hypoplasia and quadrupedal locomotion in a consanguineous kindred. Genome Res. 2011;21(12):1995–2003. doi: 10.1101/gr.126110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Komara M, John A, Suleiman J, Ali BR, Al-Gazali L. Clinical and molecular delineation of dysequilibrium syndrome type 2 and profound sensorineural hearing loss in an inbred Arab family. Am J Med Genet A. 2016;170a(2):540–543. doi: 10.1002/ajmg.a.37421. [DOI] [PubMed] [Google Scholar]
- 130.Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. 2017;541(7635):87–91. doi: 10.1038/nature20790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O’Connor Emer, Vandrovcova Jana, Bugiardini Enrico, Chelban Viorica, Manole Andreea, Davagnanam Indran, Wiethoff Sarah, Pittman Alan, Lynch David S, Efthymiou Stephanie, Marino Silvia, Manzur Adnan Y, Roberts Mark, Hanna Michael G, Houlden Henry, Matthews Emma, Wood Nicholas W. Mutations in XRCC1 cause cerebellar ataxia and peripheral neuropathy. Journal of Neurology, Neurosurgery & Psychiatry. 2018;89(11):1230–1232. doi: 10.1136/jnnp-2017-317581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36(9):1008–1013. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- 133.Parisi M, Glass I, et al. Joubert syndrome and related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle: University of Washington, Seattle University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 134.Pearl PL, Gibson KM, Acosta MT, Vezina LG, Theodore WH, Rogawski MA, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology. 2003;60(9):1413–1417. doi: 10.1212/01.wnl.0000059549.70717.80. [DOI] [PubMed] [Google Scholar]
- 135.Trettel F, Malaspina P, Jodice C, Novelletto A, Slaughter CA, Caudle DL, et al. Human succinic semialdehyde dehydrogenase. Molecular cloning and chromosomal localization. Adv Exp Med Biol. 1997;414:253–260. [PubMed] [Google Scholar]
- 136.Imbach T, Burda P, Kuhnert P, Wevers RA, Aebi M, Berger EG, et al. A mutation in the human ortholog of the Saccharomyces cerevisiae ALG6 gene causes carbohydrate-deficient glycoprotein syndrome type-Ic. Proc Natl Acad Sci U S A. 1999;96(12):6982–6987. doi: 10.1073/pnas.96.12.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Morava E, Tiemes V, Thiel C, Seta N, de Lonlay P, de Klerk H, et al. ALG6-CDG: a recognizable phenotype with epilepsy, proximal muscle weakness, ataxia and behavioral and limb anomalies. J Inherit Metab Dis. 2016;39(5):713–723. doi: 10.1007/s10545-016-9945-x. [DOI] [PubMed] [Google Scholar]
- 138.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 139.Onat OE, Gulsuner S, Bilguvar K, Nazli Basak A, Topaloglu H, Tan M, et al. Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur J Hum Genet. 2013;21(3):281–285. doi: 10.1038/ejhg.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Alsahli S, Alrifai MT, Al Tala S, Mutairi FA, Alfadhel M. Further delineation of the clinical phenotype of cerebellar ataxia, mental retardation, and disequilibrium syndrome type 4. J Cent Nerv Syst Dis. 2018;10:1179573518759682. doi: 10.1177/1179573518759682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Boukhris A, Schule R, Loureiro JL, Lourenco CM, Mundwiller E, Gonzalez MA, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93(1):118–123. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pomponio RJ, Reynolds TR, Cole H, Buck GA, Wolf B. Mutational hotspot in the human biotinidase gene causes profound biotinidase deficiency. Nat Genet. 1995;11(1):96–98. doi: 10.1038/ng0995-96. [DOI] [PubMed] [Google Scholar]
- 143.Wolf B. Biotinidase deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean, LJH, Stephens K et al., editors. GeneReviews((R)). Seattle (WA)1993. [PubMed]
- 144.Klockars T, Savukoski M, Isosomppi J, Peltonen L. Positional cloning of the CLN5 gene defective in the Finnish variant of the LINCL. Mol Genet Metab. 1999;66(4):324–328. doi: 10.1006/mgme.1999.2832. [DOI] [PubMed] [Google Scholar]
- 145.Yoshida K, Furihata K, Takeda S, Nakamura A, Yamamoto K, Morita H, et al. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat Genet. 1995;9(3):267–272. doi: 10.1038/ng0395-267. [DOI] [PubMed] [Google Scholar]
- 146.Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, Miao J, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science. 1996;271(5256):1731–1734. doi: 10.1126/science.271.5256.1731. [DOI] [PubMed] [Google Scholar]
- 147.Leegwater PA, Vermeulen G, Konst AA, Naidu S, Mulders J, Visser A, et al. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29(4):383–388. doi: 10.1038/ng764. [DOI] [PubMed] [Google Scholar]
- 148.Fogli A, Schiffmann R, Bertini E, Ughetto S, Combes P, Eymard-Pierre E, et al. The effect of genotype on the natural history of eIF2B-related leukodystrophies. Neurology. 2004;62(9):1509–1517. doi: 10.1212/01.wnl.0000123259.67815.db. [DOI] [PubMed] [Google Scholar]
- 149.Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 150.Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 151.Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell. 1996;86(5):811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 152.Doi H, Koyano S, Miyatake S, Nakajima S, Nakazawa Y, Kunii M, et al. Cerebellar ataxia-dominant phenotype in patients with ERCC4 mutations. J Hum Genet. 2018;63(4):417–423. doi: 10.1038/s10038-017-0408-5. [DOI] [PubMed] [Google Scholar]
- 153.Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D, et al. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet. 2008;83(5):643–648. doi: 10.1016/j.ajhg.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Steinfeld R, Grapp M, Kraetzner R, Dreha-Kulaczewski S, Helms G, Dechent P, et al. Folate receptor alpha defect causes cerebral folate transport deficiency: a treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am J Hum Genet. 2009;85(3):354–363. doi: 10.1016/j.ajhg.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Grapp M, Just IA, Linnankivi T, Wolf P, Lucke T, Hausler M, et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain. 2012;135(Pt 7):2022–2031. doi: 10.1093/brain/aws122. [DOI] [PubMed] [Google Scholar]
- 156.Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26(3):370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- 157.Kuhlenbaumer G, Timmerman V, Bomont P. Giant axonal neuropathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K et al., editors. GeneReviews((R)). Seattle (WA) 1993.
- 158.Nishimoto J, Nanba E, Inui K, Okada S, Suzuki K. GM1-gangliosidosis (genetic beta-galactosidase deficiency): identification of four mutations in different clinical phenotypes among Japanese patients. Am J Hum Genet. 1991;49(3):566–574. [PMC free article] [PubMed] [Google Scholar]
- 159.Karimzadeh P, Naderi S, Modarresi F, Dastsooz H, Nemati H, Farokhashtiani T, et al. Case reports of juvenile GM1 gangliosidosisis type II caused by mutation in GLB1 gene. BMC Med Genet. 2017;18(1):73. doi: 10.1186/s12881-017-0417-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Myerowitz R, Costigan FC. The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the alpha-chain of beta-hexosaminidase. J Biol Chem. 1988;263(35):18587–18589. [PubMed] [Google Scholar]
- 161.Gort L, de Olano N, Macias-Vidal J, Coll MA, Spanish GMWG. GM2 gangliosidoses in Spain: analysis of the HEXA and HEXB genes in 34 Tay-Sachs and 14 Sandhoff patients. Gene. 2012;506(1):25–30. doi: 10.1016/j.gene.2012.06.080. [DOI] [PubMed] [Google Scholar]
- 162.O’Dowd BF, Klavins MH, Willard HF, Gravel R, Lowden JA, Mahuran DJ. Molecular heterogeneity in the infantile and juvenile forms of Sandhoff disease (O-variant GM2 gangliosidosis) J Biol Chem. 1986;261(27):12680–12685. [PubMed] [Google Scholar]
- 163.Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet. 2010;87(2):282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Matsukawa T, Koshi KM, Mitsui J, Bannai T, Kawabe M, Ishiura H, et al. Slowly progressive d-bifunctional protein deficiency with survival to adulthood diagnosed by whole-exome sequencing. J Neurol Sci. 2017;372:6–10. doi: 10.1016/j.jns.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 165.Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007;39(3):366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- 166.Schule R, Schlipf N, Synofzik M, Klebe S, Klimpe S, Hehr U, et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry. 2009;80(12):1402–1404. doi: 10.1136/jnnp.2008.167528. [DOI] [PubMed] [Google Scholar]
- 167.Van Bogaert P, Azizieh R, Desir J, Aeby A, De Meirleir L, Laes JF, et al. Mutation of a potassium channel-related gene in progressive myoclonic epilepsy. Ann Neurol. 2007;61(6):579–586. doi: 10.1002/ana.21121. [DOI] [PubMed] [Google Scholar]
- 168.Kousi M, Anttila V, Schulz A, Calafato S, Jakkula E, Riesch E, et al. Novel mutations consolidate KCTD7 as a progressive myoclonus epilepsy gene. J Med Genet. 2012;49(6):391–399. doi: 10.1136/jmedgenet-2012-100859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis. 2008;3:21. doi: 10.1186/1750-1172-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Leegwater PA, Yuan BQ, van der Steen J, Mulders J, Konst AA, Boor PK, et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68(4):831–838. doi: 10.1086/319519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nasca A, Scotton C, Zaharieva I, Neri M, Selvatici R, Magnusson OT, et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum Mutat. 2017;38(8):970–977. doi: 10.1002/humu.23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gal A, Balicza P, Weaver D, Naghdi S, Joseph SK, Varnai P, et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol Med. 2017;9(7):967–984. doi: 10.15252/emmm.201607058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sharp D, Blinderman L, Combs KA, Kienzle B, Ricci B, Wager-Smith K, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993;365(6441):65–69. doi: 10.1038/365065a0. [DOI] [PubMed] [Google Scholar]
- 174.Bonten E, van der Spoel A, Fornerod M, Grosveld G, d’Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996;10(24):3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- 175.Lai SC, Chen RS, Wu Chou YH, Chang HC, Kao LY, Huang YZ, et al. A longitudinal study of Taiwanese sialidosis type 1: an insight into the concept of cherry-red spot myoclonus syndrome. Eur J Neurol. 2009;16(8):912–919. doi: 10.1111/j.1468-1331.2009.02622.x. [DOI] [PubMed] [Google Scholar]
- 176.Dorboz I, Aiello C, Simons C, Stone RT, Niceta M, Elmaleh M, et al. Biallelic mutations in the homeodomain of NKX6-2 underlie a severe hypomyelinating leukodystrophy. Brain. 2017;140(10):2550–2556. doi: 10.1093/brain/awx207. [DOI] [PubMed] [Google Scholar]
- 177.Chelban V, Patel N, Vandrovcova J, Zanetti MN, Lynch DS, Ryten M, et al. Mutations in NKX6-2 cause progressive spastic ataxia and hypomyelination. Am J Hum Genet. 2017;100(6):969–977. doi: 10.1016/j.ajhg.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 179.Nadjar Y, Hutter-Moncada AL, Latour P, Ayrignac X, Kaphan E, Tranchant C, et al. Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J Rare Dis. 2018;13(1):175. doi: 10.1186/s13023-018-0913-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290(5500):2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 181.Schaaf CP, Blazo M, Lewis RA, Tonini RE, Takei H, Wang J, et al. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol Genet Metab. 2011;103(4):383–387. doi: 10.1016/j.ymgme.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 182.Bonneau D, Colin E, Oca F, Ferre M, Chevrollier A, Gueguen N, et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain. 2014;137(Pt 10):e301. doi: 10.1093/brain/awu184. [DOI] [PubMed] [Google Scholar]
- 183.Shimozawa N, Suzuki Y, Orii T, Moser A, Moser HW, Wanders RJ. Standardization of complementation grouping of peroxisome-deficient disorders and the second Zellweger patient with peroxisomal assembly factor-1 (PAF-1) defect. Am J Hum Genet. 1993;52(4):843–844. [PMC free article] [PubMed] [Google Scholar]
- 184.Sevin C, Ferdinandusse S, Waterham HR, Wanders RJ, Aubourg P. Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene. Orphanet J Rare Dis. 2011;6:8. doi: 10.1186/1750-1172-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.van den Brink DM, Brites P, Haasjes J, Wierzbicki AS, Mitchell J, Lambert-Hamill M, et al. Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet. 2003;72(2):471–477. doi: 10.1086/346093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mihalik SJ, Morrell JC, Kim D, Sacksteder KA, Watkins PA, Gould SJ. Identification of PAHX, a Refsum disease gene. Nat Genet. 1997;17(2):185–189. doi: 10.1038/ng1097-185. [DOI] [PubMed] [Google Scholar]
- 187.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38(7):752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Salih M, Mundwiller E, Khan A, Al Drees A, Elmalik S, Hassan H, et al. PLA2G6 gene mutations cause evolving spinocerebellar ataxia influenced by the genotype. J Neurol. 2013;260:S10–SS1. [Google Scholar]
- 189.Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat Genet. 1997;16(1):88–92. doi: 10.1038/ng0597-88. [DOI] [PubMed] [Google Scholar]
- 190.Schiff M, Roda C, Monin ML, Arion A, Barth M, Bednarek N, et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet. 2017;54(12):843–851. doi: 10.1136/jmedgenet-2017-104903. [DOI] [PubMed] [Google Scholar]
- 191.Meneret A, Gaudebout C, Riant F, Vidailhet M, Depienne C, Roze E. PRRT2 mutations and paroxysmal disorders. Eur J Neurol. 2013;20(6):872–878. doi: 10.1111/ene.12104. [DOI] [PubMed] [Google Scholar]
- 192.Delcourt M, Riant F, Mancini J, Milh M, Navarro V, Roze E, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J Neurol Neurosurg Psychiatry. 2015;86(7):782–785. doi: 10.1136/jnnp-2014-309025. [DOI] [PubMed] [Google Scholar]
- 193.Hu H, Matter ML, Issa-Jahns L, Jijiwa M, Kraemer N, Musante L, et al. Mutations in PTRH2 cause novel infantile-onset multisystem disease with intellectual disability, microcephaly, progressive ataxia, and muscle weakness. Ann Clin Transl Neurol. 2014;1(12):1024–1035. doi: 10.1002/acn3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Picker-Minh S, Mignot C, Doummar D, Hashem M, Faqeih E, Josset P, et al. Phenotype variability of infantile-onset multisystem neurologic, endocrine, and pancreatic disease IMNEPD. Orphanet J Rare Dis. 2016;11(1):52. doi: 10.1186/s13023-016-0433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Agamy O, Ben Zeev B, Lev D, Marcus B, Fine D, Su D, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet. 2010;87(4):538–544. doi: 10.1016/j.ajhg.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.van Dijk T, Vermeij JD, van Koningsbruggen S, Lakeman P, Baas F, Poll-The BT. A SEPSECS mutation in a 23-year-old woman with microcephaly and progressive cerebellar ataxia. J Inherit Metab Dis. 2018;41(5):897–898. doi: 10.1007/s10545-018-0151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, et al. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet. 1999;23(4):462–465. doi: 10.1038/70585. [DOI] [PubMed] [Google Scholar]
- 198.Aula N, Salomaki P, Timonen R, Verheijen F, Mancini G, Mansson JE, et al. The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am J Hum Genet. 2000;67(4):832–840. doi: 10.1086/303077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Barmherzig R, Bullivant G, Cordeiro D, Sinasac DS, Blaser S, Mercimek-Mahmutoglu S. A new patient with intermediate severe Salla disease with hypomyelination: a literature review for Salla disease. Pediatr Neurol. 2017;74:87–91 e2. doi: 10.1016/j.pediatrneurol.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 200.Seidner G, Alvarez MG, Yeh JI, O’Driscoll KR, Klepper J, Stump TS, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998;18(2):188–191. doi: 10.1038/ng0298-188. [DOI] [PubMed] [Google Scholar]
- 201.Hully M, Vuillaumier-Barrot S, Le Bizec C, Boddaert N, Kaminska A, Lascelles K, et al. From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet. 2015;58(9):443–454. doi: 10.1016/j.ejmg.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 202.Johnson JO, Gibbs JR, Megarbane A, Urtizberea JA, Hernandez DG, Foley AR, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain. 2012;135(Pt 9):2875–2882. doi: 10.1093/brain/aws161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Foley AR, Menezes MP, Pandraud A, Gonzalez MA, Al-Odaib A, Abrams AJ, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137(Pt 1):44–56. doi: 10.1093/brain/awt315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guissart C, Drouot N, Oncel I, Leheup B, Gershoni-Barush R, Muller J, et al. Genes for spinocerebellar ataxia with blindness and deafness (SCABD/SCAR3, MIM# 271250 and SCABD2) Eur J Hum Genet. 2016;24(8):1154–1159. doi: 10.1038/ejhg.2015.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, Arcos-Burgos M, et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet. 2004;36(9):999–1002. doi: 10.1038/ng1405. [DOI] [PubMed] [Google Scholar]
- 206.Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. 2015;47(8):926–932. doi: 10.1038/ng.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hammer MB, Ding J, Mochel F, Eleuch-Fayache G, Charles P, Coutelier M, et al. SLC25A46 mutations associated with autosomal recessive cerebellar ataxia in North African families. Neurodegener Dis. 2017;17(4–5):208–212. doi: 10.1159/000464445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142(2):203–217. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wheeler PG, Ng BG, Sanford L, Sutton VR, Bartholomew DW, Pastore MT, et al. SRD5A3-CDG: expanding the phenotype of a congenital disorder of glycosylation with emphasis on adult onset features. Am J Med Genet A. 2016;170(12):3165–3171. doi: 10.1002/ajmg.a.37875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43(3):259–263. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]
- 211.Morino H, Miyamoto R, Ohnishi S, Maruyama H, Kawakami H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. 2014;14:5. doi: 10.1186/1471-2377-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ardissone A, Granata T, Legati A, Diodato D, Melchionda L, Lamantea E, et al. Mitochondrial complex III deficiency caused by TTC19 defects: report of a novel mutation and review of literature. JIMD Rep. 2015;22:115–120. doi: 10.1007/8904_2015_419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet. 2014;95(6):637–648. doi: 10.1016/j.ajhg.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Vodopiutz J, Seidl R, Prayer D, Khan MI, Mayr JA, Streubel B, et al. WDR73 mutations cause infantile neurodegeneration and variable glomerular kidney disease. Hum Mutat. 2015;36(11):1021–1028. doi: 10.1002/humu.22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Strom TM, Hortnagel K, Hofmann S, Gekeler F, Scharfe C, Rabl W, et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet. 1998;7(13):2021–2028. doi: 10.1093/hmg/7.13.2021. [DOI] [PubMed] [Google Scholar]
- 216.Mallaret M, Lagha Boukbiza O, Drouot N, Renaud M, Klein FAC, Anheim M, et al. The tumor suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Mov Disord. 2013;28:S398. doi: 10.1093/brain/awt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mignot C, Lambert L, Pasquier L, Bienvenu T, Delahaye-Duriez A, Keren B, et al. WWOX-related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J Med Genet. 2015;52(1):61–70. doi: 10.1136/jmedgenet-2014-102748. [DOI] [PubMed] [Google Scholar]
- 218.Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7(4):327–340. doi: 10.1016/S1474-4422(08)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Coutelier Marie, Hammer Monia B., Stevanin Giovanni, Monin Marie-Lorraine, Davoine Claire-Sophie, Mochel Fanny, Labauge Pierre, Ewenczyk Claire, Ding Jinhui, Gibbs J. Raphael, Hannequin Didier, Melki Judith, Toutain Annick, Laugel Vincent, Forlani Sylvie, Charles Perrine, Broussolle Emmanuel, Thobois Stéphane, Afenjar Alexandra, Anheim Mathieu, Calvas Patrick, Castelnovo Giovanni, de Broucker Thomas, Vidailhet Marie, Moulignier Antoine, Ghnassia Robert T., Tallaksen Chantal, Mignot Cyril, Goizet Cyril, Le Ber Isabelle, Ollagnon-Roman Elisabeth, Pouget Jean, Brice Alexis, Singleton Andrew, Durr Alexandra. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurology. 2018;75(5):591. doi: 10.1001/jamaneurol.2017.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nemeth AH, Kwasniewska AC, Lise S, Parolin Schnekenberg R, Becker EB, Bera KD, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain. 2013;136(Pt 10):3106–3118. doi: 10.1093/brain/awt236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sun Miao, Johnson Amy Knight, Nelakuditi Viswateja, Guidugli Lucia, Fischer David, Arndt Kelly, Ma Lan, Sandford Erin, Shakkottai Vikram, Boycott Kym, Chardon Jodi Warman, Li Zejuan, del Gaudio Daniela, Burmeister Margit, Gomez Christopher M., Waggoner Darrel J., Das Soma. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genetics in Medicine. 2018;21(1):195–206. doi: 10.1038/s41436-018-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Valence Stéphanie, Cochet Emmanuelle, Rougeot Christelle, Garel Catherine, Chantot-Bastaraud Sandra, Lainey Elodie, Afenjar Alexandra, Barthez Marie-Anne, Bednarek Nathalie, Doummar Diane, Faivre Laurence, Goizet Cyril, Haye Damien, Heron Bénédicte, Kemlin Isabelle, Lacombe Didier, Milh Mathieu, Moutard Marie-Laure, Riant Florence, Robin Stéphanie, Roubertie Agathe, Sarda Pierre, Toutain Annick, Villard Laurent, Ville Dorothée, Billette de Villemeur Thierry, Rodriguez Diana, Burglen Lydie. Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genetics in Medicine. 2018;21(3):553–563. doi: 10.1038/s41436-018-0089-2. [DOI] [PubMed] [Google Scholar]
- 223.Fogel BL, Lee H, Deignan JL, Strom SP, Kantarci S, Wang X, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014;71(10):1237–1246. doi: 10.1001/jamaneurol.2014.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kang Ce, Liang Christina, Ahmad Kate E., Gu Yufan, Siow Sue-Faye, Colebatch James G., Whyte Scott, Ng Karl, Cremer Philip D., Corbett Alastair J., Davis Ryan L., Roscioli Tony, Cowley Mark J., Park Jin-Sung, Sue Carolyn M., Kumar Kishore R. High Degree of Genetic Heterogeneity for Hereditary Cerebellar Ataxias in Australia. The Cerebellum. 2018;18(1):137–146. doi: 10.1007/s12311-018-0969-7. [DOI] [PubMed] [Google Scholar]
- 225.Jiang B, Glover JN, Weinfeld M. Neurological disorders associated with DNA strand-break processing enzymes. Mech Ageing Dev. 2017;161(Pt A):130–140. doi: 10.1016/j.mad.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tahbaz N, Subedi S, Weinfeld M. Role of polynucleotide kinase/phosphatase in mitochondrial DNA repair. Nucleic Acids Res. 2012;40(8):3484–3495. doi: 10.1093/nar/gkr1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol basis Dis. 2017;1863(6):1539–1555. doi: 10.1016/j.bbadis.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 228.Almajan ER, Richter R, Paeger L, Martinelli P, Barth E, Decker T, et al. AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J Clin Invest. 2012;122(11):4048–4058. doi: 10.1172/JCI64604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Levytskyy RM, Germany EM, Khalimonchuk O. Mitochondrial quality control proteases in neuronal welfare. J NeuroImmune Pharmacol. 2016;11(4):629–644. doi: 10.1007/s11481-016-9683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Choy KR, Watters DJ. Neurodegeneration in ataxia-telangiectasia: multiple roles of ATM kinase in cellular homeostasis. Dev Dyn. 2018;247(1):33–46. doi: 10.1002/dvdy.24522. [DOI] [PubMed] [Google Scholar]
- 231.Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, et al. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet. 1999;8(3):425–430. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 232.Abeti R, Parkinson MH, Hargreaves IP, Angelova PR, Sandi C, Pook MA, et al. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia’. Cell Death Dis. 2016;7:e2237. doi: 10.1038/cddis.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cuddihy SL, Ali SS, Musiek ES, Lucero J, Kopp SJ, Morrow JD, et al. Prolonged alpha-tocopherol deficiency decreases oxidative stress and unmasks alpha-tocopherol-dependent regulation of mitochondrial function in the brain. J Biol Chem. 2008;283(11):6915–6924. doi: 10.1074/jbc.M702572200. [DOI] [PubMed] [Google Scholar]
- 234.Garcia-Diaz B, Barca E, Balreira A, Lopez LC, Tadesse S, Krishna S, et al. Lack of aprataxin impairs mitochondrial functions via downregulation of the APE1/NRF1/NRF2 pathway. Hum Mol Genet. 2015;24(16):4516–4529. doi: 10.1093/hmg/ddv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Stefely JA, Licitra F, Laredj L, Reidenbach AG, Kemmerer ZA, Grangeray A, et al. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity. Mol Cell. 2016;63(4):608–620. doi: 10.1016/j.molcel.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lariviere R, Gaudet R, Gentil BJ, Girard M, Conte TC, Minotti S, et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum Mol Genet. 2015;24(3):727–739. doi: 10.1093/hmg/ddu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Bradshaw TY, Romano LE, Duncan EJ, Nethisinghe S, Abeti R, Michael GJ, et al. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum Mol Genet. 2016;25(15):3232–3244. doi: 10.1093/hmg/ddw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Anding AL, Wang C, Chang TK, Sliter DA, Powers CM, Hofmann K, et al. Vps13D encodes a ubiquitin-binding protein that is required for the regulation of mitochondrial size and clearance. Curr Biol. 2018;28(2):287–95 e6. doi: 10.1016/j.cub.2017.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Aradjanski M, Dogan SA, Lotter S, Wang S, Hermans S, Wibom R, et al. DARS2 protects against neuroinflammation and apoptotic neuronal loss, but is dispensable for myelin producing cells. Hum Mol Genet. 2017;26(21):4181–4189. doi: 10.1093/hmg/ddx307. [DOI] [PubMed] [Google Scholar]
- 240.Bratic A, Clemente P, Calvo-Garrido J, Maffezzini C, Felser A, Wibom R, et al. Mitochondrial polyadenylation is a one-step process required for mRNA integrity and tRNA maturation. PLoS Genet. 2016;12(5):e1006028. doi: 10.1371/journal.pgen.1006028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lu YW, Claypool SM. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rzem R, Achouri Y, Marbaix E, Schakman O, Wiame E, Marie S, et al. A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS One. 2015;10(3):e0119540. doi: 10.1371/journal.pone.0119540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Rao L, Sha Y, Eissa NT. The E3 ubiquitin ligase STUB1 regulates autophagy and mitochondrial biogenesis by modulating TFEB activity. Mol Cell Oncol. 2017;4(6):e1372867. doi: 10.1080/23723556.2017.1372867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Regal JA, Festerling TA, Buis JM, Ferguson DO. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet. 2013;22(25):5146–5159. doi: 10.1093/hmg/ddt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cohen S, Puget N, Lin YL, Clouaire T, Aguirrebengoa M, Rocher V, et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat Commun. 2018;9(1):533. doi: 10.1038/s41467-018-02894-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Fogel BL, Cho E, Wahnich A, Gao F, Becherel OJ, Wang X, et al. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum Mol Genet. 2014;23(18):4758–4769. doi: 10.1093/hmg/ddu190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hirasawa M, Xu X, Trask RB, Maddatu TP, Johnson BA, Naggert JK, et al. Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol Cell Neurosci. 2007;35(1):161–170. doi: 10.1016/j.mcn.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kakegawa W, Miyoshi Y, Hamase K, Matsuda S, Matsuda K, Kohda K, et al. D-serine regulates cerebellar LTD and motor coordination through the delta2 glutamate receptor. Nat Neurosci. 2011;14(5):603–611. doi: 10.1038/nn.2791. [DOI] [PubMed] [Google Scholar]
- 249.Sugawara T, Hisatsune C, Le TD, Hashikawa T, Hirono M, Hattori M, et al. Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintaining the spine morphology of Purkinje cells in adult mice. J Neurosci. 2013;33(30):12186–12196. doi: 10.1523/JNEUROSCI.0545-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Perkins E, Suminaite D, Jackson M. Cerebellar ataxias: beta-III spectrin’s interactions suggest common pathogenic pathways. J Physiol. 2016;594(16):4661–4676. doi: 10.1113/JP271195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Razafsky D, Hodzic D. A variant of Nesprin1 giant devoid of KASH domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol Dis. 2015;78:57–67. doi: 10.1016/j.nbd.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Itoh M, Li S, Ohta K, Yamada A, Hayakawa-Yano Y, Ueda M, et al. Cayman ataxia-related protein is a presynapse-specific caspase-3 substrate. Neurochem Res. 2011;36(7):1304–1313. doi: 10.1007/s11064-011-0430-5. [DOI] [PubMed] [Google Scholar]
- 253.Bossi S, Musante I, Bonfiglio T, Bonifacino T, Emionite L, Cerminara M, et al. Genetic inactivation of mGlu5 receptor improves motor coordination in the Grm1(crv4) mouse model of SCAR13 ataxia. Neurobiol Dis. 2018;109(Pt A):44–53. doi: 10.1016/j.nbd.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 254.Dietrich CJ, Morad M. Synaptic acidification enhances GABAA signaling. J Neurosci. 2010;30(47):16044–16052. doi: 10.1523/JNEUROSCI.6364-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Praschberger R, Lowe SA, Malintan NT, Giachello CNG, Patel N, Houlden H, et al. Mutations in Membrin/GOSR2 reveal stringent secretory pathway demands of dendritic growth and synaptic integrity. Cell Rep. 2017;21(1):97–109. doi: 10.1016/j.celrep.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Husain N, Yuan Q, Yen YC, Pletnikova O, Sally DQ, Worley P, et al. TRIAD3/RNF216 mutations associated with Gordon Holmes syndrome lead to synaptic and cognitive impairments via arc misregulation. Aging Cell. 2017;16(2):281–292. doi: 10.1111/acel.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Sakurai M, Sekiguchi M, Zushida K, Yamada K, Nagamine S, Kabuta T, et al. Reduction in memory in passive avoidance learning, exploratory behaviour and synaptic plasticity in mice with a spontaneous deletion in the ubiquitin C-terminal hydrolase L1 gene. Eur J Neurosci. 2008;27(3):691–701. doi: 10.1111/j.1460-9568.2008.06047.x. [DOI] [PubMed] [Google Scholar]
- 258.Duncan EJ, Lariviere R, Bradshaw TY, Longo F, Sgarioto N, Hayes MJ, et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum Mol Genet. 2017;26(16):3130–3143. doi: 10.1093/hmg/ddx197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Menade M, Kozlov G, Trempe JF, Pande H, Shenker S, Wickremasinghe S, et al. Structures of ubiquitin-like (Ubl) and Hsp90-like domains of sacsin provide insight into pathological mutations. J Biol Chem. 2018;293(33):12832–12842. doi: 10.1074/jbc.RA118.003939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ichhaporia Viraj P., Kim Jieun, Kavdia Kanisha, Vogel Peter, Horner Linda, Frase Sharon, Hendershot Linda M. SIL1, the endoplasmic-reticulum-localized BiP co-chaperone, plays a crucial role in maintaining skeletal muscle proteostasis and physiology. Disease Models & Mechanisms. 2018;11(5):dmm033043. doi: 10.1242/dmm.033043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23(4):1013–1024. doi: 10.1093/hmg/ddt497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Lenz D, McClean P, Kansu A, Bonnen PE, Ranucci G, Thiel C, et al. SCYL1 variants cause a syndrome with low gamma-glutamyl-transferase cholestasis, acute liver failure, and neurodegeneration (CALFAN) Genet Med. 2018;20(10):1255–1265. doi: 10.1038/gim.2017.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Patron M, Sprenger HG, Langer T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. 2018;28(3):296–306. doi: 10.1038/cr.2018.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Wanitchakool P, Ousingsawat J, Sirianant L, Cabrita I, Faria D, Schreiber R, et al. Cellular defects by deletion of ANO10 are due to deregulated local calcium signaling. Cell Signal. 2017;30:41–49. doi: 10.1016/j.cellsig.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 265.Synofzik M, Helbig KL, Harmuth F, Deconinck T, Tanpaiboon P, Sun B, et al. De novo ITPR1 variants are a recurrent cause of early-onset ataxia, acting via loss of channel function. Eur J Hum Genet. 2018;26(11):1623–1634. doi: 10.1038/s41431-018-0206-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, et al. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell. 2015;60(1):47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gotzl JK, Colombo AV, Fellerer K, Reifschneider A, Werner G, Tahirovic S, et al. Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol Neurodegener. 2018;13(1):48. doi: 10.1186/s13024-018-0281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24(41):9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Bryant D, Liu Y, Datta S, Hariri H, Seda M, Anderson G, et al. SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum Mol Genet. 2018;27(11):1927–1940. doi: 10.1093/hmg/ddy101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ulatowski L, Parker R, Warrier G, Sultana R, Butterfield DA, Manor D. Vitamin E is essential for Purkinje neuron integrity. Neuroscience. 2014;260:120–129. doi: 10.1016/j.neuroscience.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Joshi A, Shaikh M, Singh S, Rajendran A, Mhetre A, Kamat SS. Biochemical characterization of the PHARC-associated serine hydrolase ABHD12 reveals its preference for very-long-chain lipids. J Biol Chem. 2018;293(44):16953–16963. doi: 10.1074/jbc.RA118.005640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2014;9:179. doi: 10.1186/s13023-014-0179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Woeste MA, Wachten D. The enigmatic role of GBA2 in controlling locomotor function. Front Mol Neurosci. 2017;10:386. doi: 10.3389/fnmol.2017.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Dutta S, Rieche F, Eckl N, Duch C, Kretzschmar D. Glial expression of Swiss cheese (SWS), the Drosophila orthologue of neuropathy target esterase (NTE), is required for neuronal ensheathment and function. Dis Model Mech. 2016;9(3):283–294. doi: 10.1242/dmm.022236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Liu Y, Zaun HC, Orlowski J, Ackerman SL. CHP1-mediated NHE1 biosynthetic maturation is required for Purkinje cell axon homeostasis. J Neurosci. 2013;33(31):12656–12669. doi: 10.1523/JNEUROSCI.0406-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Abrams CK. Diseases of connexins expressed in myelinating glia. Neurosci Lett. 2019;695:91–99. doi: 10.1016/j.neulet.2017.05.037. [DOI] [PubMed] [Google Scholar]
- 277.Schirmer L, Mobius W, Zhao C, Cruz-Herranz A, Ben Haim L, Cordano C, et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. Elife. 2018;7. 10.7554/eLife.36428. [DOI] [PMC free article] [PubMed]
- 278.Tingaud-Sequeira A, Raldua D, Lavie J, Mathieu G, Bordier M, Knoll-Gellida A, et al. Functional validation of ABHD12 mutations in the neurodegenerative disease PHARC. Neurobiol Dis. 2017;98:36–51. doi: 10.1016/j.nbd.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 279.Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hubner CA, et al. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci. 2007;27(24):6581–6589. doi: 10.1523/JNEUROSCI.0338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Duchesne A, Vaiman A, Frah M, Floriot S, Legoueix-Rodriguez S, Desmazieres A, et al. Progressive ataxia of Charolais cattle highlights a role of KIF1C in sustainable myelination. PLoS Genet. 2018;14(8):e1007550. doi: 10.1371/journal.pgen.1007550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Takanashi J, Osaka H, Saitsu H, Sasaki M, Mori H, Shibayama H, et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain and Development. 2014;36(3):259–263. doi: 10.1016/j.braindev.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 282.Ady V, Toscano-Marquez B, Nath M, Chang PK, Hui J, Cook A, et al. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J Physiol. 2018;596(17):4253–4267. doi: 10.1113/JP275902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Cavallin M, Rujano MA, Bednarek N, Medina-Cano D, Bernabe Gelot A, Drunat S, et al. WDR81 mutations cause extreme microcephaly and impair mitotic progression in human fibroblasts and Drosophila neural stem cells. Brain. 2017;140(10):2597–2609. doi: 10.1093/brain/awx218. [DOI] [PubMed] [Google Scholar]
- 284.Kizhakkedath P, Loregger A, John A, Bleijlevens B, Al-Blooshi AS, Al-Hosani AH, et al. Impaired trafficking of the very low density lipoprotein receptor caused by missense mutations associated with dysequilibrium syndrome. Biochim Biophys Acta. 2014;1843(12):2871–2877. doi: 10.1016/j.bbamcr.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 285.Renaud M, Tranchant C, Martin JVT, Mochel F, Synofzik M, van de Warrenburg B, et al. A recessive ataxia diagnosis algorithm for the next generation sequencing era. Ann Neurol. 2017;82(6):892–899. doi: 10.1002/ana.25084. [DOI] [PubMed] [Google Scholar]