
Figure 1.
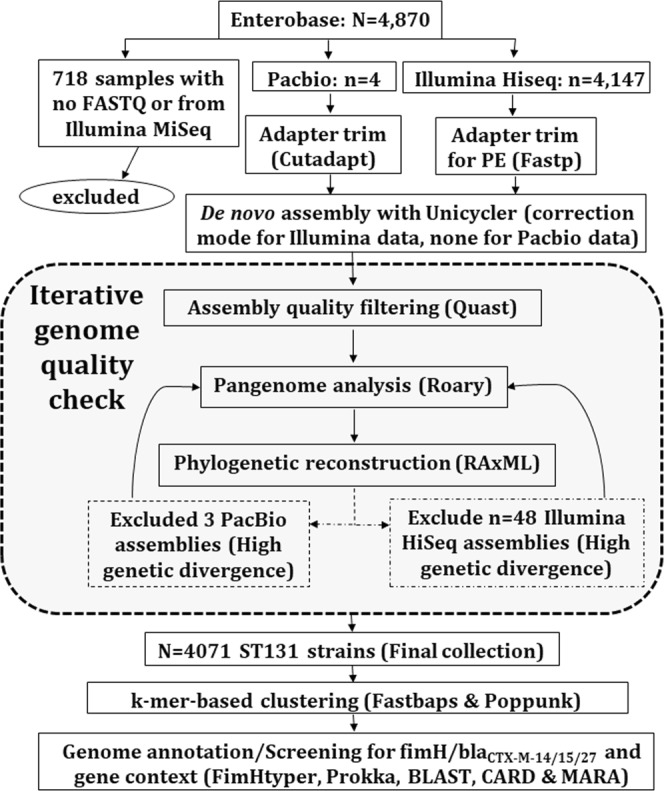
Methods summary: 4,870 read libraries were downloaded from Enterobase. 718 uninformative ones were excluded. Of those assessed, four were long read libraries (PacBio) and the rest were short paired-end reads (Illumina HiSeq). The adapters of the four PacBio and 4,147 Illumina reads were trimmed using Cutadapt and Fastp, respectively. The resulting adapter-free reads were assembled using Unicycler. An iterative genome quality check eliminated three PacBio and 77 Illumina libraries, yielding 4,071 as the final collection. Cleaned reads after Quast filtering were examined with Roary using Prokka annotation to evaluate the pangenomic diversity. Phylogenetic construction was performed by RAxML on the core genome. The assembled genomes were annotated and screened for AMR genes (including blaCTX-M-14/15/27) and their context. Genetically distinct clusters from the phylogeny were determined using Fastbaps. Distances between the core and accessory genomes of isolate pairs were estimated using Poppunk based on k-mer differences.