

Abstract
Epigenetic changes may contribute substantially to risks of diseases of aging. Previous studies reported seven methylation variable positions (MVPs) robustly associated with incident type 2 diabetes mellitus (T2DM). However, their causal roles in T2DM are unclear. In an incident T2DM case-cohort study nested within the population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk cohort, we used whole blood DNA collected at baseline, up to 11 years before T2DM onset, to investigate the role of methylation in the etiology of T2DM. We identified 15 novel MVPs with robust associations with incident T2DM and robustly confirmed three MVPs identified previously (near to TXNIP, ABCG1, and SREBF1). All 18 MVPs showed directionally consistent associations with incident and prevalent T2DM in independent studies. Further conditional analyses suggested that the identified epigenetic signals appear related to T2DM via glucose and obesity-related pathways acting before the collection of baseline samples. We integrated genome-wide genetic data to identify methylation-associated quantitative trait loci robustly associated with 16 of the 18 MVPs and found one MVP, cg00574958 at CPT1A, with a possible direct causal role in T2DM. None of the implicated genes were previously highlighted by genetic association studies, suggesting that DNA methylation studies may reveal novel biological mechanisms involved in tissue responses to glycemia.
Introduction
Type 2 diabetes mellitus (T2DM) is a major and increasing public health problem. Genome-wide studies have identified more than 240 genetic variants (1) that are robustly associated with T2DM. However, these only explain a minor portion of T2DM susceptibility variance (2,3). Environmental factors, including diet and physical activity, and also early life factors during fetal and early postnatal development are reported to contribute to the etiology of T2DM. Epigenetic variation can occur as a result of genetic and/or environmental factors (4). DNA methylation (DNAm) at cytosine-guanine dinucleotides (CpG sites) is the most commonly studied epigenetic mechanism to date, due to its role in expression regulation and available assays to quantify DNAm intensity at multiple sites across the epigenome that are applicable to large-scale studies. Unlike genotypic variation, DNAm intensity patterns are liable to change over time, with age or following disease or other exposure, and therefore disease-associated changes may be either causal or consequential (5).
Previous epigenome-wide association studies (EWAS) have identified seven methylation variable positions (MVPs) that are significantly associated (P < 1.0 × 10−7) with incident T2DM (6,7). However, the causal role of those markers in T2DM is unclear. Here, we aimed to elucidate DNAm determinants of T2DM by performing an EWAS for incident T2DM in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk study (8). By further integrating genome-wide genetic array data, we aimed to identify methylation quantitative trait loci (methQTLs) for any T2DM-associated MVPs in order to assess the likely causal role of DNAm markers in T2DM through Mendelian randomization analyses (9).
Research Design and Methods
Cohort Descriptions
The discovery phase EWAS was undertaken in an incident T2DM case-cohort study nested within the EPIC-Norfolk study (8), a prospective cohort study that recruited 25,639 individuals aged between 40 and 79 years at baseline in 1993–1997. The cohort was representative of the general population of England and Wales for age, sex, anthropometric measures, blood pressure, and serum lipids but differed in that 99.7% of the cohort were of European descent. We defined a random subcohort of the whole EPIC-Norfolk study population excluding known prevalent case subjects of diabetes at baseline using the same definitions as used in the InterAct Project (10) who had available genotype data. Incident T2DM cases were ascertained from multiple sources: two follow-up health and lifestyle questionnaires providing self-reported information on doctor-diagnosed diabetes or medications, medications brought to the second clinical exam, and medical record linkage. Record linkage to external sources included the listing of any EPIC-Norfolk participant in the general practice diabetes register, local hospital diabetes register, hospital admissions data with screening for diabetes-related admissions, and Office of National Statistics mortality data with coding for diabetes. Participants who self-reported a history of diabetes that could not be confirmed against any other sources were not considered confirmed case subjects. Follow-up was censored at date of diagnosis of T2DM, 31 July 2006, or date of death—whichever came first. By definition in a case-cohort design, there are case subjects within and outside the random subcohort, but for the purposes of this analysis, we considered them in the incident case set only, with noncase subjects forming the comparison group. BMI and HbA1c levels were measured for each participant at baseline (Table 1). All participants in the EPIC-Norfolk study gave signed informed consent, and the study was approved by the local research ethics committee.
Table 1.
Baseline characteristics of participants in the EPIC-Norfolk, LOLIPOP, and FHS study samples
| EPIC-Norfolk, discovery phase | LOLIPOP, confirmation phase | FHS, confirmation phase | ||||
|---|---|---|---|---|---|---|
| Incident T2DM | Noncase | Incident T2DM | Noncase | Prevalent T2DM | Noncase | |
| N | 563 | 701 | 1,074 | 1,590 | 403 | 2,204 |
| Female sex, n (%) | 474 (84) | 407 (58) | 352 (36.3) | 507 (31.8) | 173 (43.0) | 1,245 (56.5) |
| Age (years) | 61.6 (8.1) | 59.1 (9.2) | 52.5 (10.2) | 49.9 (9.8) | 69.3 (8.4) | 65.8 (8.9) |
| Ethnicity | European | European | Indian Asian | Indian Asian | European | European |
| HbA1c (%) | 6.5 (1.3) | 5.5 (0.33) | 5.77 (0.49) | 5.37 (0.48) | 6.67 (1.15) | 5.55 (0.27) |
| HbA1c (mmol/mol) | 47.4 (14.2) | 36.2 (3.6) | 40 (5.4) | 35 (5.2) | 49 (12.6) | 37 (3) |
| BMI (kg/m2) | 29.2 (4.5) | 25.6 (3.6) | 28.9 (4.6) | 26.7 (3.9) | 31.6 (6.2) | 27.7 (5.0) |
Data are means (SD) unless otherwise indicated.
Confirmation of top signals from the discovery EWAS was sought in two further studies. The London Life Sciences Prospective Population (LOLIPOP) study is a prospective population study of Indian Asian (N = 17,606) and European (N = 7,766) individuals, recruited at age 35–75 years from the lists of 58 family doctors in west London, U.K., between 1 May 2002 and 12 September 2008. Indian Asians had all four grandparents born on the Indian subcontinent (India, Pakistan, Sri Lanka, or Bangladesh). The LOLIPOP study is approved by the National Research Ethics Service (07/H0712/150), and all participants gave written informed consent at enrolment. The LOLIPOP nested case-control study of incident T2DM has previously been described (6). Briefly, at follow-up, on 31 December 2013, individuals with T2DM were identified by primary care electronic health records and structured queries. Participants with incident T2DM were defined as those who did not have T2DM at baseline but who developed the disease during follow-up. Control subjects were identified from a random subset of 7,640 participants with a clinical assessment of fasting blood glucose concentration and HbA1c and questionnaire assessment between 11 January 2010 and 31 December 2013.
The Framingham Heart Study (FHS) is a community-based longitudinal study of participants living in and near Framingham, MA, at the start of the study in 1948 (11). The Offspring cohort comprised the children and spouses of the original FHS participants, as previously described (12). Briefly, enrollment for the Offspring cohort began in 1971 (N = 5,124), and in-person evaluations occurred approximately every 4–8 years thereafter. The current analysis was limited to participants from the Offspring cohort who survived until the eighth examination cycle (2005–2008) and consented to genetics research. DNAm data of peripheral blood samples collected at the eighth examination cycle were available in 2,741 participants. Prevalent T2DM was defined as having fasting glucose ≥7 mmol/L or as reporting taking T2DM medication at any examination cycle up to the eighth examination. All participants provided written informed consent at the time of each examination visit. The study protocol was approved by the institutional review board at Boston University Medical Center (Boston, MA).
Methylation Array Profiling
In all studies, DNAm intensity was measured using the Illumina Infinium Human Methylation 450K BeadChip array (12-sample array for FHS and 96-sample array for EPIC-Norfolk and LOLIPOP). Bisulfite conversion of DNA was performed using the EZ DNAm kit (Zymo Research, Orange, CA).
For 1,378 EPIC-Norfolk participants, DNAm was measured in DNA extracted from whole blood samples collected at baseline. Converted DNA was assayed by PCR and gel electrophoresis. Each 96-well DNA sample plate contained two duplicate samples. The average correlation between the duplicate samples was 98%.
In LOLIPOP, DNAm was measured among the first 1,074 Indian Asian participants with incident T2DM and 1,590 matched Indian Asian control subjects. Control subjects were matched to case subjects by age (5-year groups) and sex. DNAm was quantified in the baseline DNA samples collected at study enrollment. Samples were analyzed in random order, with masking to case-control status.
In FHS, peripheral blood samples were collected at the eighth examination (2005–2008). Genomic DNA was extracted from buffy coat using the Gentra Puregene DNA extraction kit (QIAGEN). Bisulphite-converted DNA samples were hybridized to the 12-sample Infinium HumanMethylation450 BeadChip array using the Infinium HD Methylation Assay protocol and Tecan robotics (Illumina, San Diego, CA). DNAm quantification was conducted in two laboratory batches.
EWAS Quality Control and Normalization
In EPIC-Norfolk, epigenome-wide DNAm data were analyzed in R (version 3.2.2). Initial quality control was performed as recommended by the array manufacturer; methylation intensity values were corrected using the Illumina background correction algorithm as implemented in minfi (13), methylation intensities with a detection P value ≥ 0.01 were set to “missing,” and methylation intensity β values were calculated for each methylation marker per sample. For duplicate samples, the sample with the lower CpG detection percentage was excluded.
Sample call rates were calculated as the proportion of missing data in each sample, by autosomal, X and Y chromosomes. For the autosomal data, 77 samples with a call rate ≤ 0.99 were excluded. All samples passed the call rate threshold on the X chromosome. For the Y chromosome, seven male samples that did not pass the call rate and two further female samples were excluded. Distributions of methylation intensities were also inspected by autosomal and sex chromosomes and separately in females and males, leading to the exclusion of two additional samples that had an unusual distribution of methylation intensities. After those quality control procedures, data on 1,290 samples remained. All further downstream analyses were restricted to autosomal methylation markers.
Marker call rates were calculated as the proportion of missing data at each CpG site. 8,775 CpGs with a call rate ≤0.95 were excluded. The R package ENmix (14) was used to identify CpG sites with multimodal distributions of methylation intensity, which typically arise from technical artifacts; 3,295 such CpG sites were excluded. A further 18,874 CpG sites with probes previously identified as mapping to more than one genomic location were also excluded (15).
To ensure reliability of the data, we repeated filtering on sample and marker call rates until all samples and all markers passed their respective call rate thresholds. After exclusion of prevalent T2DM case subjects at baseline, the final data set comprised 1,264 samples (563 incident T2D case subjects, including 22 case subjects from the subcohort, and 701 noncase subjects) with methylation intensities at 442,920 autosomal CpG sites. Quantile normalization of methylation intensity β values was applied separately to the different subgroups of markers based on color channel, probe type, and methylated/unmethylated subtypes as proposed by Lehne et al. (16)
In LOLIPOP, DNAm data were analyzed in R (version 2.15) using minfi (13) and other R scripts. Marker intensities were normalized by quantile normalization as previously described (6).
In FHS, DNAm data were normalized using the Dasen methodology implemented in the wateRmelon package (17). Sample exclusion criteria included poor single nucleotide polymorphism (SNP) matching of control positions, missing rate >1%, outliers from multidimensional scaling, and sex mismatch. Probes were excluded if missing rate was >20%. Data from laboratory batches were pooled, leaving up to 2,635 samples and 443,304 CpG probes for analysis. Additional information on DNAm, normalization, and quality control is available the previously published work by Aslibekyan et al. (18). Differences in DNAm data generation, quality control, and statistical models are summarized in Supplementary Table 1.
EWAS Statistical Analyses
In EPIC-Norfolk, to identify MVPs associated with incident T2DM, we performed a logistic regression model for each methylation marker with incident T2DM status, with adjustment for age, sex, estimated cell counts, and sample plate using the EWAC pipeline. A conservative multiple test–corrected P value threshold was applied (P < 1 × 10−7). Different methylation profiles have been observed between the different cell types in whole blood (19), and blood-based profile of DNAm was shown to predict the underlying distribution of cell types (20). To correct for cell composition variability (21), we estimated first the proportions of different cell types (CD4+ and CD8+ T-cell subtypes, natural killer cells, monocytes, granulocytes, and B cells) from DNAm data using the algorithm described by Houseman et al. (22) as implemented in the R package minfi (13). These cell count estimates were then used as covariates in the epigenome-wide regression models for incident T2DM.
We used STRING (23) to perform gene set enrichment on the significant genes associated with the 18 significant MVPs identified in the EWAS. We also performed a modified version of the MAGENTA (24) pipeline to identify the pathways associated with genes at the loci of the significant MVPs. Since MAGENTA uses SNP data to identify loci, we assigned to each CpG a “nearest SNP” based on HapMap3 data and using build 36 positions for both the CpG site and the SNPs (average distance to the nearest SNP = 4,175 base pairs [bp] [interquartile range 1,375–4,859]; 1,707 of 466,039 CpGs were not assigned a SNP). In effect, rather than using a SNP P value to rank genes to assess enrichment, we used the P value from the methylation site to run MAGENTA.
For LOLIPOP, an epigenome-wide association of DNAm was performed in Indian Asians with incident T2DM who were identified from the 8-year follow-up of the study. Differential white blood cell (lymphocyte, monocyte, and granulocyte) count was available for all participants, and epigenome-wide methylation scores were used to impute a further four lymphocyte subsets (CD4, CD8, natural killer, and B cells). Principal components analysis was performed to quantify latent structure in the data, including batch effects. Associations between incident T2DM and the 18 significant MVPs identified in EPIC-Norfolk were tested using logistic regression including intensity values from Infinium HumanMethylation450 BeadChip assay control probes, bisulfite conversion batch, measured white cells, and imputed white cell subsets and the first five principal components as covariates. Association results were corrected for the genomic control inflation factor. For testing of the predictive ability of the 18 markers for incident T2DM, univariate logistic regressions were run for each of the 18 markers to obtain individual effect sizes (β values) for incident T2DM. A weighted methylation risk score was subsequently calculated from these β values, and receiver operating curve analyses were performed to provide estimates for area under the curve (AUC).
In FHS, association between each identified MVP (associated with incident T2DM in EPIC-Norfolk) was tested for association with prevalent diabetes and glycemic traits (fasting glucose, fasting insulin, and HbA1c). The analysis of glycemic traits included only individuals without diabetes. Fasting insulin was natural log transformed. Random effects statistical models were used to analyze the data to account for sibling correlation and included adjustments for age, sex, white blood cell counts, technical covariates, batch effects, and BMI, with DNAm as the dependent variable.
We also examined each T2DM-associated MVP for additional cross-sectional association with type 1 diabetes mellitus (T1DM) in an earlier EWAS of 52 monozygous twin pairs discordant for T1DM, in cell-sorted peripheral blood mononuclear cells (monocytes, B cells, or T cells) (25). As T2DM and T1DM have largely differing aetiologies, MVPs that are consistently associated with both outcomes may indicate metabolic effects of diabetes on DNAm.
Other Tissues
The relevance of changes in DNAm intensity in whole blood to other tissues was tested by analysis of genome-wide DNAm data, generated using the Illumina Infinium Human Methylation 450K BeadChip array, from human liver, adipose tissue, and skeletal muscle, as previously published (26). Human liver DNAm data were from participants of the Kuopio Obesity Surgery Study (KOBS); 35 with T2DM and 60 without (27). Data on adipose tissue (14 pairs), skeletal muscle (17 pairs), and blood (19 pairs) were from monozygotic twins discordant for T2DM (26,28,29). Adipose tissue and skeletal muscle from the same individual were available for most of these twin pairs (16 pairs in blood/muscle and 14 pairs in blood/fat); concordance in DNAm intensity across these tissues was tested for each highlighted MVP by Spearman correlation tests. We further tested cross-tissue correlations in DNAm at T2DM-associated MVPs between blood and other tissues of relevance to T2DM etiology, liver, and pancreas in publicly available Infinium HumanMethylation450 BeadChip array data from six cadavers sampled within 12 h postmortem (mean [SD] age 65.5 [7.2] years) (30).
Mendelian Randomization Analyses
We performed bidirectional Mendelian randomization analyses to test whether any T2DM-associated MVPs had a causal effect on T2DM or are a consequence of metabolic differences that had originated before the baseline measurement in this study. To predict the causal effect of each of T2DM-associated MVPs on T2DM, methQTLs associated with each MVP (FDR <0.05) in whole blood in 3,841 adults of European descent were identified using the BIOS QTL browser (31). To run Mendelian randomization analyses, we converted the Z score for each methQTL to β and SE using the following formulas (32):
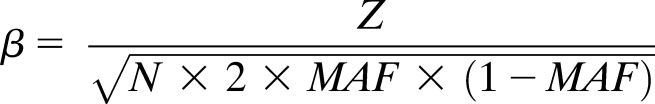 |
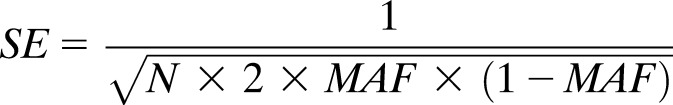 |
where N is the sample size and MAF is the minor allele frequency. We then tested these methQTLs in Mendelian randomization analyses (9) for T2DM. Genetic associations with T2DM were estimated in 69,677 case and 551,081 control subjects from the UK Biobank study (33), the EPIC-InterAct study (10) and the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium (2). A summary statistics method (inverse variance weighted) that combines all the SNPs for each MVP as a genetic instrument was used to predict the effect of that MVP on T2DM (34). To ensure that the instruments are independent, we performed clumping. The Egger regression for Mendelian randomization was also used to assess the sensitivity of the results to violations of Mendelian randomization assumptions. Mendelian randomization analyses were run using the R package TwoSampleMR (35).
For the reverse direction causal assessment, we tested SNPs with previously reported associations with T2DM (2) or related metabolic phenotypes (BMI [36], fasting glucose [37], 2-h glucose [38], fasting insulin [39], fasting insulin adjusted for BMI [37], insulin resistance [40], insulin secretion [41], and waist-hip-ratio adjusted for BMI [42]) to test whether these traits have causal effects on methylation intensity at any T2DM-associated MVP. We used summary statistics methods (inverse variance weighted and Egger tests) that combine all the SNPs for each trait as a genetic instrument to predict the effect of that trait on each T2DM-associated MVP (34) in the cohort control samples of EPIC-Norfolk (N = 613), in which genotype data were generated using the Affymetrix Axiom UK Biobank chip. All genotypes passed standard quality control criteria as specified by the Affymetrix best practice pipeline, and SNPs with MAF <5% in this sample were excluded.
Data and Resource Availability
Full summary data from the discovery EWAS for incident T2DM in the EPIC-Norfolk study are available from https://www.repository.cam.ac.uk/; BIOS QTL browser, http://bbmri.researchlumc.nl/atlas/; GoDMC, http://www.godmc.org.uk/.
Results
MVPs Associated With Incident T2DM
In the EPIC-Norfolk study, we identified 18 MVPs that are associated with incident T2DM at P < 1 × 10−7, including 15 novel signals (Table 2). None of these was reported to have a SNP on the target CpG (15). The two strongest associations were the previously reported signals at TXNIP (cg19693031; P = 2.7 × 10−21) and ABCG1 (cg06500161; P = 6.4 × 10−14) (6,7). We confirmed a third previously reported signal at SREBF1 (cg11024682; P = 6.0 × 10−10) and provide supportive evidence for an additional signal at PROC (cg09152259; P = 4.2 × 10−4) that had previously not been considered to be true due to lack of replication in Europeans (Supplementary Table 2).
Table 2.
MVPs associated with incident T2DM at P < 1.0E-07 in EPIC-Norfolk (N = 1,264)
| CpG identifier | Chr | Position | OR | 95% CI | P | FDR | Gene name | Gene position |
|---|---|---|---|---|---|---|---|---|
| cg19693031 | 1 | 144152909 | 0.52 | 0.46–0.6 | 2.7E-21 | 1.3E-15 | TXNIP | 3′ UTR |
| cg06500161 | 21 | 42529656 | 1.65 | 1.45–1.89 | 6.4E-14 | 1.5E-08 | ABCG1 | Body |
| cg14476101 | 1 | 120057515 | 0.67 | 0.59–0.76 | 2.8E-10 | 3.9E-05 | PHGDH | Body |
| cg14020176 | 17 | 70276580 | 1.63 | 1.4–1.9 | 3.3E-10 | 3.9E-05 | SLC9A3R1 | 3′ UTR |
| cg11024682 | 17 | 17670819 | 1.56 | 1.35–1.79 | 6.0E-10 | 5.7E-05 | SREBF1 | Body |
| cg06397161 | 22 | 38090005 | 1.51 | 1.32–1.73 | 4.5E-09 | 3.3E-04 | SYNGR1 | Body; TSS200 |
| cg00574958 | 11 | 68364198 | 0.69 | 0.61–0.78 | 5.2E-09 | 3.3E-04 | CPT1A | 5′ UTR |
| cg06235429 | 11 | 67129690 | 1.49 | 1.3–1.7 | 5.5E-09 | 3.3E-04 | NDUFV1 | TSS1500 |
| cg05778424 | 17 | 52524507 | 1.69 | 1.42–2.02 | 7.4E-09 | 3.9E-04 | AKAP1 | 5′ UTR |
| cg11376147 | 11 | 57017774 | 0.68 | 0.59–0.77 | 1.3E-08 | 6.0E-04 | SLC43A1 | Body |
| cg04816311 | 7 | 1033176 | 1.51 | 1.31–1.75 | 1.7E-08 | 7.2E-04 | C7orf50 | Body |
| cg02711608 | 19 | 51979804 | 0.69 | 0.6–0.79 | 4.5E-08 | 1.5E-03 | SLC1A5 | 1st exon; 5′ UTR |
| cg08309687 | 21 | 34242466 | 0.68 | 0.6–0.78 | 4.5E-08 | 1.5E-03 | ||
| cg13514042 | 7 | 1158728 | 1.42 | 1.25–1.61 | 4.5E-08 | 1.5E-03 | ||
| cg08994060 | 10 | 6254032 | 0.65 | 0.55–0.76 | 5.2E-08 | 1.6E-03 | PFKFB3 | Body |
| cg01676795 | 7 | 75424284 | 1.56 | 1.33–1.84 | 6.5E-08 | 1.8E-03 | POR | Body |
| cg25130381 | 1 | 27313308 | 1.49 | 1.29–1.73 | 6.7E-08 | 1.8E-03 | SLC9A1 | Body |
| cg11183227 | 15 | 89256411 | 1.49 | 1.29–1.72 | 7.0E-08 | 1.8E-03 | MAN2A2 | Body |
Position: by HapMap build 37. OR: odds ratio per 1 SD in methylation intensity. Genes: gene names in which the CpG falls from 1,500 bp upstream of the transcriptional start site to the end of the 3′ UTR as in Illumina’s Infinium Human Methylation 450K BeadChip manifest file. Chr, chromosome; FDR, false discovery rate.
We sought confirmation of the top 18 MVPs in data on 1,074 incident T2DM cases and 1,590 control samples from the LOLIPOP study and in cross-sectional data from FHS (403 with prevalent T2DM and 2,204 control subjects) (Table 3). All 18 MVPs showed directionally consistent associations with incident T2DM (14 at P < 0.05) and prevalent T2DM (16 at P < 0.05).
Table 3.
Confirmation of the top 18 T2DM-associated MVPs in LOLIPOP and FHS
| CpG identifier | Chr | Gene | Discovery, incident T2DM | LOLIPOP, incident T2DM | FHS, prevalent T2DM* | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | OR | 95% CI | P | β | SE | P | |||
| cg19693031 | 1 | TXNIP | 0.52 | 0.46–0.6 | 0.68 | 0.62–0.75 | 1.2E-14 | −2.6E-02 | 2.7E-03 | 1.6E-21 |
| cg06500161 | 21 | ABCG1 | 1.65 | 1.45–1.89 | 1.44 | 1.31–1.58 | 2.6E-14 | 1.5E-02 | 1.8E-03 | 7.1E-17 |
| cg14476101 | 1 | PHGDH | 0.67 | 0.59–0.76 | 0.81 | 0.75–0.89 | 3.0E-06 | −1.6E-02 | 3.6E-03 | 1.5E-05 |
| cg14020176 | 17 | SLC9A3R1 | 1.63 | 1.4–1.9 | 1.14 | 1–1.29 | 4.3E-02 | 5.4E-03 | 1.5E-03 | 3.9E-04 |
| cg11024682 | 17 | SREBF1 | 1.56 | 1.35–1.79 | 1.40 | 1.26–1.57 | 2.2E-09 | 8.6E-03 | 1.6E-03 | 5.4E-08 |
| cg06397161 | 22 | SYNGR1 | 1.51 | 1.32–1.73 | 1.17 | 1.06–1.28 | 1.1E-03 | 9.6E-03 | 2.2E-03 | 1.6E-05 |
| cg00574958 | 11 | CPT1A | 0.69 | 0.61–0.78 | 0.80 | 0.74–0.88 | 1.1E-06 | −6.7E-03 | 7.9E-04 | 4.8E-17 |
| cg06235429 | 11 | NDUFV1 | 1.49 | 1.3–1.7 | 1.11 | 1–1.24 | 5.8E-02 | 2.4E-03 | 1.3E-03 | 6.5E-02 |
| cg05778424 | 17 | AKAP1 | 1.69 | 1.42–2.02 | 1.44 | 1.21–1.71 | 3.5E-05 | 4.9E-03 | 1.6E-03 | 2.5E-03 |
| cg11376147 | 11 | SLC43A1 | 0.68 | 0.59–0.77 | 0.85 | 0.74–0.97 | 1.5E-02 | −3.2E-03 | 1.2E-03 | 8.4E-03 |
| cg04816311 | 7 | C7orf50 | 1.51 | 1.31–1.75 | 1.13 | 1–1.27 | 4.4E-02 | 2.0E-02 | 3.2E-03 | 8.4E-10 |
| cg02711608 | 19 | SLC1A5 | 0.69 | 0.6–0.79 | 0.84 | 0.76–0.93 | 9.7E-04 | −7.9E-03 | 1.7E-03 | 2.0E-06 |
| cg08309687 | 21 | — | 0.68 | 0.6–0.78 | 0.82 | 0.74–0.91 | 1.9E-04 | −7.8E-03 | 3.0E-03 | 1.0E-02 |
| cg13514042 | 7 | — | 1.42 | 1.25–1.61 | 1.04 | 0.94–1.15 | 4.4E-01 | 1.8E-04 | 1.4E-03 | 9.0E-01 |
| cg08994060 | 10 | PFKFB3 | 0.65 | 0.55–0.76 | 0.81 | 0.72–0.92 | 6.6E-04 | −1.6E-02 | 2.5E-03 | 8.5E-10 |
| cg01676795 | 7 | POR | 1.56 | 1.33–1.84 | 1.09 | 0.95–1.26 | 2.2E-01 | 9.2E-03 | 2.4E-03 | 1.2E-04 |
| cg25130381 | 1 | SLC9A1 | 1.49 | 1.29–1.73 | 1.23 | 1.09–1.39 | 1.2E-03 | 6.5E-03 | 1.7E-03 | 1.7E-04 |
| cg11183227 | 15 | MAN2A2 | 1.49 | 1.29–1.72 | 1.08 | 0.97–1.2 | 1.9E-01 | 4.6E-03 | 2.0E-03 | 2.2E-02 |
MVPs and individual cells with confirmed association P < 0.05 appear in boldface type. FHS: 403 case and 2,204 control subjects). LOLIPOP: 1,074 case and 1,590 control subjects. OR: odds ratio for T2DM per 1 SD in methylation intensity. Chr, chromosome.
In FHS, β indicates difference in percentage DNAm intensity between case and control subjects, with adjustment for age, sex, principal components 1–3 (calculated from methylation data), batch, and family structure.
Novel MVPs associated with incident T2DM include cg14476101 (P = 2.8 × 10−10), located in the gene body of PHGDH, which encodes phosphoglycerate dehydrogenase, an enzyme involved in the synthesis of l-serine and other amino acids, and cg00574958 (P = 5.2 × 10−9) in the 5′ UTR (untranslated region) of CPT1A, which encodes an enzyme that initiates mitochondrial oxidation of long-chain fatty acids (Supplementary Table 11). Four of the 18 MVPs were located within solute carrier family genes (SLC1A5, SLC43A1, SLC9A1, and SLC9A3R1), which encode plasma membrane proteins that regulate cell transport of amino acids and other metabolites.
To systematically explore the biological pathways implicated by T2DM-associated methylation signals, we first tested the 18 MVPs for gene set enrichment using STRING and identified significant enrichment for three pathways: “positive regulation of cholesterol biosynthetic process” (indicated by MVPs at ABCG1, SREBF1, and POR), “carnitine metabolic process” (indicated by CPT1A and POR), and “AMPK signaling” (indicated by PFKFB3, CPT1A, and SREBF1). We then tested the full EWAS data set in a modified MAGENTA pipeline and identified significant enrichment for T2DM-associated methylation signals in 10 pathways (Supplementary Table 4), including “insulin receptor signaling,” “IGF-1 signalling,” “erythropoietin signaling,” “JAK signaling,” and “integrin signaling.”
MVPs Associated With Glycemic Traits
In nondiabetic control FHS samples, all 18 T2DM-associated MVPs showed directionally concordant associations with fasting glucose, fasting insulin levels and BMI, and 16 of the 18 MVPs showed directionally concordant associations with HbA1c (Supplementary Table 5). In additional conditional models in the EPIC-Norfolk discovery sample, the associations of all individual 18 MVPs with incident T2DM were markedly attenuated when models were further adjusted for baseline BMI and HbA1c (median attenuation 49% [Supplementary Table 3]), indicating that these DNAm intensity changes largely reflect baseline differences between future incident T2DM cases and other cohort participants.
Furthermore, among 52 monozygous twin pairs discordant for T1DM, 7 of the 18 T2DM-associated MVPs showed cross-sectional differences in DNAm intensity in peripheral white blood cells (monocytes, B cells, or T cells) between the T1DM-affected and -unaffected twins, consistent with an effect of glycemia on DNAm intensity (at TXNIP, SLC9A3R1, SREBF1, CPT1A, C7orf50, PFKFB3, and cg08309687) (Supplementary Table 6).
Relevance of Whole Blood MVPs to Other Tissues
To explore the possible relevance of changes in DNAm intensity in whole blood to other tissues, relevant to T2DM pathogenesis, we examined these 18 MVPs in liver, adipose tissue, and skeletal muscle from individuals with and without T2DM. Nominal associations (P < 0.05) were found with only our 2 strongest whole blood MVP signals: cg06500161 at ABCG1 in adipose tissue (as previously published [26]) and cg19693031 at TXNIP in skeletal muscle (Table 4). Furthermore, at 12 of the 18 MVPs there was evidence for a positive correlation in DNAm intensity between whole blood and liver, pancreas, adipose tissue, or muscle (Supplementary Table 7).
Table 4.
Analysis of the top 18 T2DM-associated MVPs in nonblood tissues
| CpG identifier | Chr | Gene | Blood, T2DM-CON | Liver, T2DM-CON | Fat | Muscle | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T2DM | CON | T2DM-CON | T2DM-CON P | T2DM-CON, consistent | T2DM | CON | T2DM-CON | T2DM-CON P | T2DM-CON, consistent | ||||||||
| OR | P | T2DM-CON | P | Consistent | |||||||||||||
| cg19693031 | 1 | TXNIP | 0.52 | 2.7E-21 | −0.041 | 0.65 | True | 0.501 | 0.499 | 0.002 | 0.71 | False | 0.594 | 0.634 | -0.040 | 0.02 | True |
| cg06500161 | 21 | ABCG1 | 1.65 | 6.4E-14 | 0.035 | 0.64 | True | 0.477 | 0.439 | 0.038 | 0.02 | True | 0.360 | 0.349 | 0.011 | 0.75 | True |
| cg14476101 | 1 | PHGDH | 0.67 | 2.8E-10 | 0.080 | 0.14 | False | 0.565 | 0.561 | 0.004 | 0.86 | False | 0.483 | 0.484 | −0.001 | 0.85 | True |
| cg14020176 | 17 | SLC9A3R1 | 1.63 | 3.3E-10 | 0.065 | 0.26 | True | 0.678 | 0.686 | −0.008 | 0.36 | False | 0.741 | 0.748 | −0.007 | 0.52 | False |
| cg11024682 | 17 | SREBF1 | 1.56 | 6.0E-10 | 0.081 | 0.24 | True | 0.646 | 0.640 | 0.007 | 0.58 | True | 0.406 | 0.391 | 0.016 | 0.96 | True |
| cg06397161 | 22 | SYNGR1 | 1.51 | 4.5E-09 | −0.145 | 0.07 | False | 0.501 | 0.516 | −0.015 | 0.09 | False | 0.562 | 0.566 | −0.005 | 0.78 | False |
| cg00574958 | 11 | CPT1A | 0.69 | 5.2E-09 | 0.059 | 0.42 | FALSE | 0.091 | 0.096 | −0.005 | 0.22 | True | 0.097 | 0.103 | −0.006 | 0.33 | True |
| cg06235429 | 11 | NDUFV1 | 1.49 | 5.5E-09 | −0.037 | 0.60 | False | 0.793 | 0.793 | 0.001 | 0.43 | True | 0.764 | 0.765 | −0.001 | 0.82 | False |
| cg05778424 | 17 | AKAP1 | 1.69 | 7.4E-09 | 0.019 | 0.75 | True | 0.583 | 0.594 | −0.011 | 0.36 | False | 0.644 | 0.648 | −0.005 | 0.64 | False |
| cg11376147 | 11 | SLC43A1 | 0.68 | 1.3E-08 | 0.076 | 0.15 | False | 0.301 | 0.305 | −0.005 | 0.54 | True | 0.282 | 0.285 | −0.003 | 0.64 | True |
| cg04816311 | 7 | C7orf50 | 1.51 | 1.7E-08 | 0.026 | 0.63 | True | 0.853 | 0.861 | −0.008 | 0.81 | False | 0.879 | 0.885 | −0.006 | 0.35 | False |
| cg02711608 | 19 | SLC1A5 | 0.69 | 4.5E-08 | 0.018 | 0.72 | False | 0.245 | 0.256 | −0.011 | 0.06 | True | 0.356 | 0.376 | −0.020 | 0.06 | True |
| cg08309687 | 21 | 0.68 | 4.5E-08 | 0.017 | 0.85 | False | 0.634 | 0.653 | −0.019 | 0.12 | True | 0.446 | 0.445 | 0.001 | 0.85 | False | |
| cg13514042 | 7 | 1.42 | 4.5E-08 | 0.130 | 0.09 | True | 0.710 | 0.706 | 0.003 | 0.71 | True | 0.732 | 0.724 | 0.007 | 0.55 | True | |
| cg08994060 | 10 | PFKFB3 | 0.65 | 5.2E-08 | −0.095 | 0.17 | True | 0.170 | 0.175 | −0.005 | 0.86 | True | 0.154 | 0.143 | 0.011 | 0.55 | False |
| cg01676795 | 7 | POR | 1.56 | 6.5E-08 | −0.090 | 0.47 | False | 0.851 | 0.852 | −0.001 | 0.86 | False | 0.890 | 0.892 | −0.002 | 0.85 | False |
| cg25130381 | 1 | SLC9A1 | 1.49 | 6.7E-08 | −0.069 | 0.14 | False | 0.621 | 0.610 | 0.011 | 0.19 | True | 0.764 | 0.776 | −0.011 | 0.40 | False |
| cg11183227 | 15 | MAN2A2 | 1.49 | 7.0E-08 | −0.105 | 0.19 | False | 0.945 | 0.946 | −0.001 | 0.63 | False | 0.906 | 0.910 | −0.004 | 0.40 | False |
Data are mean β unless otherwise indicated. CON, control subjects.
Causal Effects on T2DM
To investigate the potential causal effects of the 18 T2DM-associated MVPs, we used the BIOS QTL browser (31) to identify methQTLs (genetic sequence variants) that are robustly associated (at P < 5 × 10−8) with DNAm intensity at any of the 18 MVPs. We found 54 methQTLs (33 cis, 21 trans), each associated with one of 16 MVPs (Supplementary Table 8). We then used these methQTLs as instrumental variables in Mendelian randomization analyses, based on aggregated publicly available GWAS data in 69,677 T2DM case and 551,081 control subjects (DIAGRAM [2], UK Biobank [33], and EPIC-InterAct [10]). Only one of the 16 T2DM-associated MVPs with an identified methQTL showed nominal evidence for a direct causal association with T2DM, cg00574958 at CPT1A (P = 0.01); however, for other MVPs the genetic-predicted effects overlapped with the observed effects in the LOLIPOP study (Fig. 1 and Supplementary Table 9).
Figure 1.

Predicted causal effects of DNAm on T2DM. The scatterplot shows the genetic-predicted effects of DNAm intensity on risk for T2DM (y-axis) plotted against observed effect estimates (from the LOLIPOP confirmation phase [x-axis]) at each of 16 top-hit MVPs (see Supplementary Table 7). Effect sizes are log–odds ratios per 1-unit change in normalized methylation intensity aligned to higher observed odds of T2DM.
We performed reverse direction causal analyses to identify causal effects of BMI and glycemic traits on methylation intensity at the 18 MVPs. Among participants without T2DM in EPIC-Norfolk (N = 613), none of the genetic instruments for the tested glycemic or metabolic traits (T2DM, BMI, fasting glucose, 2-h glucose, fasting insulin, fasting insulin adjusted for BMI, insulin resistance, insulin secretion, and waist-to-hip ratio adjusted for BMI) showed a consistent association with any of the 18 T2DM-associated MVPs (Supplementary Table 10).
Prediction of T2DM
In the LOLIPOP study sample, which was independent of the discovery EWAS, the top 18 T2DM-associated MVPs in aggregate showed no predictive ability for incident T2DM (AUC = 0.53). Furthermore, the addition of these 18 MVPs did not improve on a prediction model based on other baseline phenotypes (BMI, HbA1c, age, sex: AUC = 0.761; BMI, HbA1c, age, sex, plus 18 MVPs: AUC = 0.762).
Discussion
In this prospective study, we substantially increased the number of MVPs in whole blood that are robustly associated with incident T2DM. Associations for 17 of the 18 MVPs were confirmed with either incident or prevalent T2DM in two independent studies, which indicates the consistency of T2DM-associated whole blood DNAm intensity changes across different settings and ethnicities. Genetic causal modeling identified evidence to support a causal effect of DNAm on T2DM at one of these MVPs, cg00574958 at CPT1A.
The prospective designs of the EPIC-Norfolk and LOLIPOP studies aimed to identify MVPs that precede the development of T2DM. However, the identified T2DM-associated DNAm intensity changes were largely attenuated by adjustment for differences in BMI and glycemia that had developed prior to the baseline measurement in the prospective studies. Our Mendelian randomization analyses failed to find evidence for direct causal effects for the majority of T2DM-associated MVPs, as indicated by no detectable genetic-predicted effect of DNAm intensity on T2DM and a wide discordance between the observed and genetic-predicted effects. Conversely, overlap between EWAS signals for T2DM and T1DM was consistent with effects of glycemia on DNAm intensity for at least 7 of the 18 T2DM-associated MVPs.
Whether or not they show directly causal associations, these novel and consistent T2DM-associated MVPs are highly informative with regard to implicated genes and biological pathways. Notably, none of the genes implicated by this EWAS were previously identified by genetic variant association studies. This stark difference may suggest that T2DM-associated DNAm intensity changes may reveal novel biological mechanisms involved in tissue responses to glycemia rather than in the pathogenesis of insulin resistance or insulin secretion, which are implicated by those genetic studies. The topmost signal, cg19693031, which lies on TXNIP, is also the most significant observation in other T2DM EWAS studies (6,7). Phosphoglycerate dehydrogenase (PHGDH) catalyzes the first and rate-limiting step in glucose-derived serine synthesis and may indicate consequent purine and deoxythymidine nucleotide synthesis in response to hyperglycemia and potential tissue proliferative responses (43). Functional variation in carnitine palmitoyltransferase 1 (CPT1A) regulates the composition of circulating polyunsaturated n-3 fatty acids and docosahexaoenic acid (44) and is reported to activate lipolysis and mitochondrial activity in brown fat (45,46) and to maintain pancreatic islet secretion of the principal hyperglycemic hormone, glucagon (47). Solute carrier family members are sodium-dependent membrane transporters that regulate intracellular cell pH, cell volume, and other cellular events such as adhesion, migration, and proliferation and also contribute to systemic homeostasis of fluid volume, acid-base balance, and electrolytes. Specifically, SLC9A3R1 (NHERF1) binds to PTEN to activate the PI3 kinase signaling cascade involved in cell survival, growth, proliferation (48) and is a key component of insulin and IGF-1 signaling pathways that we found enriched for T2DM EWAS associations. These highlighted pathways could potentially contribute to the pathogenesis of micro- and macrovascular complications of hyperglycemia. PFBK3, a regulator of glycolysis and insulin signaling in mice, was recently highlighted by a SNP association with late-onset autoimmune diabetes, and we here provide independent evidence to support its role in human glucose regulation (49).
We recognize a number of limitations of our study. Both of the prospective study samples displayed large differences in baseline glycemia and BMI between incident T2DM case and noncase subjects. This nested prospective study design aimed to identify interactions between genetic factors and baseline lifestyle factors measured prior to the development of clinically diagnosed T2DM (10). Since it is impossible to develop T2DM except by passing through a phase of nondiabetic hyperglycemia, it is inevitable that people who go on to develop incident diabetes in a cohort study will have raised glucose levels at baseline if follow-up is of short or medium duration. Future studies that have samples stored many years prior to disease onset would be required to identify when in the development of diabetes the T2DM-MVP associations become apparent. Secondly, our assessments of other, nonblood, tissues were limited in the range of tissues and numbers of samples available. Despite concordant changes in DNAm intensity between whole blood and various tissues relevant to T2DM pathogenesis at 12 of the 18 T2DM-associated MVPs, nominal differences in DNAm were found only for our strongest two MVPs, which suggests that larger study samples are needed. We recognize that whole blood is not a tissue of interest to the pathogenesis of T2DM; however, current, and most likely future, large-scale EWAS are confined to such samples, and functional insights will depend on follow-up of whole blood signals in other tissues (50,51). The same issue of appropriate tissue of interest limits our genetic modeling approach, which identified genetic markers of DNAm intensity in peripheral blood. Furthermore, the sample size for this approach (N = 3,841 in BIOS QTL [31] and N = 613 in the EPIC-Norfolk cohort control group) is relatively small compared with data on QTLs for gene expression in peripheral blood (N = 8,086 in the study by Westra et al. [52]). Hence, we found only nominal evidence for a causal effect of DNAm at only 1 of the 18 T2DM-associated MVPs, at CPT1A, and for several MVPs the genetic-predicted effects were overlapping with the observed effects. Similarly, a recent large EWAS for BMI found a causal role of methylation at only one MVP (cg26663590 at NFATC2IP) (53). There are various possible conceptualizations of the functional interplay between SNP, MVP, and T2DM, which provide alternative explanations other than SNP-to-DNAm-to-T2D (54), but they do not limit the statistical detection of apparent causal signals. Future, larger reference data on QTLs for DNAm intensity in whole blood are being generated (Genetics of DNA Methylation Consortium [GoDMC]), which will allow more powerful tests for causality, although their relevance to DNAm in tissues of interest remains an important question. Finally, the determinants of the identified T2DM-associated MVPs remain unknown. Again, larger reference panels of GWAS and DNAm array data, as well as new methods to integrate findings across multiple methQTLs for each MVP, will inform future causal analyses. Future studies are needed to identify the potential lifestyle and developmental determinants of these T2DM-associated MVPs.
In conclusion, we identified several robust and consistent DNAm markers for incident T2DM. These appear to be related to T2DM via glucose and obesity-related pathways that had their effects before the collection of baseline samples in these cohort studies, which commenced in midlife. These associations indicate several plausible biological mechanisms involved in tissue responses and comorbidities of hyperglycemia.
Supplementary Material
Article Information
Acknowledgments. The authors are grateful for all of the participants and staff of the EPIC-Norfolk, LOLIPOP, and FHS cohorts. The authors thank Dr. Stephen Burgess (University of Cambridge) for advice on methQTLs and Dr. Jan Bert van Klinken (Leiden University) for advice on the BIOS QTL data as well as Stephen Sharp and Dr. Jian’an Luan (University of Cambridge) for advice on statistical analyses and Ylva Wessman, Per-Anders Jansson, and Emma Nilsson (Lund University Diabetes Center) for help with the study on twin pairs discordant for T2DM.
Funding. EPIC-Norfolk is supported by program grants from the Medical Research Council (MRC) [G9502233, G9502233, and G9502233] and Cancer Research UK [C864/A8257]. The generation and management of the Illumina Infinium Human Methylation 450K BeadChip array data in this cohort are supported through the MRC Cambridge initiative in metabolomic science [MR/L00002/1]. The genome-wide genotyping data in EPIC-Norfolk was funded by MRC award MC_PC_13048. This work is also supported by MRC program grants MC_UU_12015/1, MC_UU_12015/2, and MC_UU_12015/5. The LOLIPOP study is supported by the National Institute for Health Research (NIHR) Comprehensive Biomedical Research Centre Imperial College Healthcare National Health Service (NHS) Trust, the British Heart Foundation (SP/04/002), the MRC (G0601966 and G0700931), the Wellcome Trust (084723/Z/08/Z, 090532, and 098381), the NIHR (RP-PG-0407-10371), the NIHR Official Development Assistance (award 16/136/68), and the European Union Seventh Framework Programme (FP7) (EpiMigrant, 279143) and Horizon 2020 Framework Programme (iHealth-T2D, 643774). We acknowledge support of the MRC-PHE Centre for Environment and Health and the NIHR Health Protection Research Unit in Health Impact of Environmental Hazards. The work was carried out in part at the NIHR/Wellcome Trust Imperial Clinical Research Facility. J.C.C. is supported by the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research Investigator (STaR) Award (NMRC/STaR/0028/2017). The FHS is supported by grants N01-HC-25195 and HHSN268201500001I. The laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and by the National Institutes of Health (NIH) Director’s Challenge Award (principal investigator: D.L.). The analytical component of this project was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and the Center for Information Technology, NIH. J.B.M. is supported by National Institute of Diabetes and Digestive and Kidney Diseases grants U01 DK078616 and K24 DK080140. Data on T1DM-discordant twin pairs arose from studies funded by the EU FP7 project BLUEPRINT (282510). The Cardiovascular Epidemiology Unit at the University of Cambridge is supported by the U.K. MRC (MR/L003120/1), British Heart Foundation (RG/13/13/30194), and NIHR (Cambridge Biomedical Research Centre at the Cambridge University Hospitals NHS Foundation Trust). Data from human tissues are from studies supported by grants from the Novo Nordisk foundation; Swedish Research Council, Region Skåne (ALF); Euoropean Foundation for the Study of Diabetes; EXODIAB; Swedish Foundation for Strategic Research (IRC15-0067); Swedish Diabetes Foundation; and Albert Påhlsson Foundation.
The views expressed are those of the authors and do not necessarily represent the views of the NHS, the NIHR, the Department of Health and Social Care, the National Heart, Lung, and Blood Institute, the NIH, or the U.S. Department of Health and Human Services. The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the manuscript.
Duality of Interest. A.Y.C. is currently employed by Merck Research Laboratories. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. A.C., F.R.D., J.R.B.P., M.L., A.Y.C., B.L., D.S.P., I.D.S., and C. Liu performed data analyses. A.C., F.R.D., and K.K.O. drafted the manuscript. A.C. constructed the figure. A.C., F.R.D., J.R.B.P., N.J.W., and K.K.O. had full access to all of the data in the study. A.C. and K.K.O. had final responsibility for the decision to submit for publication. L.A.L., N.D.K., R.A.S., K.-T.K., N.G.F., C.La., M.M.M., D.L., S.B., R.D.L., J.D., J.B.M., J.S.K., M.-F.H., J.C.C., N.J.W., and K.K.O. contributed to the data collection. K.-T.K., N.G.F., J.D., J.B.M., J.S.K., C.Lin., M.-F.H., J.C.C., N.J.W., and K.K.O. contributed to the study design. All authors contributed to data interpretation and revisions of the manuscript. A.C. and K.K.O. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db18-0290/-/DC1.
References
- 1.Mahajan A, Taliun D, Thurner M, et al. . Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris AP, Voight BF, Teslovich TM, et al.; Wellcome Trust Case Control Consortium; Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium . Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahajan A, Go MJ, Zhang W, et al.; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium . Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 2014;46:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007;128:669–681 [DOI] [PubMed] [Google Scholar]
- 5.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011;12:529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambers JC, Loh M, Lehne B, et al. . Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol 2015;3:526–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soriano-Tárraga C, Jiménez-Conde J, Giralt-Steinhauer E, et al.; GENESTROKE Consortium . Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet 2016;25:609–619 [DOI] [PubMed] [Google Scholar]
- 8.Day N, Oakes S, Luben R, et al. . EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br J Cancer 1999;80(Suppl. 1):95–103 [PubMed] [Google Scholar]
- 9.Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 2013;42:1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langenberg C, Sharp S, Forouhi NG, et al.; InterAct Consortium . Design and cohort description of the InterAct Project: an examination of the interaction of genetic and lifestyle factors on the incidence of type 2 diabetes in the EPIC Study. Diabetologia 2011;54:2272–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawber TR, Meadors GF, Moore FE Jr. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health 1951;41:279–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol 1979;110:281–290 [DOI] [PubMed] [Google Scholar]
- 13.Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. . Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30:1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Z, Niu L, Li L, Taylor JA. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res 2016;44:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naeem H, Wong NC, Chatterton Z, et al. . Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genomics 2014;15:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehne B, Drong AW, Loh M, et al. . A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol 2015;16:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 2013;14:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslibekyan S, Demerath EW, Mendelson M, et al. . Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring) 2015;23:1493–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baron U, Türbachova I, Hellwag A, et al. . DNA methylation analysis as a tool for cell typing. Epigenetics 2006;1:55–60 [DOI] [PubMed] [Google Scholar]
- 20.Koestler DC, Christensen B, Karagas MR, et al. . Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics 2013;8:816–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2014;15:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houseman EA, Accomando WP, Koestler DC, et al. . DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szklarczyk D, Franceschini A, Wyder S, et al. . STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segrè AV, Groop L, Mootha VK, Daly MJ, Altshuler D; DIAGRAM Consortium; MAGIC investigators . Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 2010;6:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paul DS, Teschendorff AE, Dang MAN, et al. . Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat Commun 2016;7:13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dayeh T, Tuomi T, Almgren P, et al. . DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016;11:482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nilsson E, Matte A, Perfilyev A, et al. . Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J Clin Endocrinol Metab 2015;100:E1491–E1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitert MD, Dayeh T, Volkov P, et al. . Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 2012;61:3322–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson E, Jansson PA, Perfilyev A, et al. . Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014;63:2962–2976 [DOI] [PubMed] [Google Scholar]
- 30.Slieker RC, Bos SD, Goeman JJ, et al. . Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 2013;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonder MJ, Luijk R, Zhernakova DV, et al.; BIOS Consortium . Disease variants alter transcription factor levels and methylation of their binding sites. Nat Genet 2017;49:131–138 [DOI] [PubMed] [Google Scholar]
- 32.Rietveld CA, Medland SE, Derringer J, et al.; LifeLines Cohort Study . GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 2013;340:1467–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen NE, Sudlow C, Downey P, et al. . UK Biobank: current status and what it means for epidemiology. Health Policy Technol 2012;1:123–126 [Google Scholar]
- 34.Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG; EPIC-InterAct Consortium . Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 2015;30:543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hemani G, Zheng J, Elsworth B, et al. . The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Locke AE, Kahali B, Berndt SI, et al.; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium . Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manning AK, Hivert M-F, Scott RA, et al.; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Multiple Tissue Human Expression Resource (MUTHER) Consortium . A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saxena R, Hivert M-F, Langenberg C, et al.; GIANT Consortium; MAGIC Investigators . Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010;42:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott RA, Lagou V, Welch RP, et al.; DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium . Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lotta LA, Gulati P, Day FR, et al. . Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 2017;49:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prokopenko I, Poon W, Mägi R, et al. . A central role for GRB10 in regulation of islet function in man. PLoS Genet 2014;10:e1004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shungin D, Winkler TW, Croteau-Chonka DC, et al.; ADIPOGen Consortium; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GEFOS Consortium; GENIE Consortium; GLGC; ICBP; International Endogene Consortium; LifeLines Cohort Study; MAGIC Investigators; MuTHER Consortium; PAGE Consortium; ReproGen Consortium . New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015;518:187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pacold ME, Brimacombe KR, Chan SH, et al. . A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol 2016;12:452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skotte L, Koch A, Yakimov V, et al. . CPT1A missense mutation associated with fatty acid metabolism and reduced height in Greenlanders. Circ Cardiovasc Genet 2017;10:e001618. [DOI] [PubMed] [Google Scholar]
- 45.Clemente FJ, Cardona A, Inchley CE, et al. . A selective sweep on a deleterious mutation in CPT1A in arctic populations. Am J Hum Genet 2014;95:584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calderon-Dominguez M, Sebastián D, Fucho R, et al. . Carnitine palmitoyltransferase 1 increases lipolysis, UCP1 protein expression and mitochondrial activity in brown adipocytes. PLoS One 2016;11:e0159399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briant LJB, Dodd MS, Chibalina MV, et al. . CPT1a-dependent long-chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Reports 2018;23:3300–3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi Y, Morales FC, Kreimann EL, Georgescu MM. PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J 2006;25:910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cousminer DL, Ahlqvist E, Mishra R, et al. . First genome-wide association study of latent autoimmune diabetes in adults reveals novel insights linking immune and metabolic diabetes. Diabetes Care. 2018;41:2396–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davegårdh C, García-Calzón S, Bacos K, Ling C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol Metab 2018;14:12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bacos K, Gillberg L, Volkov P, et al. . Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun 2016;7:11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westra H-J, Peters MJ, Esko T, et al. . Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013;45:1238–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wahl S, Drong A, Lehne B, et al. . Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017;541:81–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in mendelian randomization. Epidemiology 2014;25:427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.