

Abstract
Drosophila melanogaster is a unique, powerful genetic model organism for studying a broad range of biological questions. Human studies that probe the genetic causes of rare and undiagnosed diseases using massive-parallel sequencing often require complementary gene function studies to determine if and how rare variants affect gene function. These studies also provide inroads to disease mechanisms and therapeutic targets. In this review we discuss strategies for functional studies of rare human variants in Drosophila. We focus on our experience in establishing a Drosophila core of the Model Organisms Screening Center for the Undiagnosed Diseases Network (UDN) and concurrent fly studies with other large genomic rare disease research efforts such as the Centers for Mendelian Genomics. We outline four major strategies that use the latest technology in fly genetics to understand the impact of human variants on gene function. We also mention general concepts in probing disease mechanisms, therapeutics and using rare disease to understand common diseases. Drosophila is and will continue to be a fundamental genetic model to identify new disease-causing variants, pathogenic mechanisms and drugs that will impact medicine.
Introduction
The fruit fly, Drosophila melanogaster, has an outstanding track record as a model organism because it allows studies related to genetics, development, neural function and maintenance, physiology, metabolism, wiring of the brain, as well as molecular and cellular mechanisms (1–3). Flies are easy to grow and have a short generation time and life span. The ease of manipulating fly genomes is unrivaled in any model organism, and fly phenotyping is integral to a vast field of fly studies, making it a uniquely powerful model (4). These features allow fly researchers to tackle simple as well as intricate questions rapidly and at low cost.
An area of research that has developed rapidly in the past 5 years is the use of Drosophila to facilitate the diagnoses of rare human diseases. Technical advancements in sequencing personal genomes using whole-exome or whole-genome sequencing technologies have revolutionized human genetics (5). According to the Congressional Orphan Drug Act of 1983, a rare disease is a condition affecting fewer than 200 000 people and with up to 7000 distinct rare diseases; approximately 30 million people in the US and 400 million worldwide suffer from a rare disease (https://rarediseases.info.nih.gov). People with rare disease often undergo a diagnostic odyssey and remain undiagnosed for many years (6). By sequencing exomes or genomes of individuals affected by rare disease, human geneticists have been able to uncover rare variants and point to candidate loci for novel gene–disease relationships. However, uncovering rare variants is only a beginning, and assessing which variant(s) is/are related to disease remains challenging. This is precisely where genetic model organisms can contribute, and Drosophila is now playing a more prominent role in experimentally determining which human variants affect protein function (7).
The National Institutes of Health (NIH) launched the Undiagnosed Diseases Network (UDN) 4 years ago (8, 9). The goal of this project is to diagnose the most difficult medical mysteries using cutting-edge medicine, genomics and experimental strategies. In 2015 we established the UDN Model Organisms Screening Center and the Drosophila Core of this center. This center initially included two cores using zebrafish (University of Oregon) and flies (Baylor College of Medicine) but has now expanded with a second center that includes a Caenorhabditis elegans core and another zebrafish core (Washington University at St Louis). In developing Drosophila studies for specific patients from the UDN we developed a battery of strategies to study undiagnosed disease. In this review we will provide an overview of the activity and achievements of this ‘UDN MOSC Drosophila Core’ at BCM during the past 3 years. In addition, the Drosophila Core also tackles rare and undiagnosed cases outside the UDN, in particular, candidate genes identified in the Centers for Mendelian Genomics (CMG), a large effort to solve Mendelian single-gene disorders (5). We also use flies to help in the diagnosis of cases from clinicians and human geneticists in various institutes in the US who have identified difficult-to-diagnose cases through clinical sequencing, Finally, through GeneMatcher, a matchmaking tool developed by the CMG to aid in matching cases with common candidate genes (10), we often collaborate with teams around the world. Prior to model organism experimentation each variant and gene is analyzed using information from human genetic and model organism databases through the MARRVEL.org tool (11). This initial analysis is often critical and lowers or increases the likelihood that pursuing a candidate gene in a model organism will be productive (12).
We developed or adopted four different conceptually distinct strategies to assess the functional impact of genes and variants depending on the conservation, the nature of the allele, availability of reagents including fly strains and human cDNAs and the outcome of initial exploratory experiments (Fig. 1) (7, 13). Importantly, in cases where the variant of interest is deleterious, we are in a position to probe into disease mechanism and provide insights useful for therapeutics. These efforts have led to discovery of novel genotype–phenotype links in human and have provided probing data to begin to understand the molecular mechanism of diseases (Table 1).
Figure 1.
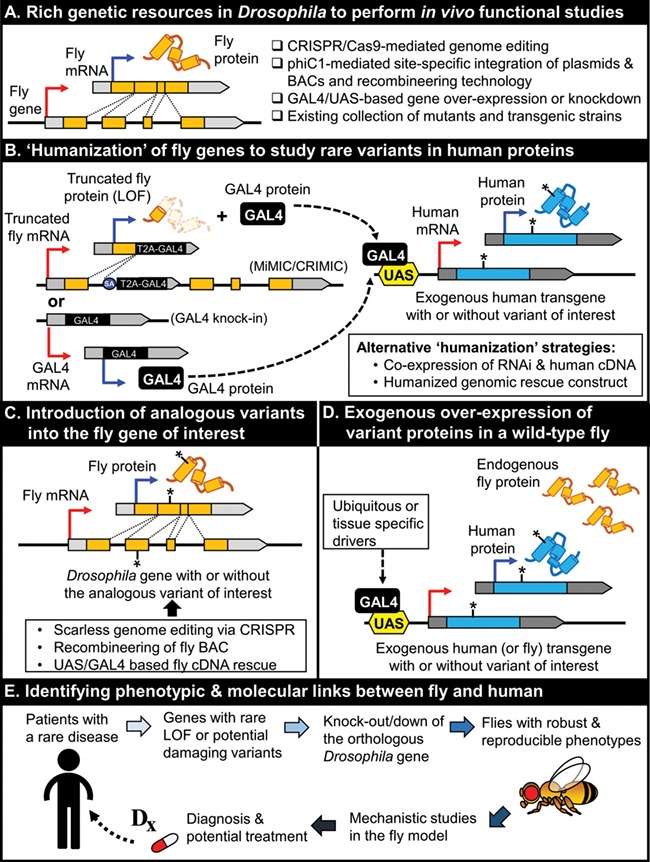
Four conceptually distinct strategies to study the effect of rare human variants linked to human disease using state-of-the-art technologies in Drosophila. (A) Rich genetic resources in Drosophila to perform in vivo functional studies. (B) ‘Humanization’ of fly genes to study variants of interest in the context of the human protein. (C) Introduction of analogous variants into the fly gene of interest. (D) Exogenous over-expression of variant proteins in a wild-type fly. (E) Studying phenotypic and molecular links between fly and human biology to provide diagnoses, understand disease mechanisms and develop potential treatments
Table 1.
Examples of novel human disease associated genes or phenotypic expansions studied by the MOSC Drosophila Core
| Human gene | Disease | Key Clinical collaborators | Fly gene | Classification | References |
|---|---|---|---|---|---|
| ANKLE2 | Primary autosomal recessive microcephaly 16 (OMIM #616681) | CMG [Baylor College of Medicine (BCM)] | Ankyrin repeat and LEM domain containing 2 (Ankle2) | Novel disease gene | (14, 32) |
| ARIH1 | Thoracic aortic aneurysms | CMG (University of Washington) |
ariadne 1 (ari-1) | Novel disease gene | (31) |
| ATAD3A | Harel-Yoon syndrome (OMIM #617183) | CMG (BCM) | belphegor (bor) | Novel disease gene | (47) |
| ATP5F1D | Mitochondrial complex V (ATP synthase) deficiency (OMIM #618120) | UDN (Stanford) | ATP synthase δ subunit (ATPsynδ) | Novel disease gene | (35) |
| CACNA1A | Early onset developmental delay, ataxia | UDN (BCM) | cacophony (cac) | Phenotypic expansion | (43) |
| DNMBP | Infantile cataracts | GeneMatcher (Univeristy of Geneva) |
still life (sif) | Novel disease gene | (49) |
| DNM1L | Encephalopathy, lethal, due to defective mitochondrial peroxisomal fission 1 (OMIM #614388) | BCM | Dynamin related protein 1 (Drp1) | Phenotypic expansion | (44) |
| EBF3 | Hypotonia, ataxia and delayed development | UDN (NIH UDP) and BCM | knot (kn) | Novel gene | (29) |
| IRF2BPL | Neurodevelopmental disorder with regression, abnormal movements, loss of speech and seizures | UDN [(Duke University, University of California, Los Angeles(UCLA)] | Protein interacting with Ttk69 and Sin3A (Pits) | Novel disease gene | (46) |
| MARK3 | Visual impairment and progressive phthisis bulbi (OMIM #618283) | GeneMatcher (University of Geneva) |
par-1 | Phenotypic expansion | |
| NR5A1 | 46XY sex reversal 3 (OMIM #612965) | UDN (BCM/UCLA) | ftz transcription factor 1 (ftz-f1) | Phenotypic expansion | (9) |
| NRDC | Severe global developmental delay and ataxia | GeneMatcher (UCLA) | Nardylisin (Nrd1) | Novel disease gene | (30) |
| OGDHL | Global developmental delay with cerebral and cerebellar atrophy and corpus callosum abnormality | CMG (BCM) | Neural conserved at 73EF (Nc73EF) | Novel disease gene | (30) |
| TBX2 | Vertebral anomalies, endocrine and T-cell dysfunction (OMIM #618223) | UDN (Duke University, UCLA) |
bifid (bi) | Novel disease gene | (15) |
| TM2D3 | Late onset Alzheimer’s disease | CHARGE consortium | almondex (amx) | Susceptibility variant | (36) |
| WDR37 | Epilepsy, developmental delay, dysmorphic features and cataracts | UDN (NIH) | CG12333 (wdr37) | Novel disease gene | (28) |
The relationship between human disease phenotypes and fly phenotypes
The most common question clinical geneticists have about fly research in human disease studies is whether human disease phenotypes can be ‘modeled’ in an invertebrate model like flies. In some cases, human disease phenotypes can be reproduced in flies. For example, fly mutants for Ankle2 have small brains and were part of our study establishing human ANKLE2 as a novel primary microcephaly gene (14). In other cases, the fly phenotype may have no obvious relationship to human disease phenotypes. For example, we used visual system abnormalities in flies to study a cardiovascular and skeletal disease in patients with TBX2 variants (15). In these scenarios, the fly phenotypes provide insight into fundamental biology, a ‘read out’ for gene function, and it often allows us to determine if the human variant is detrimental to protein function or not in vivo.
Regardless of the genetic strategy used, the first step is to determine the fly loss-of-function (LOF) phenotype. This provides the basis for the interpretation of all subsequent observed phenotypes in variants. Even when the removal of the gene leads to viable and fertile flies that do not display obvious morphological defects, assays for behavioral defects such as locomotion, climbing, flight, seizure-like bang sensitivity, learning and memory, mating behavior and measurement of neuronal activity via electrophysiology recordings (e.g. electroretinograms, neuromuscular junction and giant fiber recordings) often point to important functions of genes (16–19). Any fly phenotype observed should be rescued using genomic plasmid or Bacterial Artificial Chromosome transgenes [e.g. P (acman) clones] (20, 21) that contain the gene of interest to ensure that other mutations present on the chromosome are not responsible for the phenotype. In summary, a comprehensive range of phenotypes are accessible in flies, and we typically initiate each project with determining the LOF phenotype to provide a context for the study of the variants.
Strategy 1: ‘Humanization’ of a fly gene to test variant function
‘Humanization’ refers to replacement of a fly gene with a human homolog/ortholog to allow the study of a human protein in vivo (22). Successful humanization allows one to test the function of missense alleles in the human protein, and this includes amino acid variants that are not conserved between flies and human.
Strategy 1A: expression of human cDNAs in the spatial and temporal expression pattern of the fly ortholog
This approach is based on the introduction of an artificial exon that has dual functions, one as a gene trap and another as a gene-specific transgene driver based on the GAL4/UAS system (Fig. 1B) (23). A Splice Acceptor (SA)-T2A-GAL4-polyA (T2A-GAL4) construct is introduced into an intron of the fly gene of interest using MiMIC transposons (24) or CRISPR (25). This produces a severe LOF mutation as well as the GAL4 protein that allows expression of an exogenous cDNA cloned downstream of UAS and a minimal promoter. Since one molecule of GAL4 is produced when one molecule of the truncated gene product is produced based on the ribosome skipping T2A sequence, the human cDNA can be expressed in the same spatial and temporal expression pattern as the fly gene of interest in a LOF mutant background (26). If the gene of interest lacks a taggable intron, one can replace the open reading frame (ORF) of the fly gene of interest with the GAL4 sequence to generate a GAL4-knock-in allele using CRISPR (27, 28). By crossing these T2A-GAL4/GAL4-knock-in alleles to UAS-fluorescent proteins, the expression pattern of the gene can be assessed, which often provides valuable information.
Once a T2A-GAL4 line is established and the gene expression pattern is documented, the LOF phenotype can be determined. Next, the corresponding human ORF under the control of the UAS is generated, and transgenic flies are created to assess whether a reference (wild-type) human protein can rescue the fly phenotype in vivo. Full or partial rescue has been observed 50–70% of the time in our hands. Note that in some cases the human cDNA may rescue one phenotype but not others. To provide accessibility of human cDNA resources to Drosophila biologists, we are currently generating an ‘off-the-shelf’ human cDNA library constructs and transgenic flies. These constructs are being distributed through Drosophila Genome Resource Center (DGRC, https://dgrc.bio.indiana.edu), and transgenic flies are being deposited into Bloomington Drosophila Stock Center (BDSC, https://bdsc.indiana.edu/) and Kyoto Stock Center (http://www.dgrc.kit.ac.jp/).
Finally, once it is established that the reference human cDNA can rescue the fly T2A-GAL4 or GAL4-knock-in allele, the patient variants can be assessed. Some examples in which this overall strategy has been successful include EBF3 (29) that causes Hypotonia, Ataxia and Delayed Development Syndrome (OMIM #617330); OGDHL, a gene linked to a novel slow progressing pediatric neurodegenerative disorder (30); and, WDR37, a gene found to be mutated in patients with dysmorphism, developmental delay and neurological issues (28).
Strategy 1B: rescuing existing mutant alleles with UAS-human cDNA using ubiquitous or tissue specific GAL4 drivers
For some genes a number of useful reagents such as LOF alleles have already been previously generated. We take advantage of these resources and express the human cDNA with ubiquitous or tissue-specific GAL4 drivers in existing strains to assess rescue. For example, to test the effect of missense variants in ARIH1 linked to familial aortic aneurysm (31) or in ANLKE2 associated with congenital microcephaly (14, 32), we utilized LOF alleles of the Drosophila orthologs from a large-scale forward genetic screen (14) and performed rescue experiments using ubiquitous drivers and corresponding human cDNA transgenes (31, 32).
Strategy 1C: rescuing RNAi-mediated knockdowns of fly genes by expressing UAS-human cDNAs
An alternative but complementary approach is to utilize GAL4/UAS-based RNA interference (RNAi) (33). Reagents to perform in vivo knockdown of nearly every fly gene are available through public stock centers including the BDSC, Vienna Drosophila Research Center (https://stockcenter.vdrc.at/control/main) and the Japanese National Institute of Genetics (https://shigen.nig.ac.jp/fly/nigfly/). RNAi allows the knock down of genes of interest and can include two or even three genes simultaneously for situations in which there is redundancy between homologous genes (34).
Introduction of the UAS-human cDNA transgene allows one to assess the rescue of the RNAi knockdown phenotype using the same driver. One caveat of this approach is that the introduction of the UAS-human cDNA may reduce the efficacy of RNAi by diluting the GAL4 that activates UAS elements and hence suppresses the phenotype by diluting the RNAi efficacy. This can be overcome by introducing a neutral UAS transgene such as UAS-β galactosidase. This approach was used to show that two missense variants in ATP5F1D linked to a novel mitochondrial disease were partial LOF alleles (35).
Strategy 1D: humanization of fly genomic rescue constructs
In some cases, expression of the human protein from a transgene can be toxic to the fly. T2A-GAL4/GAL4-knock-in lines allow expression of human cDNAs in the same spatial and temporal fashion as the fly gene, but the level of expression is often higher than the endogenous protein due to GAL4 amplification. For smaller (<5 kb) single-exon genes, it is relatively easy to ‘humanize’ the fly gene using a genomic rescue construct by replacing the ORF of the fly gene with the human ORF. Using this strategy, a rare missense variant associated with increased risk of late-onset Alzheimer’s disease (AD) in TM2D3 was shown to be damaging (36). Interestingly, this variant affected an amino acid that is not conserved between human and other model organisms, showing the value of this strategy.
Strategy 2: testing conserved variants of a human gene in the fly ortholog
Humanization experiments can fail for numerous reasons. Even when the overall homology or domain conservation is high (37), a human protein expressed in a fly tissue may not properly interact with other fly proteins that form a complex, may be localized to a different subcellular component, may be unstable, may not rescue the lack of specific isoforms or may be toxic. If the variant of interest from the patient affects an evolutionarily conserved amino acid, one can introduce the analogous variant into the fly ortholog and test function in the context of a fly protein. Here we describe three strategies using this approach (Fig. 1C): 1) CRISPR /Cas9-mediated variant knock-in in the endogenous fly gene, 2) expression of wild-type and variant fly cDNAs in mutant backgrounds and 3) genomic rescue experiments on mutant alleles in which disease associated variants are introduced in a genomic transgene.
Strategy 2A: knock-in of analogous variants into the endogenous fly gene using scarless CRISPR editing
Although it is possible to generate knock-in alleles of the analogous variant in the fly genome, the technology is still labor intensive, even when using the latest CRISPR/Cas9-mediated genome-editing technologies. We developed a two-step scarless gene editing system that allows the study of the variant of interest in the endogenous genomic context (38). In a first step, the genomic clone containing the gene of interest is replaced with a dominant marker flanked by two new PAM sequences. In a second step, using gRNAs against the newly introduced PAM sequences, the variant sequences in the genomic context are introduced by a donor DNA and by screening against the dominant marker. This leads to the scarless replacement of the gene. We used this approach to study variants in the fly Nmnat gene, the fly homolog of the Leber congenital amaurosis 9 gene (NMNAT1, OMIM #608553). This technology also allows monitoring of the effect of the introduced variants on protein stability and distribution when the donor DNA is tagged with Green Fluorescent Protein (GFP) or other tags (38).
Strategy 2B: rescue experiments using fly cDNA constructs harboring analogous variants
Similar to performing a rescue experiment using UAS-human cDNAs, we can generate UAS-fly cDNAs with an analogous mutation to the human variant. Most fly cDNAs have been shown to rescue the phenotypes seen in LOF alleles (25), and a large collection of high-quality fly cDNA constructed by the Berkeley Drosophila Genome Project, known as the Gold Collection, is available from DGRC (39). A collection of fly transgenic stocks carrying UAS-fly cDNAs is also available via FlyORF (40), By expressing reference and variant fly cDNAs in T2A-GAL4/GAL4-knock-in flies or other mutant animals using specific GAL4 drivers, one can assess the impact of the disease-associated variant, as shown for Ankle2 (32).
Strategy 2C: rescue experiments using fly genomic construct harboring analogous variants
For genes that encode large proteins, a full length cDNA may not be available, or the number of splice isoforms and their requirement in different cells preclude rescue with a single cDNA. Analogous variant can then be introduced in a fly genomic construct. For example, some large genes like those that encode ion channels often have numerous isoforms: Ca2+-channels (cacaphony has 18 isoforms), K+-channels (shaker has 14) and Na+-channels (paralytic has 60) (41). Attempts to rescue LOF alleles of these genes with single fly cDNAs have failed, likely because a single isoform cannot rescue loss of many isoforms or their expression levels must be tightly regulated. Genomic rescue constructs can be modified with various site directed mutagenesis protocols, gene synthesis or recombineering techniques to generate reference and variant forms of the construct (21, 42). Using this approach, we characterized a rare missense variant in CACNA1A that was associated with a unique cerebellar degeneration phenotype in a patient with severe early onset developmental delay and congenital ataxia (43).
Strategy 3: ectopic expression of human or fly cDNAs using GAL4 drivers
In some cases expression of cDNAs in a wild-type background induces phenotypes. These phenotypes can be the basis of a simple assay to assess the function of rare human variants (Fig. 1D). For example, if GAL4-driven expression of the reference human protein causes lethality whereas the variant does not, the variant is likely to be a LOF function allele. Although this assay is testing variant function in a non-endogenous context, it can provide functional information to facilitate diagnosis. We have used this strategy to determine the functional impact of disease associated variants in DNM1L (44, 45), TBX2 (15), IRF2BPL (46) and NR5A1 (9) by over-expressing the human variants of interest and comparing it with reference transgenes. Similarly, we have also successfully demonstrated the damaging nature of variants in ATAD3A (47) and MARK3 (48) by testing the variant in the context of the fly protein through over-expression experiments. If possible, this approach should be complemented with other experiments, and one should try to obtain phenotypic data that can be related to the loss or gain of function of the fly gene.
Strategy 4: LOF experiments to identify phenotypic links between flies and human
When the human variants are typical LOF alleles based on the nature of the variant (e.g. nonsense, frameshift and gene deletions), Drosophila LOF alleles can be used to explore potential pathogenic mechanisms of the disease (Fig. 1E). For example, LOF mutations in the fly Nardilysin (Nrd1) gene were identified in a genetic screen (14), and these alleles cause a slow progressive neurodegenerative phenotype due to mTORC1 activation and decreased autophagy (30). Subsequently, an individual with a neurodegenerative disorder and acquired microcephaly with a de novo nonsense variant in the human ortholog, NRDC, was identified, indicating that NRDC likely plays a role in neuronal maintenance in human as well. In another example, RNAi-mediated reduction of still life (sif) during the development of cone cells in flies was shown to affect the development of the lens and impairs the function of the photoreceptors. Interestingly, identification of biallelic variants in DNMBP, the human ortholog of sif, in patients with infantile cataracts indicates that this gene plays a conserved role in eye development in vertebrates (49). These and other examples illustrate the value of studying the LOF phenotype of fly genes in depth to discover conserved molecular functions of orthologous genes to better understand disease mechanisms.
Concluding Remarks
While a precise molecular diagnosis provides a degree of relief and hope to patients who have undergone a diagnostic odyssey, further functional studies are required to understand how the variant of interest causes disease. Identification of the cellular and biochemical pathways that are at the root of phenotypes is the most straightforward way to identify possible drug targets. The genetic resources available to fly researchers allow functional studies of most genes or proteins in a temporal and spatially precisely controlled manner at an unprecedented speed (29, 47, 49–53). Once the molecular players and potential drug targets are identified FDA (Food and Drug Administration)-approved drugs can quickly be tested for repurposing. These studies have already led to altered medical management for some patients (43, 50).
Finally, an important theme that has emerged from functional studies of rare disease is that by studying genes that are affected in pediatric neurological cases (more than 40% of the cases in the UDN) we can obtain insight into more common adult onset neurodegenerative diseases like AD (52, 53) and Parkinson’s disease (50, 51). Hence, rare disease research can facilitate common disease studies, and Drosophila provides a platform for development of new drugs and approaches to facilitate the speed of discovery.
Funding
National Institutes of Health (U54NS093793 to H.J.B., M.F.W. and S.Y., R01GM067858 and R24OD022005 to H.J.B.)
Acknowledgements
We acknowledge the National Institute of Health of the United States for supporting the Model Organisms Screening Centers for the Undiagnosed Diseases Network and the initiatives to generate useful genetic resources for the global Drosophila community. We thank the contribution of rare disease patients, family members and healthcare providers who participate in clinical research programs including but not limited to the Undiagnosed Diseases Network and the Centers for Mendelian Genomics. HJB is an Investigator of the Howard Hughes Medical Institute.
References
- 1. Bellen H.J., Tong C. and Tsuda H. (2010) 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat. Rev. Neurosci., 11, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee T. (2017) Wiring the Drosophila brain with individually tailored neural lineages. Curr. Biol., 27, R77–R82. [DOI] [PubMed] [Google Scholar]
- 3. Robie A.A., Hirokawa J., Edwards A.W., Umayam L.A., Lee A., Phillips M.L., Card G.M., Korff W., Rubin G.M., Simpson J.H. et al. (2017) Mapping the neural substrates of behavior. Cell, 170, 393–406e328. [DOI] [PubMed] [Google Scholar]
- 4. Hales K.G., Korey C.A., Larracuente A.M. and Roberts D.M. (2015) Genetics on the fly: a primer on the Drosophila model system. Genetics, 201, 815–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Posey J.E., O’Donnell-Luria A.H., Chong J.X., Harel T., Jhangiani S.N., Coban Akdemir Z.H., Buyske S., Pehlivan D., Carvalho C.M.B., Baxter S. et al. (2019) Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med., 21, 798–812. (https://www.ncbi.nlm.nih.gov/pubmed/30655598). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reuter C.M., Brimble E., DeFilippo C., Dries A.M., Undiagnosed Diseases N., Enns G.M., Ashley E.A., Bernstein J.A., Fisher P.G. and Wheeler M.T. (2018) A new approach to rare diseases of children: the Undiagnosed Diseases Network. J. Pediatr., 196, 291–297e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wangler M.F., Yamamoto S., Chao H.T., Posey J.E., Westerfield M., Postlethwait J., Members of the Undiagnosed Diseases (UDN), Hieter P., Boycott K.M., Campeau P.M. et al. (2017) Model organisms facilitate rare disease diagnosis and therapeutic research. Genetics, 207, 9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramoni R.B., Mulvihill J.J., Adams D.R., Allard P., Ashley E.A., Bernstein J.A., Gahl W.A., Hamid R., Loscalzo J., McCray A.T. et al. (2017) The Undiagnosed Diseases Network: accelerating discovery about health and disease. Am. J. Hum. Genet., 100, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Splinter K., Adams D.R., Bacino C.A., Bellen H.J., Bernstein J.A., Cheatle-Jarvela A.M., Eng C.M., Esteves C., Gahl W.A., Hamid R. et al. (2018) Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med., 379, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sobreira N., Schiettecatte F., Valle D. and Hamosh A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Al-Ouran R., Hu Y., Kim S.Y., Wan Y.W., Wangler M.F., Yamamoto S., Chao H.T., Comjean A., Mohr S.E. et al. (2017) MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am. J. Hum. Genet., 100, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J., Network U.D., Liu Z., Bellen H.J. and Yamamoto S. (2019) Navigating MARRVEL, a web-based tool that integrates human genomics and model organism genetics information J Vis Exp, in press. [DOI] [PMC free article] [PubMed]
- 13. Senturk M. and Bellen H.J. (2018) Genetic strategies to tackle neurological diseases in fruit flies. Curr. Opin. Neurobiol., 50, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto S., Jaiswal M., Charng W.L., Gambin T., Karaca E., Mirzaa G., Wiszniewski W., Sandoval H., Haelterman N.A., Xiong B. et al. (2014) A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell, 159, 200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu N., Schoch K., Luo X., Pena L.D.M., Bhavana V.H., Kukolich M.K., Stringer S., Powis Z., Radtke K., Mroske C. et al. (2018) Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum. Mol. Genet., 27, 2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiu J.C., Low K.H., Pike D.H., Yildirim E. and Edery I. (2010) Assaying locomotor activity to study circadian rhythms and sleep parameters in Drosophila. J. Vis. Exp., 43, 2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Branson K., Robie A.A., Bender J., Perona P. and Dickinson M.H. (2009) High-throughput ethomics in large groups of Drosophila. Nat. Methods, 6, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deal S.L. and Yamamoto S. (2018) Unraveling novel mechanisms of neurodegeneration through a large-scale forward genetic screen in Drosophila. Front. Genet., 9, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harnish J.M., Deal S.L., Chao H.T., Wangler M.F. and Yamamoto S. (2019) In vivo functional study of disease-associated rare human variants using Drosophila. JoVE, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Venken K.J., Carlson J.W., Schulze K.L., Pan H., He Y., Spokony R., Wan K.H., Koriabine M., de Jong P.J., White K.P. et al. (2009) Versatile P (acman) BAC libraries for transgenesis studies in Drosophila melanogaster. Nat. Methods, 6, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venken K.J., He Y., Hoskins R.A. and Bellen H.J. (2006) P (acman): a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science, 314, 1747–1751. [DOI] [PubMed] [Google Scholar]
- 22. Bellen H.J. and Yamamoto S. (2015) Morgan’s legacy: fruit flies and the functional annotation of conserved genes. Cell, 163, 12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brand A.H. and Perrimon N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development, 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 24. Venken K.J., Schulze K.L., Haelterman N.A., Pan H., He Y., Evans-Holm M., Carlson J.W., Levis R.W., Spradling A.C., Hoskins R.A. et al. (2011) MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods, 8, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee P.T., Zirin J., Kanca O., Lin W.W., Schulze K.L., Li-Kroeger D., Tao R., Devereaux C., Hu Y., Chung V. et al. (2018) A gene-specific T2A-GAL4 library for Drosophila. Elife, 7, e35574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diao F. and White B.H. (2012) A novel approach for directing transgene expression in Drosophila: T2A-Gal4 in-frame fusion. Genetics, 190, 1139–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salazar J.L. and Yamamoto S. (2018) Integration of drosophila and human genetics to understand notch signaling related diseases. Adv. Exp. Med. Biol., 1066, 141–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanca O., Andrews J.C., Lee P.T., Patel C., Braddock S.R., Slavotinek A.M., Cohen J., Gubbels C.S., Aldinger K.A., Williams J. et al. (2019) De novo variants in WDR37 are associated with epilepsy, colobomas, dysmorphism, developmental delay, intellectual disability, and cerebellar hypoplasia. Am. J. Hum. Genet., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chao H.T., Davids M., Burke E., Pappas J.G., Rosenfeld J.A., McCarty A.J., Davis T., Wolfe L., Toro C., Tifft C. et al. (2017) A Syndromic neurodevelopmental disorder caused by de novo variants in EBF3. Am. J. Hum. Genet., 100, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoon W.H., Sandoval H., Nagarkar-Jaiswal S., Jaiswal M., Yamamoto S., Haelterman N.A., Putluri N., Putluri V., Sreekumar A., Tos T. et al. (2017) Loss of nardilysin, a mitochondrial co-chaperone for alpha-Ketoglutarate dehydrogenase, promotes mTORC1 activation and neurodegeneration. Neuron, 93, 115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan K.L., Haelterman N.A., Kwartler C.S., Regalado E.S., Lee P.T., Nagarkar-Jaiswal S., Guo D.C., Duraine L., Wangler M.F., University of Washington Center for Mendelian, G et al. (2018) Ari-1 regulates myonuclear organization together with parkin and is associated with aortic aneurysms. Dev. Cell, 45, 226–244e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Link N., Chung H., Jolly A., Withers M., Tepe B., Arenkiel B.R., Shah P.S., Krogan N.J., Aydin H., Geckinli B.B. et al. (2019) Ankle2, a target of Zika virus, controls asymmetric cell division of neuroblasts and uncovers a novel microcephaly pathway. Biorxiv. [DOI] [PMC free article] [PubMed]
- 33. Heigwer F., Port F. and Boutros M. (2018) RNA interference (RNAi) screening in Drosophila. Genetics, 208, 853–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qiao H.H., Wang F., Xu R.G., Sun J., Zhu R., Mao D., Ren X., Wang X., Jia Y., Peng P. et al. (2018) An efficient and multiple target transgenic RNAi technique with low toxicity in drosophila. Nat. Commun., 9, 4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olahova M., Yoon W.H., Thompson K., Jangam S., Fernandez L., Davidson J.M., Kyle J.E., Grove M.E., Fisk D.G., Kohler J.N. et al. (2018) Biallelic mutations in ATP5F1D, which encodes a subunit of ATP synthase, cause a metabolic disorder. Am. J. Hum. Genet., 102, 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jakobsdottir J., van der Lee S.J., Bis J.C., Chouraki V., Li-Kroeger D., Yamamoto S., Grove M.L., Naj A., Vronskaya M., Salazar J.L. et al. (2016) Rare functional variant in TM2D3 is associated with late-onset Alzheimer’s disease. PLoS Genet., 12, e1006327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N. and Mohr S.E. (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics, 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li-Kroeger D., Kanca O., Lee P.T., Cowan S., Lee M.T., Jaiswal M., Salazar J.L., He Y., Zuo Z. and Bellen H.J. (2018) An expanded toolkit for gene tagging based on MiMIC and scarless CRISPR tagging in drosophila. Elife, 7, e38709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stapleton M., Carlson J., Brokstein P., Yu C., Champe M., George R., Guarin H., Kronmiller B., Pacleb J., Park S. et al. (2002) A drosophila full-length cDNA resource. Genome Biol., 3, RESEARCH0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bischof J., Bjorklund M., Furger E., Schertel C., Taipale J. and Basler K. (2013) A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development, 140, 2434–2442. [DOI] [PubMed] [Google Scholar]
- 41. Thurmond J., Goodman J.L., Strelets V.B., Attrill H., Gramates L.S., Marygold S.J., Matthews B.B., Millburn G., Antonazzo G., Trovisco V. et al. (2019) FlyBase 2.0: the next generation. Nucleic Acids Res., 47, D759–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Venken K.J. and Bellen H.J. (2012) Genome-wide manipulations of Drosophila melanogaster with transposons, Flp recombinase, and PhiC31 integrase. Methods Mol. Biol., 859, 203–228. [DOI] [PubMed] [Google Scholar]
- 43. Luo X., Rosenfeld J.A., Yamamoto S., Harel T., Zuo Z., Hall M., Wierenga K.J., Pastore M.T., Bartholomew D., Delgado M.R. et al. (2017) Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet., 13, e1006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Assia Batzir N., Bhagwat P., Eble T., Liu P., Eng C., Elsea S.H., Robak L.A., Scaglia F., Goldman A., Dhar S.U. et al. (2019) De novo missense variant in the GTPase effector domain (GED) of DNM1L leads to static encephalopathy and seizures. Cold Spring Harb Mol Case Stud, 5, a003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chao Y.H., Robak L.A., Xia F., Koenig M.K., Adesina A., Bacino C.A., Scaglia F., Bellen H.J. and Wangler M.F. (2016) Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in drosophila. Hum. Mol. Genet., 25, 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marcogliese P.C., Shashi V., Spillmann R.C., Stong N., Rosenfeld J.A., Koenig M.K., Martinez-Agosto J.A., Herzog M., Chen A.H., Dickson P.I. et al. (2018) IRF2BPL is associated with neurological phenotypes. Am. J. Hum. Genet., 103, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harel T., Yoon W.H., Garone C., Gu S., Coban-Akdemir Z., Eldomery M.K., Posey J.E., Jhangiani S.N., Rosenfeld J.A., Cho M.T. et al. (2016) Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am. J. Hum. Genet., 99, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ansar M., Chung H., Waryah Y.M., Makrythanasis P., Falconnet E., Rao A.R., Guipponi M., Narsani A.K., Fingerhut R., Santoni F.A. et al. (2018) Visual impairment and progressive phthisis bulbi caused by recessive pathogenic variant in MARK3. Hum. Mol. Genet., doi: 10.1093/hmg/ddy180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ansar M., Chung H.L., Taylor R.L., Nazir A., Imtiaz S., Sarwar M.T., Manousopoulou A., Makrythanasis P., Saeed S., Falconnet E. et al. (2018) Bi-allelic loss-of-function variants in DNMBP cause infantile cataracts. Am. J. Hum. Genet., 103, 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin G., Lee P.T., Chen K., Mao D., Tan K.L., Zuo Z., Lin W.W., Wang L. and Bellen H.J. (2018) Phospholipase PLA2G6, a parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide kevels, similar to alpha-Synuclein gain. Cell Metab., 28, 605–618. [DOI] [PubMed] [Google Scholar]
- 51. Lin G., Wang L., Marcogliese P.C. and Bellen H.J. (2019) Sphingolipids in the pathogenesis of Parkinson’s disease and parkinsonism. Trends Endocrinol. Metab., 30, 106–117. [DOI] [PubMed] [Google Scholar]
- 52. Liu L., MacKenzie K.R., Putluri N., Maletic-Savatic M. and Bellen H.J. (2017) The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab., 26, 719–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu L., Zhang K., Sandoval H., Yamamoto S., Jaiswal M., Sanz E., Li Z., Hui J., Graham B.H., Quintana A. et al. (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell, 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]