

Abstract
N-methyl-D-aspartate receptors (NMDARs) mediate slow excitatory postsynaptic transmission in central nervous system, thereby exerting a critical role in neuronal development and brain function. Rare genetic variants in the GRIN genes encoding NMDAR subunits segregated with neurological disorders. Here we summarize the clinical presentations for 18 patients harboring 12 de novo missense variants in GRIN1, GRIN2A, GRIN2B that alter residues in the M2 reentrant loop, a region that lines the pore and is intolerant to missense variation. These de novo variants were identified in children with a set of neurological and neuropsychiatric conditions. Evaluation of the receptor cell surface expression, pharmacological properties, and biophysical characteristics show that these variants can have modest changes in agonist potency, proton inhibition, and surface expression. However, voltage-dependent magnesium inhibition is significantly reduced in all variants. The NMDARs hosting a single copy of a mutant subunit showed a dominant reduction in magnesium inhibition for some variants. These variant NMDARs also show reduced calcium permeability and single channel conductance, as well as altered open probability. The data suggest that M2 missense variants increase NMDAR charge transfer in addition to varied and complex influences on NMDAR functional properties, which may underlie the patients’ phenotypes.
Keywords: glutamate receptor, GluN, transmembrane domain, channelopathy, missense variants, neurological diseases, epilepsy, intellectual disability, autism, movement disorder, translation study
Graphical Abstract
1. Background
N-methyl-D-aspartate receptors (NMDARs), as glutamate-gated ion channels, mediate a slow component of the excitatory synaptic current and play a key role in synaptic plasticity, memory/learning, and normal neuronal development. NMDAR dysfunction has been suggested in various pathological conditions, including epilepsy, Alzheimer disease, Huntington disease, Parkinson disease, pain, stroke, and schizophrenia (Paoletti, Bellone, & Zhou, 2013; Regan, Romero-Hernandez, & Furukawa, 2015; S. F. Traynelis et al., 2010). The hetero-tetrameric NMDARs are composed of two GluN1 and two GluN2 subunits. The glycine-binding GluN1 is a product of a single GRIN1 gene with eight RNA splice variants expressed throughout the central nervous system, while the glutamate-binding GluN2 subunits (GluN2A, 2B, 2C, and 2D) are encoded by four genes (GRIN2A, 2B, 2C and 2D), which show diverged regional and developmental expression patterns as well as distinct pharmacological and biophysical properties (Akazawa, Shigemoto, Bessho, Nakanishi, & Mizuno, 1994; Jantzie et al., 2015; Law et al., 2003; Monyer, Burnashev, Laurie, Sakmann, & Seeburg, 1994; Paoletti et al., 2013; S. F. Traynelis et al., 2010; Watanabe, Inoue, Sakimura, & Mishina, 1993; Wyllie, Livesey, & Hardingham, 2013). All NMDAR subunits share a similar architecture, containing four semi-autonomous domains: an extracellular amino-terminal domain (ATD), a bi-lobed agonist binding domain (ABD), a pore-forming transmembrane domains (TMD) comprising three transmembrane helices (M1, M3, M4) and a reentrant loop (M2), and an intracellular carboxy-terminal domain (CTD) (S. F. Traynelis et al., 2010). Activation of NMDARs requires both glutamate and glycine binding, which promotes ABD clamshell closure around the agonist, and subsequent conformational changes in linkers and the transmembrane helices, resulting in the opening of a cation-selective pore (S. F. Traynelis et al., 2010).
Recent progress in the next-generation sequencing technologies have allowed the identification of a significant number of clinically relevant de novo rare variants in NMDAR GRIN genes in patients with various neurological and neuropsychiatric disorders, including epilepsy, autism, intellectual disability, and developmental delay (Burnashev & Szepetowski, 2015; Hu, Chen, Myers, Yuan, & Traynelis, 2016; Soto, Altafaj, Sindreu, & Bayés, 2014; XiangWei, Jiang, & Yuan, 2018; Yuan, Low, Moody, Jenkins, & Traynelis, 2015). Clinically relevant rare variants reside in all NMDAR GluN subunits and across the whole gene (ATD, ABD, TMD, and CTD), but are enriched in the ABD and the TMD regions (XiangWei et al., 2018). Analysis of the genetic tolerance to missense variants in GRIN1/GluN1, GRIN2A/GluN2A, and GRIN2B/GluN2B subunits in the healthy population indicates that different regions/domains showed different degrees of intolerance, and certain sub-regions in ABD and TMD-linkers are most intolerant (Amengual-Gual, Sanchez Fernandez, & Wainwright, 2018; Ogden et al., 2017; Swanger et al., 2016), suggesting genetic variants identified in these regions have a greater likelihood to be disease-associated (J. Traynelis et al., 2017). In the present study, we describe 18 patients with 12 unique missense variants located in the TMD-M2 reentrant loop that forms the ion channel pore, which is homologous to the P loop in potassium channels (Kuner, Seeburg, & Guy, 2003; Sobolevsky, Rosconi, & Gouaux, 2009; S. F. Traynelis et al., 2010) and plays important roles in magnesium block, calcium permeability, and channel gating in NMDARs (Beck, Wollmuth, Seeburg, Sakmann, & Kuner, 1999; Burnashev, Monyer, Seeburg, & Sakmann, 1992; Kuner & Schoepfer, 1996; S. F. Traynelis et al., 2010; Williams et al., 1998). Probands with these missense variants present with various neurodevelopmental diseases, including seizures, intellectual disability, developmental delay, hypotonia/hypertonia, autism, and/or speech disorder. We summarize the clinical phenotypes and investigate how these disease-related rare variants in the M2 pore loop region influence NMDAR function and expression.
2. Methods
2.1. Ethics, Consent and Permissions
Written informed consents were obtained from the parents of all patients reported. This study was approved by the Medical Ethics Committee and the Institutional Review Boards of Emory University School of Medicine and University of Leipzig Hospitals and Clinics. All data of this study were analyzed anonymously. All functional studies were performed according to the guidelines of Emory University and Stony Brook University School of Medicine.
2.2. Mutagenesis
Human cDNAs encoding human GluN1-1a (GenBank accession numbers NP_015566; hereafter GluN1), human GluN2A (NP_000824), and human GluN2B (NP_000825) were used for studies involving di-heteromeric variants or wild type (WT) NMDARs. The cDNA fragments of the full open reading frames for human NMDAR GluN subunits were obtained from the I.M.A.G.E. Consortium (Carlsbad, USA) and Origene (Rockville, USA), and assembled and subcloned into pCI-neo, the mammalian expression vector (U47120) (Hedegaard, Hansen, Andersen, Bräuner-Osborne, & Traynelis, 2012). The rat cDNAs were for GluN1-1a (i.e., GluN1; GenBank accession numbers U08261), GluN2A (D13211), and GluN2B (U11419) were used for the experiments involving tri-heteromeric variants. Missense mutations were introduced into NMDAR cDNAs using Quikchange protocol from Stratagene (W. Chen et al., 2017). The rat cDNAs used for experiments involving tri-heteromeric variants were generated by adding the C1 or C2 coiled-coil domains to GluN2A, and then creating chimeric subunits by replacing the CTD of GluN2B with that of GluN2A-C1 and GluN2A-C2, as previously described (Hansen, Ogden, Yuan, & Traynelis, 2014; Yi, Zachariassen, Dorsett, & Hansen, 2018; Yuan et al., 2014). For receptor expression in Xenopus laevis oocytes, the cRNA synthesis in vitro was performed on Not I-linearized templates of cDNA constructs (Ambion, Austin, TX, USA) (W. Chen et al., 2017).
2.3. Two-electrode voltage-clamp (TEVC) current recordings from Xenopus laevis oocytes
Unfertilized Xenopus oocytes (defolliculated stage V-VI) were prepared from commercially available ovaries (Xenopus one Inc, Dexter, MI, USA) (W. XiangWei et al., 2019), and were injected with cRNA encoding either WT or mutant NMDAR subunits, and TEVC current recordings were performed (W. Chen et al., 2017). Briefly, cRNA (GluN1:GluN2 ratio 1:1; 5-10 ng in 50 nl of water) was injected into each oocyte, which was maintained at 15-19 °C in Barth’s culture medium (in mM: 2.4 NaHCO3, 88 NaCl, 1 KCl, 0.41 CaCl2, 0.33 Ca(NO3)2, 0.82 MgSO4, 5 HEPES; pH was adjusted to 7.4 with NaOH), plus gentamicin sulfate (0.1 mg/mL) and streptomycin (1 μg/mL). Oocytes were transferred to a recording chamber 2-4 days after injection, and perfused with extracellular oocyte recording solution (in mM: 90 NaCl, 1 KCl, 0.5 BaCl2, 10 HEPES, 0.01 EDTA, pH 7.4; except no EDTA for experiments measuring Mg2+ sensitivity). A computer-controlled 8-modular valve positioner controls the solution exchange (Digital MVP Valve, Hamilton, USA). Electrodes were prepared from borosilicate glass (#TW150F-4, World Precision Instruments, Sarasota, FL, USA) by a glass micropipette puller (dual-stage, PC-10, Narishige, Japan). Current responses were recorded under voltage clamp mode (VHOLD: −40 mV; unless otherwise stated). The recordings were made by current and voltage electrodes (filled with 0.3M and 3M KCl, respectively), using an amplifier (model OC-725C, Warner Instruments, Hamden, USA). A custom software written in LabWindows/CVI (National Instruments, Austin, TX, USA) was used to low-pass filter (10 Hz) and digitize (20 Hz) current responses. Maximal concentrations of agonists (100 μM glutamate and 100 μM glycine) were used in all Xenopus oocyte recordings unless otherwise stated. The agonist (glutamate and glycine) concentration-response curves were fitted by
| Equation 1 |
where EC50 is the glutamate or glycine concentration that evoked a half-maximal current response and nH is the Hill slope. Mg2+ potency (IC50 values) was generated by fitting the concentration-response curves by
| Equation 2 |
where IC50 is the Mg2+ concentration that evokes a half-maximal response and minimum is the residual blocking at a saturating concentration (e.g. 1 mM) of Mg2+. The voltage dependence and affinity were evaluated by fitting the data generated in the presence of Mg2+ with the Woodhull equation (McTague, Howell, Cross, Kurian, & Scheffer, 2016)
| Equation 3 |
where IUNBLOCKED is the current response at a holding potential in the presence of Mg2+, I is the current response in the absence of Mg2+ at a given holding potential, [Mg2+]o is the extracellular Mg2+ concentration (1000 μM), KD,0 mV is the KD in the absence of an applied electric field, z is the valence (2 for Mg2+), δ is the effective fraction of the electric field at the binding site, V is the voltage, Vrev is the reversal potential, and F, R, and T follow their usual meanings. The Woodhull equation was not adjusted for permeation by the blocking ion. The channel open probability (POPEN) was estimated (Yuan, Erreger, Dravid, & Traynelis, 2005) from the degree of current potentiation of NMDARs by MTSEA (2-aminoethyl methanethiosulfonate hydrobromide) (Toronto Research Chemicals, Toronto, Ontario, Canada) according to
| Equation 4 |
where γ is the chord conductance for NMDAR channels before and after MTSEA treatment and Potentiation is the amplitude of current response after MTSEA modification divided by the amplitude of current response before the treatment (Yuan et al., 2005).
2.4. Whole-cell voltage-clamp current recordings and single channel patch clamp recordings from temporarily-transfected HEK cells
HEK 293 cells (ATCC CRL-1573; Manassas, VA, USA) were plated on glass coverslips pre-treated with 100 μg/ml poly-D-lysine and cultured in culture media (DMEM/GlutaMax (GIBCO, 15140-122), 10% fetal bovine serum, 10 U/ml penicillin-streptomycin) at 37°C and 5% CO2. The calcium phosphate precipitation method (C. Chen & Okayama, 1987) was used to transiently transfected the HEK cells with plasmid cDNAs encoding WT or mutant receptors along with GFP to allow identifying the HEK cells expressing either WT or mutant receptors. A total of 0.2 μg/μL cDNA was used at a cDNA ratio for GluN1:GluN2A:GFP of 1:1:5 and for GluN1:GluN2B:GFP cDNA of 1:1:1. After 4 hours, the culture medium was exchanged and 200 μM D,L-APV and 200 μM 7-chlorokynuernic acid (7-CKA) added to reduce cell death caused by excessive activation of the NMDARs. The whole-cell voltage-clamp or single channel outside-out patch recordings were performed 18-24 hours following the transfection.
HEK293 cells on one glass coverslip were perfused in a recording chamber at 1.5-2 mL/min with external recording solution that contained (in mM) 3 KCl, 150 NaCl, 0.01 EDTA, 1.0 CaCl2, 10 HEPES, and 2.0 D-mannitol, with the pH adjusted to 7.4 by NaOH. Recording electrodes (resistance: 3-4 MΩ) were made using thin-walled filamented borosilicate glass pipettes (#TW150F-4, World Precision Instruments, Sarasota, FL, USA) prepared in two-steps by a vertical puller (Narishige P-10, Japan). The electrodes were filled with the internal pipette solution that contained (in mM) 110 D-gluconic acid, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 Na2ATP, 0.3 Na2GTP (pH adjusted to 7.35 with CsOH; and osmolality to ∼305 mOsmol/kg). The whole cell current responses to external application of maximally-effective concentrations of agonists (1000 μM glutamate and 100 μM glycine) at VHOLD of −60 mV were acquired by a patch-clamp amplifier (Axopatch 200B, Molecular Devices, CA, USA), anti-alias filtered at 8 kHz (−3 dB, 8 pole Bessel filter; Frequency Devices, IL, USA), and digitized at 20 kHz using a data acquisition system (Digidata 1440A, Molecular Devices, CA, USA) controlled by Clampex 10.3 (Molecular Devices, CA, USA). The deactivation rate following glutamate removal were fitted by a two-component exponential function using a non-linear least squares algorithm (ChanneLab, Synaptosoft, USA) with
| Equation 5 |
The weighted deactivation tau (τw) was calculated by
| Equation 6 |
Excised outside-out patches were obtained from transfected HEK cells. The same recording solution in whole-cell recordings was used, except the concentrations of CaCl2 was reduced to 0.5 mM and the pH was adjusted to 7.4. The recording electrodes for outside-out single channel recordings were prepared from thick-walled filamented borosilicate glass (#GC150F-10, Warner Instruments, Hamden, USA) using a Flaming/Brown horizontal puller (P-1000; Sutter Instrument, Novato, USA) and filled with the internal solution same as in whole-cell voltage-clamp recordings. Electrodes were coated with Sylgard silicone elastomer (Dow) and pipette tips (6-10 MΩ) were fire-polished before use. Maximally-effective concentrations of agonists (1000 μM glutamate and 100 μM glycine) were used to activate the WT or mutant NMDARs. Unitary currents were recorded (VHOLD: −80 mV) using a Warner PC501A amplifier, anti-aliased low pass filtered at 8 kHz (−3 dB Bessel 8-pole; Frequency Devices, Ottawa, IL, USA), digitized at 40 kHz, and analyzed by the time course fitting method after digital filtration (4 kHz; SCAN, http://www.ucl.ac.uk/Pharmacology/dcpr95.html). Open time histograms were generated from an imposed resolution for open and closed times (50 μs) (Colquhoun & Hawkes, 1990). The amplitude/chord conductance distributions were obtained by fitting the sum of multiple Gaussian components and the distributions of the open and shut duration fitted to the sum of multiple exponential components by the maximum likelihood (ChanneLab, Synaptosft, Decatur, USA). Since outside-out patches in this study likely contained more than one single active channel, we only analyzed the data stretches that contained a single active channel to define the channel open times and chord conductance.
2.5. Relative calcium permeability
The relative Ca2+ permeability (PCa/PNa) of NMDAR-mediated currents was evaluated by measuring alterations in reversal potentials (ΔErev) when switching from a Na+-based reference solution to the same solution but supplemented with 10 mM Ca2+ (Jatzke, Watanabe, & Wollmuth, 2002). The whole-cell voltage-clamp current recordings were conducted on transiently transfected HEK 293 cells with a pipette filled with internal solution (in mM: 140 KCl, 10 HEPES, 10 BAPTA, and pH 7.2 with KOH). The reversal or zero potential of the glutamate/glycine activated NMDAR current was determined by plotting peak current response amplitudes, obtained by 5 mV voltage steps increments, against voltage and fitting them with a fourth-order polynomial (Wollmuth & Sakmann, 1998). The control response was obtained by recording the NMDAR-mediated current response in the control solution containing 10 mM HEPES, 140 mM NaCl, 0 mM CaCl2, and the reversal potentials were averaged for the control reponses recorded before (pre) and after (post) exposure to the test solution containing 10 mM HEPES, 140 mM NaCl and 10 mM CaCl2. The Lewis equation was used to determine permeability ratios (PCa/PNa) from ΔErev for individual Ca2+ concentrations.
| Equation 7 |
All reversal potentials were corrected for the liquid junction potentials.
2.6. Beta-lactamase (β-lac) reporter assay
HEK 293 cells in 96-well plates were transfected with cDNA encoding WT β-lac-GluN1 and WT GluN2A or GluN2B, WT GluN1 and WT β-lac-GluN2A or WT β-lac-GluN2B, or β-lac tagged M2 pore loop variants using Fugene6 (Promega) (Swanger et al., 2016). Cells without WT GluN1, WT GluN2A, or WT GluN2B were treated as a negative control for surface β-lac activity. Competitive NMDA receptor antagonists (100 ¼M D,L-APV and 100 ¼M 7-CKA) were supplemented in the medium at the time of transfection. For each condition, surface and total levels were assessed in 4 wells each. After 24 hrs, the transfected cells were rinsed with HBSS (Hank’s Balanced Salt Solution + 10 mM HEPES), and then a nitrocefin (100 ¼L of 100 ¼M; Millipore, Burlington , USA) solution in HBSS with HEPES was added to the wells to measure the surface levels (Lam, Beerepoot, Angers, & Salahpour, 2013; Swanger et al., 2016). To define total levels, the cells were first rinsed with HBSS+HEPES, then lysed by trituration in 50 ¼L H2O and incubated for 30 minutes before adding nitrocefin (50 ¼L of 200 ¼M). The absorbance was measured at 30°C every min for 30 min by a microplate reader (at 468 nm; SpectraMax M2, San Jose, USA). The rate of increase in absorbance was obtained from the linear fitted slope of the data. The data was excluded from the analyses when the surface level exceeds the total level.
2.7. Evaluation of charge transfer
Charge transfer was estimated from the product of peak amplitude of whole cell current response and the deactivation time course (weighed tau, τw) for current responses to prolonged application of agonist (e.g. glutamate) divided by the cell capacitance. The relative change (in fold) in synaptic and non-synaptic charge transfer was evaluated by following equations modified from Swanger et al. (Swanger et al., 2016).
| Equation 8 |
| Equation 9 |
| Equation 10 |
where [glutamate] is 1 × 10−3 M and 1 × 10−7 M for RGLU, Synaptic and RGLU, Non-synaptic, respectively, [glycine] is 3 × 10−6 M, and nH is the Hill slope. τ is the mean weighted deactivation time constant, P is the channel open probability, Surf is surface protein levels, Mg is percentage inhibition by 1 mM Mg2+ at VHOLD: −60 mV, and RGLY and RGLU are relative response in a given glutamate or glycine concentration. We assessed weighted τw, Po, Surf, and Mg for the variants and presented them as a ratio to the WT receptors (τwMUT/τwWT, PwMUT/PWT,SurfMUT/SurfWT, MgMUT/MgWT)
Unless otherwise stated, all reagents used in this study were from Sigma. Data were presented as mean ± S.E.M and statistical significance was analyzed by one-way ANOVA post hoc test (Dunnett’s multiple comparisons test). The Holm-Bonferroni correction was used to control the family wise error rate (FWER). Log(EC50) and Log(IC50) were used in the statistical analysis. Unless otherwise stated, significance was set at p < 0.05 for all statistical tests. ANOVA F-statistics and p values for pairwise comparisons are presented in the Supp. Tables S2, S3, S4, S6, S8. Error bars in all figures represent S.E.M.
3. Results
3.1. Variants in the M2 reentrant pore loop associated with neurological and neuropsychiatric disorders
Twelve distinct de novo missense variants within the sequence encoding the M2 pore loop in NMDAR GRIN genes were identified in 18 probands with neurological and neuropsychiatric conditions (Table 1; Supp. Table S1; Figure. 1). These include three GRIN1 variants with one variant leading to GluN1-p.G618R and two distinct variants (c.1858 G>C and c.1858 G>A) leading to the same amino-acid change GluN1-p.G620R identified in two unrelated patients with developmental delay/intellectual disability and hypotonia (W. Chen et al., 2017; Lemke et al., 2016), three GRIN2A variants corresponding to GluN2A-p.L611Q, GluN2A-p.N614S, and GluN2A-p.N615K found in patients with epilepsy, developmental delay/intellectual disability, autism, and/or speech disorder (Allen et al., 2016; Endele et al., 2010; Farwell et al., 2015; Strehlow et al., 2018; von Stülpnagel et al., 2017), and seven GRIN2B variants encoding GluN2B-p.W607C, GluN2B-p.G611V, GluN2B-p.N615I, GluN2B-p.N615K, GluN2B-p.N616K, GluN2B-p.V618G, and GluN2B-p.V620M found in patients with developmental delay/intellectual disability, and/or epilepsy, and/or autism spectrum disorders (Lemke et al., 2014; Platzer et al., 2017; Retterer et al., 2016; Yavarna et al., 2015). All 18 patients showed a degree of intellectual disability or developmental delay. In addition, 50% (9 of 18) of the patients presented with seizures, with onset from 3 days to 7 months. The seizure types include infantile spasms, focal seizures, myoclonus, generalized tonic clonic seizures, and status epilepticus. Two patients (11%) showed autistic features, eleven patients (61%) had hypotonia, and two patients (11%) presented with hypertonia. Nine patients (50%) presented with language problems and six patients (33%) showed dysmorphic features. Twelve variants from 18 patients (94%) are confirmed de novo, while the origin of the variant GluN2A-p.N615K (Patient #11 in Table 1; Supp. Table S1) was unclear, since the patient’s father was not tested, although her mother was negative and the precise mutation was observed to have independently arisen de novo in another patient.
Table 1.
Patients’ and variants’ information*
| Gender | Variant | Gene | Genotype | Amino acid changes | Origin | Phenotype | Source | |
|---|---|---|---|---|---|---|---|---|
| Patient-1 | M | GluN1-G618R | GRIN1 | c.1852 G>C | p.Gly618Arg | De novo | DD/ID, hypotonia, language problem | Lemke, et al. |
| Patient-2 | M | GluN1-G620R | GRIN1 | c.1858 G>A | p.Gly620Arg | De novo | DD/ID, hypotonia, dysmorphic features, language problem | Chen, et al. |
| Patient-3 | F | GluN1-G620R | GRIN1 | c.1858 G>C | p.Gly620Arg | De novo | DD/ID, hypotonia, dysmorphic features, language problem | Chen, et al. |
| Patient-4 | M | GluN1-G620R | GRIN1 | c.1858 G>C | p.Gly620Arg | De novo | DD/ID, hypotonia | Lemke, et al. |
| Patient-5 | NA | GluN2A-L611Q | GRIN2A | c.1832T>A | p.Leu611Gln | De novo | Epi, DD/ID | this study; Strehlow, et al. |
| Patient-6 | NA | GluN2A-N614S | GRIN2A | c.1841A>G | p.Asn614Ser | De novo | DD/ID, hypotonia, language problem, ASD | Strehlow, et al. |
| Patient-7 | NA | GluN2A-N614S | GRIN2A | c.1841A>G | p.Asn614Ser | De novo | Epi, language problem | Farwell, et al. |
| Patient-8 | M | GluN2A-N614S | GRIN2A | c.1841A>G | p.Asn614Ser | De novo | Epi, DD/ID, hypotonia, language problem | von Stülpnagel, et al. |
| Patient-9 | NA | GluN2A-N614S | GRIN2A | c.1841A>G | p.Asn614Ser | De novo | DD/ID, hypertonia, language problem | Strehlow, et al. |
| Patient-10 | F | GluN2A-N615K | GRIN2A | c.1845C>A | p.Asn615Lys | De novo | Epi,DD,ID, hypotonia, language problem | Endele, et al. |
| Patient-11 | F | GluN2A-N615K | GRIN2A | c.1845C>A | p.Asn615Lys | n.d. | Epi, DD/ID, hypotonia, dysmorphic features, language problem | Allen, et al. |
| Patient-12 | NA | GluN2B-W607C | GRIN2B | c.1821G>T | p.Trp607Cys | De novo | DD/ID, dysmorphic features | Yavarna, et al. |
| Patient-13 | NA | GluN2B-G611V | GRIN2B | c.1832G>T | p.Gly611Val | De novo | Epi, ID, dysmorphic features | Platzer, et al. |
| Patient-14 | F | GluN2B-N615I | GRIN2B | c.1844A>T | p.Asn615Ile | De novo | ASD, Epi, hypotonia, ID | Lemke, et al. |
| Patient-15 | NA | GluN2B-N615K | GRIN2B | c.1845C>G | p.Asn615Lys | De novo | DD/ID | Platzer, et al. |
| Patient-16 | F | GluN2B-N616K | GRIN2B | c.1848C>G | p.Asn616Lys | De novo | Epi, ID, dysmorphic features, hypertonia | Platzer, et al. |
| Patient-17 | M | GluN2B-V618G | GRIN2B | c.1853T>G | p.Val618Gly | De novo | Epi, ID, hypotonia | Lemke, et al. |
| Patient-18 | M | GluN2B-V620M | GRIN2B | c.1858G>A | p.Val620Met | De novo | DD/ID, hypotonia | this study; Platzer, et al. ; Retterer, et al. |
detailed patients information provided in Supp.Table S1. ASD, autism spectrum disorder; DD, developmental delay; Epi, epilepsy; ID, intellectual disability; n.d.: not determined, mother (−), father not tested.
Figure. 1. Locations of disease-associated missense variants in the M2 re-entrant pore loop.
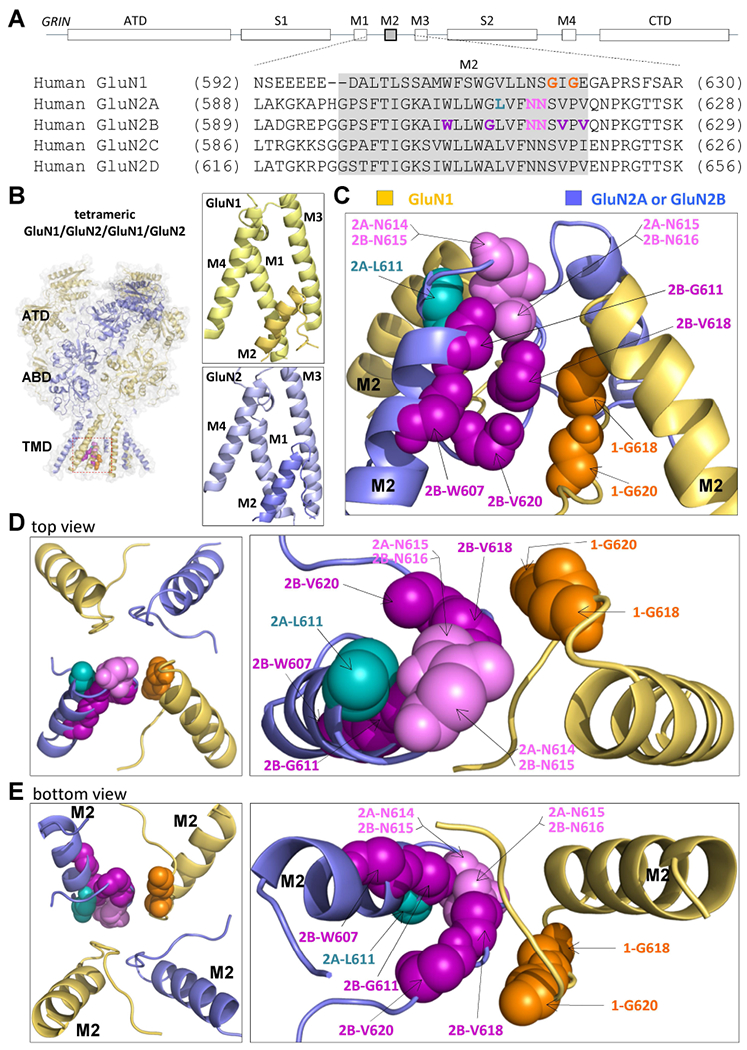
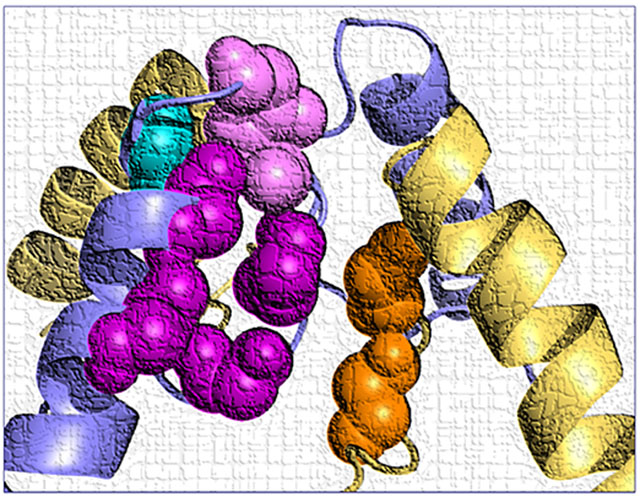
(A) A linear schematic showing domain architecture of the GRIN/GluN and protein sequence alignment for membrane associated pore loop M2 across all NMDA receptor GluN subunits. ATD, amino terminal domain; S1 and S2, first and second polypeptide sequences comprising the agonist binding domain (ABD); M1, M3, and M4, transmembrane helices within the transmembrane domain (TMD); M2: re-entrant pore loop; CTD, carboxy terminal domain. (B,C,D,E) A homology model of a tetrameric GluN1/GluN2A receptor built from GluN1/GluN2B (Karakas & Furukawa, 2014; Lee et al., 2014). The GluN1 subunit is orange and the GluN2A subunit is blue. (B) Right panels showed position of M2 re-entrant pore loop relative to M1, M3, and M4 TMDs in a single GluN1 (upper) and GluN2 (lower) subunit. The residues harboring missense mutations are highlighted by different colors in side (C), top (D), and bottom view (E).
Tolerance analysis of genetic variation adopting the published Missense Tolerance Ratio (MTR: see http://mtr-viewer.mdhs.unimelb.edu.au/mtr-viewer/) (J. Traynelis et al., 2017) across GRIN1, GRIN2A, and GRIN2B genes reveals regional variation that is consistent with strong purifying selection acting upon the second half of the M2 pore loop (Figure. 2). This is consistent with the observed depletion of missense variants in the gnomAD database (Genome Aggregation Database, Cambridge, MA, http://gnomad.broadinstitute.org/, accessed on 01-16-2019) in the M2 pore reentrant loop in GRIN1, GRIN2A and GRIN2B genes. The over-representation of disease-ascertained pathogenic-reported de novo missense variants in the M2 pore loop across the collection of the GRIN1, GRIN2A and GRIN2B genes—particularly in the second half of the M2 pore loop—coupled with the diminished missense variation in this region in the gnomAD reference cohort that excludes pediatric neurological conditions is unlikely to be due to chance (Figure. 2; Table 2), consistent with the idea that missense de novo rare variants in this region may be harmful (Fisher’s exact test p = 1.7×10−14, Table 2). This is also consistent with earlier scanning mutagenesis and covalent modification of certain residues within this region, which also suggested it was intolerant to change (Kashiwagi, Pahk, Masuko, Igarashi, & Williams, 1997; Wollmuth, Kuner, & Sakmann, 1998).
Figure. 2. Tolerance analysis of genetic variation within M2 re-entrant pore loop.
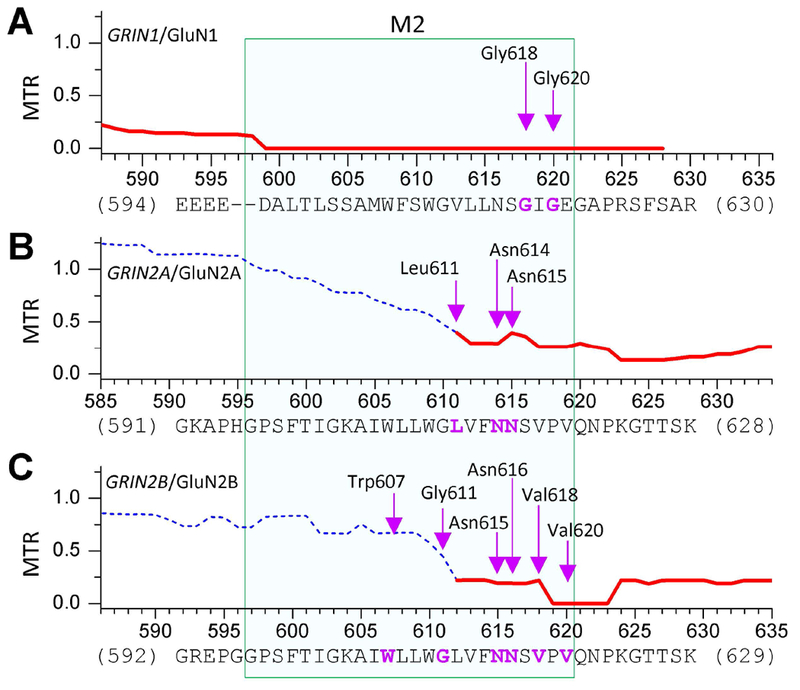
The missense tolerance ratios as calculated from the Observed/Expected mutant ratios, which are shown or GRIN1/GluN1 (A), GRIN2A/GluN2A (B), and GRIN2B/GluN2B (C). Ratios below the 10th percentile are shown as solid lines, and indicate the regions under purifying selection (J. Traynelis et al., 2017). Residues hosting the missense variants in M2 pore loop were indicated by arrows.
Table 2.
Enrichment of patient-ascertained pathogenic-reported genetic variation in M2 pore loop in GRIN1, GRIN2A, and GRIN2B
| M2 region missense (subset of total) |
Total missense |
P-value* | ||||
|---|---|---|---|---|---|---|
| Residues | gnomAD | Disease | gnomAD | Disease | ||
| GRIN1 | D599-E621 | 0 | 2 | 171 | 15 | 0.0061 |
| GRIN2A | G596-V619 | 3 | 3 | 645 | 18 | 0.0003 |
| GRIN2B | G597-V620 | 1 | 7 | 442 | 23 | 2.1×10−9 |
Fisher’s exact test, αcorrected = .016
gnomAD data based on release 2.1 (excluding filtered out variants). GRIN1 = NP_015566, GRIN2A = NP_000824 and GRIN2B = NP_000825. The total disease-related missense variants were from missense de novo variants in Swanger 2016, and were topped up with additionally published mutations since then adopting the same criteria(Swanger et al., 2016).
3.2. M2 pore loop variants moderately change agonist potency and proton sensitivity
We first evaluated the effects of M2 pore loop variants on agonist potency by TEVC current recordings from Xenopus oocytes expressing GluN1 variants with wild type (WT) GluN2A or GluN2B, and GluN2A or GluN2B variants with WT GluN1. The half-maximally effective concentration (EC50) of agonists was obtained by the analysis of the glutamate or glycine concentration-effect relationship in the presence of maximally-effective concentrations (100 μM) of glycine or glutamate. The M2 variant-containing NMDARs exhibited up to 2-fold changes in glutamate potency, with EC50 values of 2.5 ¼M for 2A-p.L611Q, 2.6 ¼M for 2A-p.N614S, and 2.8 ¼M for 2A-p.N615K, compared to 4.9 ¼M for WT GluN2A receptors (Table 3). Similarly, evaluation of the potency of glycine for the M2 pore loop variants in the presence of maximally-effective concentrations of glutamate (100 μM) showed up to a 3-fold increase in glutamate potency, i.e. with EC50 values of 0.14 ¼M of 2B-p.N616K compared to 0.43 ¼M for WT GluN2B receptors, respectively (Table 3). These data suggest that these M2 pore loop variants have a modest effect on agonist potency.
Table 3.
Summary of pharmacological data
| Glutamate EC50, μM (n) | Glycine EC50, μM (n) | %, pH6.8/pH7.6γ | |
|---|---|---|---|
| WT GluN1/WT 2A | 4.9 ± 0.35 (26) | 1.1 ± 0.06 (23) | 44 ± 1.8 (29) |
| 1-G618R/2A | 4.8 ± 0.28 (10) | 1.7 ± 0.07 (9)* | 38 ± 3.1 (12) |
| 1-G620R/2A | 6.2 ± 0.42 (11) | 1.3 ± 0.10 (12) | 25 ± 1.8 (10)* |
| 2A-L611Q | 2.5 ± 0.16 (21)* | 0.83 ± 0.08 (16)* | 63 ± 2.3 (11)* |
| 2A-N614S | 2.6 ± 0.38 (11)* | 0.61 ± 0.03 (11)* | 57 ± 2.8 (12)* |
| 2A-N615K | 2.8 ± 0.23 (12)* | 0.75 ± 0.08 (13)* | 44 ± 1.8 (10) |
| WT GluN1/WT 2B | 1.5 ± 0.09 (25) | 0.43 ± 0.04 (40) | 17 ± 0.7 (31) |
| 1-G618R/2B | 1.3 ± 0.10 (12) | n.a. | 21 ± 1.4 (15)* |
| 1-G620R/2B | 1.9 ± 0.22 (12) | 0.43 ± 0.03 (9) | 18 ± 1.2 (10) |
| 2B-W607C | 2.0 ± 0.22 (12)* | 0.44 ± 0.05 (10) | 11 ± 0.70 (12)* |
| 2B-G611V | 1.7 ± 0.33 (12) | 0.28 ± 0.04 (8) | 12 ± 2.0 (10) |
| 2B-N615I | 1.4 ± 0.16 (10) | 0.26 ± 0.02 (11)* | 19 ± 0.6 (6) |
| 2B-N615K | 1.3 ± 0.16 (11) | 0.22 ± 0.03 (12)* | 16 ± 3.8 (7) |
| 2B-N616K | 1.1 ± 0.19 (8) | 0.14 ± 0.01 (7)* | 17 ± 1.7 (12) |
| 2B-V618G | 1.6 ± 0.15 (8) | 0.47 ± 0.02 (15) | 10 ± 0.68 (10)* |
| 2B-V620M | 1.1 ± 0.11 (12) | 0.58 ± 0.07 (21) | 24 ± 1.8 (10)* |
Data were generated by oocytes recordings with −40 mV of holding potential, and were expressed as Mean ± SEM (n).
percentage of the current at pH 6.8 compared to that at pH 7.6.
p < 0.05 LogEC50 compared by one way ANOVA, with Dunnett’s multiple comparisons test.
Statistical analysis is given in Supp. Table S2.
We subsequently assessed the effects of the M2 pore loop variants on NMDAR proton sensitivity by comparing current response amplitudes recorded in oocytes at pH 6.8 to those recorded at pH 7.6. When co-expressed with WT GluN2A, the GluN1-p.G620R variant showed an enhanced proton inhibition, which presented as less current remaining at pH 6.8 vs pH 7.6 compared to WT NMDARs (Table 3); a similar result was found with GluN2B-p.W607C and GluN2B-p.V618G. By contrast, NMDARs that contained GluN2A-p.L611Q, GluN2A-p.N614S, or GluN2B-p.V620M showed reduced proton sensitivity compared to the corresponding WT receptors (Table 3; one way ANOVA).
3.3. M2 pore loop variants strongly reduce magnesium sensitivity
One of the most important properties of NMDARs function is negative regulation by endogenous extracellular magnesium ions (Paoletti et al., 2013; S. F. Traynelis et al., 2010). It has been shown that the residues within the M2 reentrant loop control voltage-dependent magnesium ion binding (Kuner & Schoepfer, 1996; Kupper, Ascher, & Neyton, 1996; Williams et al., 1998; Wollmuth et al., 1998). Two experiments were conducted to assess the effects of the M2 variants on magnesium inhibition. At a holding potential of −60 mV, the concentration-response curves (Table 4; Figure. 3A,C) showed a reduced potency for magnesium inhibition for the M2 pore loop variants with IC50 values ranging from 154 ¼M to over 1,000 ¼M compared to the 23-24 ¼M of WT GluN1/GluN2A and GluN1/GluN2B receptors (Table 4; Figure. 3A,C). Co-expression of GluN1 with GluN2B-p.W607C, -p.N615I, -p.N615K, -p.N616K, -p.V618G showed virtually no detectable inhibition at 1 mM extracellular magnesium; a similar result was found for GluN1-p.G620R/GluN2B receptors. Indeed, these variants exhibited an increase in current at high magnesium concentrations (0.3-1 mM), consistent with unopposed positive allosteric modulation by Mg2+ in the absence of channel block described by Paoletti et al. (Paoletti, Neyton, & Ascher, 1995) (Table 4; Figure. 3C). The current-voltage (I-V) curves generated by applying voltage steps (−90 mV to +30 mV) in Xenopus oocytes expressing WT or mutant receptors also revealed significantly decreased magnesium block of NMDARs containing M2 pore loop variants (Figure. 3B,D; Table 4). The block by 1 mM magnesium was strongly reduced in the mutant receptors, reflected by 2.5- to 21-fold more current (relative response at VHOLDING −60 mV normalized to the current at +30 mV) in the GluN2A variants or GluN1 variants co-expressed with GluN2A compared to the WT receptors. Similarly, there was 2.5- to 21-fold more current at −60 mV in the GluN2B variants or GluN1 variants co-expressed with GluN2B (Table 4). The current-voltage curves generated in the presence of 1 mM magnesium by the Woodhull equation (see Methods) revealed a change in affinity for Mg2+ in the absence of an electric field, KD,0 mV, from 1.5 mM in WT GluN1/GluN2A to 12 mM in GluN2A-p.L611Q and from 1.9 mM in WT GluN1/GluN2B to up to 34 mM in the variant GluN2B receptors (Table 4). The electric field felt by blocking ion can be assessed by the product zδ, which was 1.93 for GluN1/GluN2A and 1.95 for GluN1/GluN2B. Multiple M2 variants reduced this value to 0 - 1.73 (Table 4), indicating the variants can alter both the affinity and the apparent voltage dependence of magnesium binding. Taken together, these data suggested that the loss of voltage-dependent magnesium inhibition was caused by disruption of binding sites of these M2 pore loop variants, in addition to likely changes in intra-pore electrostatics.
Table 4.
Summary of magnesium data
| IC50, μM (n)a | % at 1 mMb | % at −60mV (n)c | KD,0 mV, mM | zδ | ||
|---|---|---|---|---|---|---|
| di-heteromeric receptors | WT GluN1/WT 2A | 23 ± 1.3 (34) | 2.2 ± 0.3 (34) | 0.08 ± 0.01 (25) | 1.52 | 1.93 |
| 1-G618R/2A | N.E. (13) | 87 ± 6.2 (13)* | 0.91 ± 0.15 (9)* | n.d. | n.d. | |
| 1-G620R/2A | > 1,000 (14)* | 50 ± 7.6 (14)* | 0.52 ± 0.05 (10)* | 1.59 | 0.64 | |
| 2A-L611Q | > 1,000 (10)* | 50 ± 2.0 (10)* | 0.78 ± 0.06 (13)* | 12.0 | 1.26 | |
| 2A-N614S | 180 ± 39 (16)* | 17 ± 2.4 (16)* | 0.18 ± 0.03 (10) | 0.80 | 0.91 | |
| 2A-N615K | > 1,000 (21) | 79 ± 3.4 (21)* | 1.7 ± 0.12 (13)* | n.d. | n.d. | |
| WT GluN1/WT 2B | 24 ± 3.1 (41) | 2.5 ± 0.5 (41) | 0.04 ± 0.005 (18) | 1.83 | 1.95 | |
| 1-G618R/2B | 278 ± 65 (13)* | 41 ± 2.8 (11)* | n.a. | n.a. | n.a. | |
| 1-G620R/2B | N.E. (10) | 97 ± 4.0 (10)* | 0.53 ± 0.04 (14)* | 4.59 | 0.99 | |
| 2B-W607C | N.E. (11) | 104 ± 5.3 (11)* | 0.37 ± 0.05 (24)* | n.d. | n.d. | |
| 2B-G611V | N.E. (11) | 73 ± 6.3 (11)* | 0.38 ± 0.06 (13)* | 3.14 | 1.24 | |
| 2B-N615I | N.E. (13) | 108 ± 2.7 (12)* | 1.3 ± 0.1 (12)* | 34.3 | 0.61 | |
| 2B-N615K | N.E. (13) | 149 ± 6.1 (13)* | 1.4 ± 0.4 (10)* | n.d. | n.d. | |
| 2B-N616K | N.E. (11) | 158 ± 8.9 (11)* | 1.5 ± 0.1 (10)* | n.d. | n.d. | |
| 2B-V618G | N.E. (11) | 139 ± 11 (11)* | 0.96 ± 0.05 (12)* | n.d. | n.d. | |
| 2B-V620M | 154 ± 7.7 (16)* | 16 ± 2.3 (15) | 0.34 ± 0.05 (12)* | 3.14 | 1.73 | |
| tri-heteromric receptors | WT N1/N1/2A | 39 ± 8.6 (10) | 2.6 ± 0.8 (10) | 0.05 ± 0.01 (16) | 4.44 | 2.26 |
| 1-G620R/N1/2A | 257 ± 35 (14)* | 23 ± 2.1 (14)* | 0.10 ± 0.01 (15) | 2.17 | 1.60 | |
| 1-G620R/1-G620R/2A | > 800 (12) | 44 ± 5.3 (12)* | 0.32 ± 0.03 (11)* | 1.61 | 1.00 | |
| N1/2A/2A | 31 ± 3.6 (36) | 1.3 ± 0.3 (34) | 0.01 ± 0.01 (30) | 1.58 | 1.96 | |
| N1-L611Q/2A | 414 ± 74 (23)* | 28 ± 2.6 (23)* | 0.34 ± 0.02 (17)* | 5.51 | 1.39 | |
| N1-L611Q/2A-L611Q | 747 ± 86 (10)* | 45 ± 2.1 (10)* | 0.61 ± 0.03 (11)* | 7.62 | 1.07 | |
| N1/2A-N614S/2A | 86 ± 9.5 (12)* | 4.9 ± 0.8 (11) | 0.07 ± 0.01 (10) | 2.50 | 1.86 | |
| N1/2A-N614S/2A-N614S | 152 ± 11 (11)* | 8.5 ± 0.8 (11) | 0.15 ± 0.01 (10) | 0.92 | 1.03 | |
| N1/2A-N615K/2A | >1,000 (12) | 54 ± 6.1 (12)* | 1.3 ± 0.11 (11)* | 33.1 | 1.50 | |
| N1/2A-N615K/2A-N615K | >1,000 (13) | 70 ± 1.9 (13)* | 1.6 ± 0.04 (8)* | n.d. | n.d. | |
| WT N1/2B/2B | 24 ± 3.4 (33) | 2.8 ± 0.5 (33) | 0.03 ± 0.005 (28) | 1.54 | 1.99 | |
| N1/2B-W607C/2B | 59 ± 10 (21)* | 8.6 ± 1.3 (21) | 0.04 ± 0.01 (23) | 2.40 | 2.04 | |
| N1/2B-W607C/2B-W607C | 105 ± 11 (19)* | 18 ± 1.5 (19)* | 0.13 ± 0.02 (25) | 7.62 | 1.07 | |
| N1/2B-G611V/2B | 307 ± 82 (15)* | 26 ± 4.1 (14)* | 0.21 ± 0.04 (25) | 5.36 | 1.68 | |
| N1/2B-G611V/2B-G611V | >1,000 (15) | 58 ± 4.0 (15)* | 0.42 ± 0.04 (16)* | 7.77 | 1.38 | |
| N1/2B-N615I/2B | 91 ± 30 (9)* | 10 ± 2.5 (9) | 0.21 ± 0.02 (20) | 7.54 | 1.83 | |
| N1/2B-N615I/2B-N615I | N.E. (12) | 91 ± 7.7 (12)* | 1.1 ± 0.11 (13)* | n.d. | n.d. | |
| N1/2B-N615K/2B | N.E. (10) | 121 ± 5.3 (10)* | 2.0 ± 0.26 (12)* | 170 | 0.79 | |
| N1/2B-N615K/2B-N615K | N.E. (7) | 149 ± 6.9 (7)* | 1.5 ± 0.31 (7)* | n.d. | n.d. | |
| N1/2B-N616K/2B | N.E. (12) | 117 ± 4.4 (12)* | 0.97 ± 0.11 (17)* | 26.3 | 1.10 | |
| N1/2B-N616K/2B-N616K | N.E. (8) | 154 ± 5.1 (8)* | 1.2 ± 0.08 (13)* | n.d. | n.d. | |
| N1/2B-V618G/2B | >800 (12) | 42 ± 2.2 (12)* | 0.33 ± 0.02 (21)* | 1.61 | 0.89 | |
| N1/2B-V618G/2B-V618G | N.E. (12) | 132 ± 3.5 (12)* | 0.82 ± 0.03 (16)* | n.d. | n.d. | |
| N1/2B-V620M/2B | 145 ± 49 (16)* | 15 ± 4.6 (17) | 0.10 ± 0.01 (15) | 3.00 | 1.81 | |
| N1/2B-V620M/2B-V620M | 216 ± 41 (14)* | 20 ± 2.4 (14)* | 0.16 ± 0.02 (12) | 3.10 | 1.62 | |
Data are Mean ± SEM (n) determined from two electrode voltage clamp recordings from Xenopus oocytes.
−60 mV of holding potential,
percentage current remaining in the presence of 1 mM Mg2+ compared to the agonist-evoked current (100 μM glutamate and glycine)
percentage current at −60 mV normalized to the current recorded at +30 mV.
KD,0 mV the affinity of Mg2+ in the absence of an electric field; zδ quantifies the voltage dependence of block as the product of charge times fraction of electric field.
n.a. not analyzed due to low current amplitude.
n.d. not determined due to lack of sufficient Mg2+ block.
N.E. no effect, <30% inhibition by 1 mM Mg2+.
p < 0.05 one way ANOVA, with Dunnett’s multiple comparisons test, controlled FWER (family wise error rate) by using the Holm-Bonferroni correction. Statistical analysis is given in Supp. Table S3.
Figure. 3. M2 pore loop variants influence magnesium sensitivity.
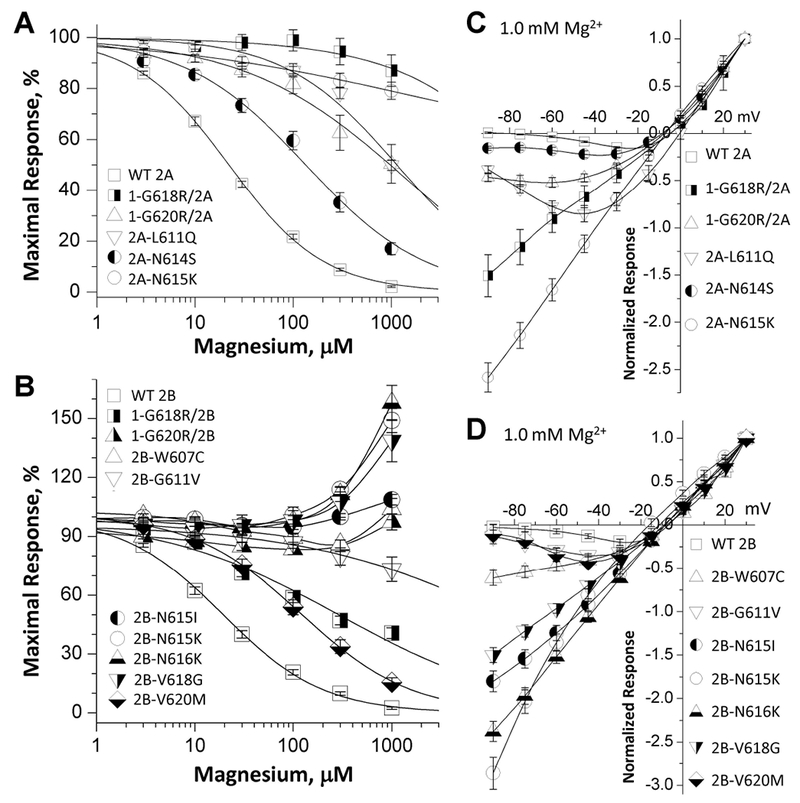
(A,B) Composite concentration-response curves for Mg2+ inhibition of di-heteromeric receptors in the presence of maximally effective concentrations of agonists (100 μM glutamate and 100 μM glycine) show decreased inhibition in all M2 variants-containing receptors. The data were generated by TEVC recordings on Xenopus oocytes at a holding potential of −60 mV. (C,D) Mg2+ current-voltage (I-V) curves for di-heteromeric receptors indicate a decreased Mg2+ inhibition of all M2 variant-containing receptors. For the I-V curve, all responses were normalized to those recorded at +30 mV. Fitted and statistical parameters are given in Table 4 and Supp. Table S3.
3.4. A single copy of M2 pore loop variant subunit reduces magnesium sensitivity
Since all patients harboring M2 pore loop variants are heterozygous and the functional NMDA receptor possesses two copies of GluN1 and two copies of GluN2 subunits, many NMDARs in these patients will have a single copy of the M2 pore loop variant. Therefore, we adapted a strategy to control receptor subunit composition on the cell surface. Using a method of engineering a pair of modified GluN subunits that contain complementary sets of coiled-coil domains C1 and C2 followed by a retention signal in endoplasmic reticulum (Hansen et al., 2014; Swanger et al., 2016; Yi et al., 2018; Yuan et al., 2014), we can control surface expression of receptors that contain 0, 1 or 2 copies of the M2 pore loop variants. For example, we are able to express GluN1/GluN2AC1/GluN2AC2 (hereafter 2A/2A), GluN1/GluN2AC1-p.L611Q/GluN2AC2 (hereafter L611Q/2A), and GluN1/GluN2AC1-p.L611Q /GluN2AC2-p.L611Q (thereafter L611Q/ L611Q) on the cell surface. Control experiments to evaluate the leak current which reflects escape of non-triheteromeric receptors from the endoplasmic reticulum retention (Hansen et al., 2014; Yi et al., 2018) suggest that less than 8% of the recorded currents arises from diheteromeric receptors that escape the engineered ER retention signal (Supp. Figure S1). We re-evaluated the concentration-response curves for Mg2+ to evaluate the effects of a single copy of each M2 pore loop variant on magnesium inhibition. The data indicated that a single copy of GluN2A-p.N615K, GluN2B-p.N615K, and GluN2B-p.N616K produced a dominant reduction in magnesium block similar to that seen with two copies of the variant subunit (Figure. 4C; Table 4). A single copy of remaining eight M2 pore loop variants produced an intermediate, but significant, reduction in magnesium block (increased IC50 values) (Figure. 4A,C; Table 4). In addition, analysis of I-V (current-voltage) curves in the presence of 1 mM magnesium revealed an intermediate or dominant reduction in magnesium block by a single copy of M2 pore loop variants (Figure. 4B,D,F; Table 4). Fitting the I-V curves with the Woodhull equation yielded an intermediate or dominant reduction in the affinity of magnesium, as well as changes in the product zδ (Table 4), indicating alteration of the apparent voltage-dependence of magnesium binding by a single copy of the M2 pore loop variants. These results suggest that a single copy of M2 pore loop variants can significantly alter receptor’s magnesium sensitivity in patients.
Figure. 4. One copy of M2 pore loop variants show either intermediate or dominant effects on Mg2+ sensitivity.
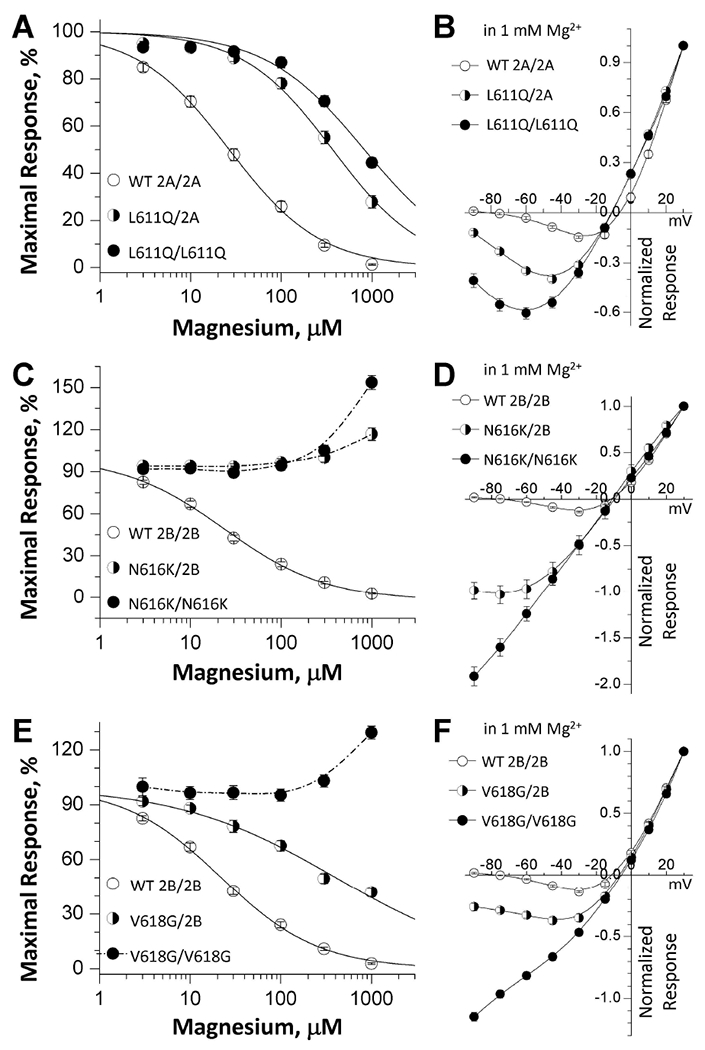
(A,C,E) Mg2+ concentration-response curves were determined by TEVC recordings from Xenopus oocytes expressing tri-heteromeric receptors with the copy number of GluN2A-p.L611Q, GluN2B-p.N616K, or GluN2B-p.V618G subunit in the tetrameric complex controlled: GluN1/GluN2A/GluN2A (2A/2A), GluN1/GluN2A-p.L611Q/GluN2A (L611Q/2A), and GluN1/GluN2A-p.L611Q /GluN2A-p.L611Q (L611Q/ L611Q) are shown in (A); GluN1/GluN2B/GluN2B (2B/2B), GluN1/GluN2B-p.N616K/GluN2B (N616K/2B), and GluN1/ GluN2B-p.N616K/GluN2B-p.N616K (N616K/N616K) are shown in (C); GluN1/GluN2B/GluN2B (2B/2B), GluN1/GluN2B-p.V618G/GluN2B (V618G/2B), and GluN1/ GluN2B-p.V618G/GluN2B-p.V618G (V618G/V618G) are shown in (E) (see Methods). (B,D,F) Mg2+ current-voltage (I-V) curves for tri-heteromeric receptors revealed an intermediate or dominant decrease in Mg2+-inhibition in the receptors containing a single copy of GluN2A-p.L611Q (B), GluN2B-p.N616K (D), and GluN2B-p.V618G (F) variants. Fitted and statistical parameters are given in Table 4 and Supp. Table S3.
3.5. M2 pore loop variants affect current amplitude and synaptic-like response time course
The deactivation response time rate can be obtained by rapid removal of agonist (e.g. glutamate) from NMDARs. It has been suggested to control the time course of the NMDAR slow component of the excitatory postsynaptic current (EPSC) at synapses (Lester, Clements, Westbrook, & Jahr, 1990). We therefore measured the current response from transiently transfected HEK 293 cells in whole cell voltage clamp current recordings following removing glutamate by a rapid solution exchange system. NMDARs containing GluN1-p.G618R, GluN1-p.G620R, GluN2A-p.N614S, GluN2B-p.W607C, GluN2B-p.G611V, GluN2B-p.N615I, GluN2B-p.N615K, and GluN2B-p.V618G all showed reduced current amplitude to prolonged (1.5 sec) application of 1000 μM glutamate and 100 μM glycine (p < 0.05, one way ANOVA) (Figure. 5A,D; Table 5). The glutamate deactivation response time course was fitted by two exponential components and compared to that for the corresponding WT receptor. The variants GluN2A-p.L611Q slowed the weighted deactivation time constant τw from 50 to 116 ms; similarly, the variant GluN2B-p.V620M slowed τw from 667 to 1065 ms (p < 0.05, one way ANOVA; Figure. 5B,C,E; Table 5). The deactivation time course for GluN2A-p.N614S and GluN1-p.G618R, when co-expressed with GluN2A or GluN2B, could not be determined due to small current. To mimic events in synaptic transmission, we assessed the effects of the M2 variants on the current responses by briefly exposing the cell to glutamate for ~5 milliseconds. M2 pore loop variants activated by brief exposure to glutamate also showed a change in current amplitudes, deactivation time course, and charge transfer similar to that observed for the response to prolonged glutamate application (Supp. Table S5). These data suggest NMDARs that contained M2 pore loop variants influence amplitude of current response and deactivation response time rate, and thus the time course of the NMDAR-mediated slow component of the EPSC.
Figure. 5. M2 pore loop variants influence current amplitude and response time course.
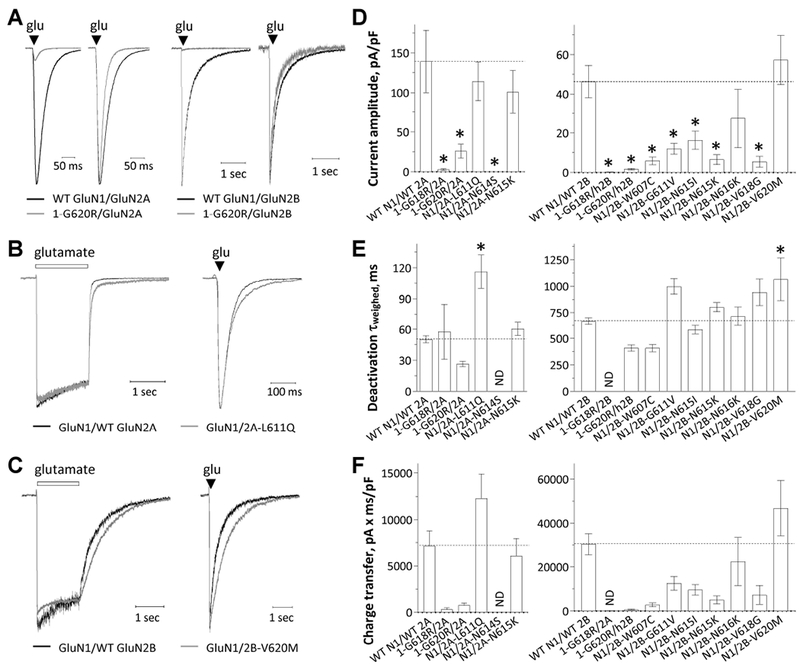
(A) Representative current response time courses are shown from a whole cell voltage clamp recording of HEK293 cells (VHOLD −60 mV) expressing GluN1-p.G620R/GluN2A (right: normalized response) or GluN1-p.G620R/GluN2B (right: normalized response) receptors in response to rapid application of brief (5 ms) duration of 1 mM glutamate (50 μM glycine was in all solutions).
(B,C) The representative current response time course from a whole cell voltage clamp recording of GluN1 /GluN2A-p.L611Q (B) and GluN1/GluN2B-p.V620M (C) receptors to rapid application of 1 mM glutamate for prolonged (left, 1.5 sec) and brief duration (right, 5 ms). Responses are shown normalized to the WT response at the moment glutamate was removed. Saturating glycine (50 ¼M) was present in all of solutions.
(D,E,F) Summary of peak amplitudes, weighted deactivation time constant, and charge transfer. *p < 0.05, one-way ANOVA, with Dunnett multiple comparisons. Fitted and statistical parameters are given in Table 5 and Supp. Tables S4, S5, S6.
Table 5.
Summary of biophysical properties and receptor surface expression
| amplitude, pA/pF | deactivation τw, ms | charge transfer, pA·ms/pF | POPEN, MTSEA | Mean open time, ms | main conductance, pS | Pca/PNa | surface/total ratio (β-lac) | |
|---|---|---|---|---|---|---|---|---|
| WT GluN1/WT 2A | 139 ± 39 (18) | 50 ± 3.4 (18) | 7,117 | 0.21 ± 0.01 (21) | 1.7 ± 0.14 (3) | 75 ± 3.6 (3) | 3.7 ± 0.43 (11) | 1.0 ± 0.11 (10) |
| 1-G618R/2A | 2.9 ± 1.0 (6)* | n.d. | n.d. | 0.10 ± 0.01 (19)* | n.a | n.a. | n.a. | 0.42 ± 0.06 (4)* |
| 1-G620R/2A | 26 ± 8.6 (12)* | 26 ± 2.5 (12) | 776 | 0.07 ± 0.01 (35)* | n.a | n.a. | −0.02 ± 0.22 (4)* | 0.13 ± 0.09 (4)* |
| 2A-L611Q | 114 ± 24 (16) | 116 ± 16 (16)* | 12,266 | 0.26 ± 0.01 (25)* | 2.8 ± 0.54 (3) | 48 ± 1.3 (3)* | 0.9 ± 0.19 (6)* | 0.84 ± 0.12 (10) |
| 2A-N614S | 0.12 ± 0.03 (8)* | n.d. | n.d. | 0.36 ± 0.02 (16)* | n.a | n.a. | n.a. | 0.19 ± 0.07 (4)* |
| 2A-N615K | 101 ± 27 (11) | 60 ± 6.4 (11) | 6,058 | 0.14 ± 0.01 (12)* | 1.1 ± 0.20 (3) | 26 ± 1.0 (3)* | −0.1 ± 0.09 (5)* | 0.84 ± 0.13 (4) |
| WT GluN1/WT 2B | 46 ± 8.2 (23) | 667 ± 33 (23) | 30,358 | 0.037 ± 0.003 (28) | 2.2 ± 0.38 (4) | 62 ± 1.4 (4) | 3.6 ± 0.29 (6) | 1.0 ± 0.10 (10) |
| 1-G618R/2B | 0.03 ± 0.16 (5)* | n.d. | n.d. | n.d. | n.a | n.a. | n.a. | 0.70 ± 0.15 (4)* |
| 1-G620R/2B | 1.8 ± 0.44 (14)* | 407 ± 27 (14) | 668 | n.d. | n.a | n.a. | n.d. | 0.37 ± 0.04 (4)* |
| 2B-W607C | 5.9 ± 1.9 (12)* | 408 ± 34 (12) | 2,829 | 0.012 ± 0.001 (12)* | 0.82 ± 0.20 (3)* | 52 ± 6.3 (3) | n.a. | 0.84 ± 0.21 (4) |
| 2B-G611V | 12 ± 2.8 (11)* | 997 ± 74 (11) | 12,535 | 0.020 ± 0.002 (17)* | 0.81 ± 0.15 (4)* | 49 ± 6.6 (4) | n.a. | 1.2 ± 0.17 (3) |
| 2B-N615I | 16 ± 4.6 (17)* | 585 ± 41 (17) | 9,600 | 0.020 ± 0.001 (20)* | n.a | n.a. | 0.1 ± 0.23 (6)* | 0.79 ± 0.26 (7) |
| 2B-N615K | 6.5 ± 2.4 (7)* | 801 ± 44 (7) | 5,085 | 0.020 ± 0.001 (21)* | n.a | n.a. | n.a. | 0.53 ± 0.08 (4)* |
| 2B-N616K | 28 ± 15 (7) | 848 ± 82 (7) | 22,420 | 0.020 ± 0.001 (17)* | n.a | n.a. | n.a. | 1.1 ± 0.06 (3) |
| 2B-V618G | 5.5 ± 2.8 (14)* | 942 ± 126 (14) | 7,270 | n.d. | 0.51 ± 0.07 (3)* | 68 ± 2.9 (3) | 2.3 ± 0.16 (5)* | 0.72 ± 0.12 (5) |
| 2B-V620M | 57 ± 13 (18) | 1065 ± 201 (18)* | 46,804 | 0.056 ± 0.006 (13)* | 2.0 ± 0.52 (3) | 64 ± 4.8 (3) | 2.1 ± 0.26 (6)* | 0.58 ± 0.11 (4) |
Data were expressed as Mean ± SEM (n).
n.a. not analyzed.
n.d. not determined due to low current amplitude.
p < 0.05 one way ANOVA, with Dunnett’s multiple comparisons test, controlled FWER (family wise error rate) by using the Holm-Bonferroni correction. Statistical analysis is given in Supp. Table S4.
3.6. M2 pore loop variants alter single channel properties
To investigate the effects of these M2 pore loop variants on single channel properties, we performed single channel recordings in outside-out patches excised from HEK cells transiently expressing wild type GluN1/GluN2A, GluN1/GluN2B, or a subset of M2 pore loop variants (Figure. 6; Supp. Figure S2; Table 5). Analysis of the pooled data of the steady-state single-channel unitary currents for WT GluN1/GluN2A presented one predominant conductance level (chord conductance 76 pS, 94%; 47 pS, 6%; n = 3 patches), assuming a reversal potential of 0 mV (Figure. 6A,B; Table 5). NMDARs containing GluN2A-p.L611Q and GluN2A-p.N615K variants showed reduced conductance levels (GluN2A-p.L611Q: 48 pS, 86%; 35 pS, 14%, n = 3 patches; GluN2A-p.N615K: 26 pS, 95%; 18 pS, 5%, n = 3 patches; p = 0.0003, compared to the WT receptors, one-way ANOVA, Dunnett’s multiple comparison test) (Figure. 6A,B; Table 5), suggesting these variants may change the ion permeation properties. No significant change on the main conductance level was detected in four GluN2B M2 pore loop variants (GluN2B-p.W607C, -p.G611V, -p.V618G, and -p.V620M, p ≥ 0.234) compared to the WT receptors (one-way ANOVA, Dunnett’s multiple comparison test) (Supp. Figure. S2; Table 5). There was a 2.8-fold decrease in mean open time for three GluN2B M2 pore loop variants (0.8 ms for GluN2B-p.W607C, -p.G611V, -p.V618G vs. 2.2 ms for WT 2B; p < 0.036, one-way ANOVA Dunnett’s multiple comparison test) for these multi-channel patches, suggesting these variants change the stability of the open pore (Supp. Figure. S2; Table 5).
Figure. 6. M2 pore loop variants influence single channel properties.
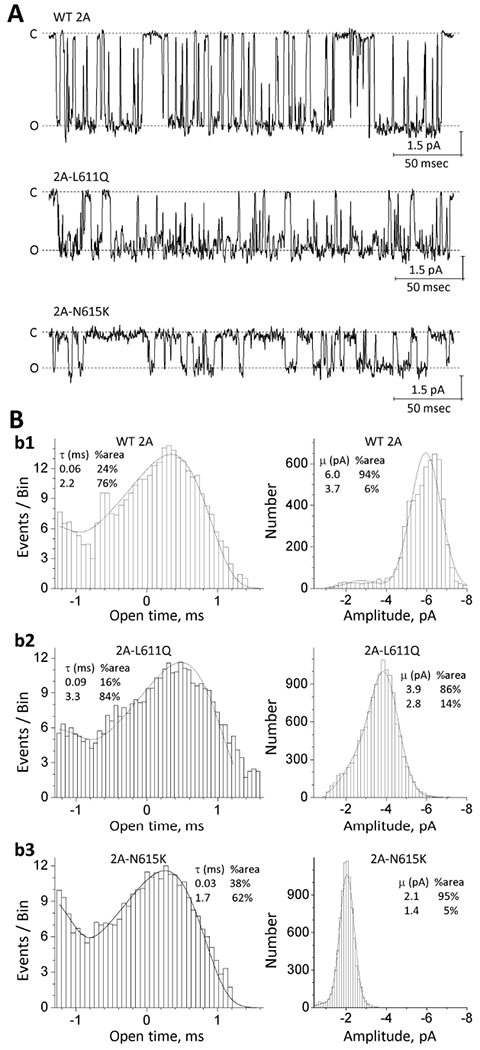
(A) Representative steady-state recordings from an outside-out patch containing GluN1/GluN2A (WT 2A), GluN1/GluN2A-p.L611Q, or GluN1/GluN2A-p.N615K NMDARs excised from transfected HEK cells. Unitary currents were activated in these excised outside-out patches by saturating concentrations of co-agonists (1 mM glutamate and 100 μM glycine) at a holding potential of −80 mV. “C” indicates the closed state and “O” indicates the open state. (B) The pooled open time duration histograms (left panels) and amplitude histograms (right panels) are shown for WT and the GluN2A M2 variants. Fitted and statistical parameters are given in Table 5 and Supp. Table S4.
To further assess the effects of these M2 pore loop variants on single channel open probability, we performed TEVC recordings on Xenopus oocytes to assess the degree of MTSEA (2-aminoethyl methanethiosulfonate hydrobromide) potentiation on the NMDARs with a mutation in the SYTANLAAF gating region in either GluN1(GluN1-A7C, referred as 1-A7C) or GluN2A (GluN2A-A7C, referred as 2A-A7C) subunit, which allows MTSEA to lock the channel open via covalent modification (W. Chen et al., 2017; Jones, VanDongen, & VanDongen, 2002; Yuan et al., 2005). Based on the degree of MTSEA potentiation of the NMDAR response to maximally effective concentration of agonist, we calculated the channel open probability. The MTSEA-induced increase in current amplitude is reciprocally related to the channel open probability before MTSEA application (W. Chen et al., 2017; Yuan et al., 2005) (see Methods). Evaluation of MTSEA potentiation of WT and variant NMDARs suggested a significant alteration of calculated receptor open probability by the M2 pore loop variants (Table 5).
3.7. M2 pore loop variants change calcium permeability
The reentrant pore loops also control Ca2+ permeability (Dingledine, Borges, Bowie, & Traynelis, 1999; Sakurada, Masu, & Nakanishi, 1993; Wollmuth & Sakmann, 1998), which is critical to their role in synaptic physiology. We therefore tested whether the M2 pore loop variants altered Ca2+ permeability using a reversal potential (Erev) approach to estimate the relative calcium permeability (Jatzke et al., 2002). We performed whole cell voltage clamp current recordings from transiently transfected HEK 293 cells, and measured reversal potentials in a control solution (0 mM Ca2+, 140 mM Na+) and a test solution (10 mM Ca2+, 140 mM Na+). The permeability ratio of Ca2+ to Na+ (PCa/PNa) was calculated from ΔErev and the Lewis equation (see Methods). Compared to the corresponding WT receptors, six M2 pore loop variants tested showed a detectable reduction of calcium permeability, ranging from a modest reduction (e.g. 35% of GluN2B-p.V618G and 41% for GluN2B-p.V620M) to strong reduction (e.g. GluN1-p.G620R/GluN2A, GluN2A-p.N615K, and GluN2B-p.N651I) (Figure. 7; Table 5). These data confirm that the residues residing in M2 pore loop control properties of the pore, and help determine subconductance states and relative Ca2+ permeability. These results suggest important consequences of M2 variants for the function of NMDA receptors in neuronal development, synaptic plasticity, and other important processes.
Figure. 7. M2 pore loop variants influence calcium permeability.
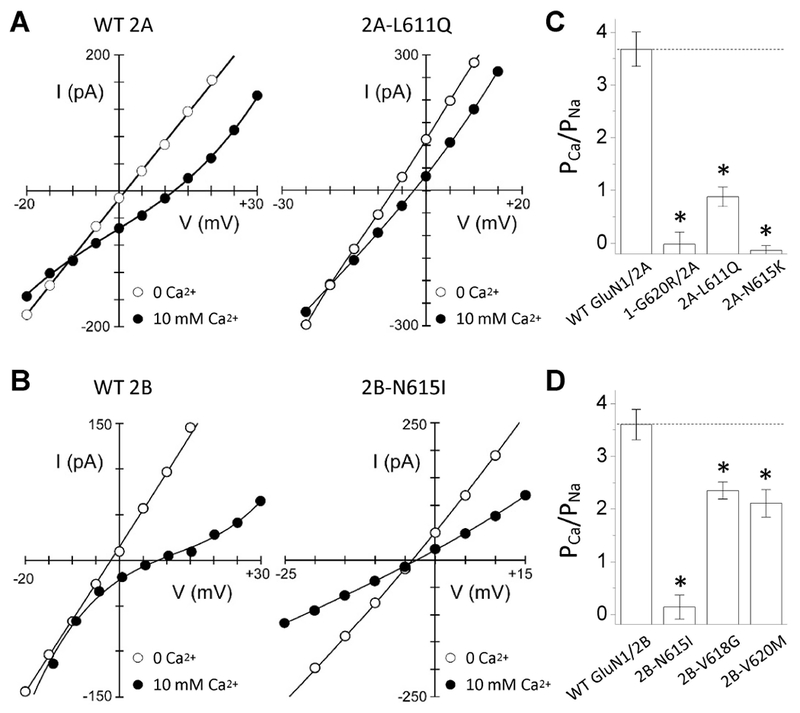
(A,B) Current-voltage (I-V) relationships in an external solution containing high Na+ (140 mM) plus 0 mM Ca2+ (open circles) or 10 mM Ca2+ (solid circles) for WT GluN1/GluN2A (A, left panel) or WT GluN1/GluN2B (B, left panel) or M2 loop variants in the same background. The Ca2+-free I-V is the average of that recorded before and after the 10 mM Ca2+ recording. We used the measured reversal potentials to calculate PCa/PNa (see Methods).
(C,D) Mean (± SEM) relative calcium permeability (PCa/PNa) was calculated from reversal potentials for (C) GluN1/GluN2A (n = 11), GluN1-p.G620R/GluN2A (n = 4), GluN1/GluN2A-p.L611Q (n = 6), and GluN1/GluN2A-p.N615K (n = 5) and (D) WT GluN1/GluN2B (n = 6), GluN1/GluN2B-p.N615I (n = 6), GluN1/GluN2B-p.V618G (n = 5), and GluN1/GluN2B-p.V620M (n = 6). M2 variant values are significantly different from their respective wild type (* p < 0.05, one way ANOVA, with Dunnett multiple comparisons). Fitted and statistical parameters are given in Table 5 and Supp. Table S4.
3.8. M2 pore loop variants influence receptor surface expression
To test if the M2 variants influence NMDAR surface expression, the cell surface and total protein levels were assessed by a reporter assay in which beta-lactamase was fused to the extracellular ATD of WT GluN1 (β-lac-GluN1), WT GluN2A (β-lac-GluN2A), WT GluN2B (β-lac-GluN2B), or the ATD of M2 pore loop variants. The fused β-lac-GluN1 protein was co-expressed with WT GluN2A or WT GluN2A, and β-lac-GluN2A or β-lac-GluN2B fusion protein was co-expressed with WT GluN1 in HEK293 cells. The surface receptor expression was determined by the beta-lactamase cleavage of the cell-impermeable chromogenic substrate nitrocefin in the extracellular solution (Lam et al., 2013; Swanger et al., 2016). NMDARs that contained each of the two GluN1 M2 pore loop variants when co-expressed with WT GluN2A or WT GluN2B indicated a significant decrease in the ratio of surface-to-total protein level, which reflects receptor trafficking efficiency, compared to WT GluN1 receptors (GluN1-p.G618R/2A: 42 ± 6% of WT, GluN1-p.G620R/2A: 13 ± 9% of WT, GluN1-p.G618R/2B: 70 ± 15% of WT, GluN1-p.G620R/2B: 37 ± 4% of WT; p < 0.001; Figure. 8A,B,E,F; Table 5). Among three GluN2A M2 pore loop variants evaluated, only 2A-N614S showed a significantly reduction of surface-to-total protein level (19 ± 7% of WT; p < 0.05; Figure. 8C,G; Table 5). However, almost all GluN2B M2 pore loop variants showed a comparable surface-to-total protein level and total protein level, except for GluN2B-p.N615K (surface-to-total: 53 ± 8% of WT; p < 0.05) and GluN2B-p.V620M (total: 57 ± 7.5% of WT; p < 0.05), which produced a significant reduction of both the surface-to-total and total protein levels, respectively (Figure. 8D,H; Table 5). These data suggest that M2 pore loop variants produce strong effects on surface expression.
Figure. 8. M2 pore loop variants influence receptor cell surface expression.
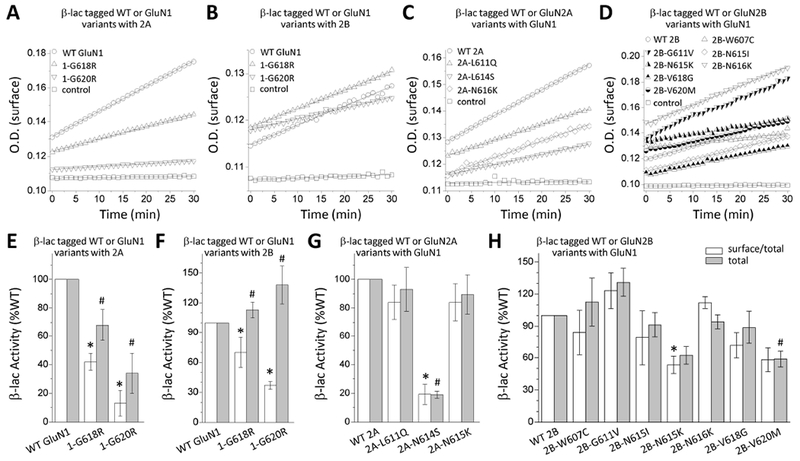
(A,B,C,D) Representative plots of nitrocefin absorbance (optical density, O.D.) as a function of time are shown for HEK293 cells expressing WT or M2 pore loop variants β-lac-GluN1 (A,B), β-lac-GluN2A (C), or β-lac-GluN2B (D). β-lac-tagged constructs were present in all conditions except control cells. (E,F,G,H) The slopes of O.D. versus time were averaged (n = 3-10 independent experiments) and graphed as percentages of WT for the ratio of surface/total. Data are expressed as mean ± SEM, and were analyzed by one-way ANOVA with Dunnett’s Multiple Comparison Test compared to WT (*p < 0.05, compared to WT surface/total ratio; #p < 0.05, compared to WT total level). Fitted and statistical parameters are given in Table 5 and Supp. Tables S4, S7, S8.
3.9. Evaluating the overall impact of M2 pore loop variants on NMDA receptor function
The functional changes caused by the rare variants identified in patients with neuropathological conditions are often conflicting, in the sense that some changes should increase current responses whereas others seem to decrease current responses. We have developed a strategy to estimate the net effect of rare variants and predict the variant’s effect on neuronal function by combining measured multiple functional parameters to evaluate alternation of synaptic and non-synaptic charge transfer mediated by NMDAR variants compared to WT receptors (Swanger et al., 2016). Our analyses indicated that most of the M2 pore loop variants showed an enhanced activity (Table 6), and revealed that functional enhancements produced by diminishing Mg2+ block more than compensate for mildly to moderately decreased surface expression, such as GluN2A-p.L611Q, GluN2B-p.N615I, -p.N615K, and -p.V620M, but not severe reductions, such as GluN1-p.G620R.
Table 6.
Predicted synaptic and non-synaptic changes of M2 variants relative to WT
| Synaptic charge transfer | Non-synaptic charge transfer | |
|---|---|---|
| WT GluN1/WT 2A | 1.0 | 1.0 |
| 1-G618R/2A | -- | -- |
| 1-G620R/2A | 0.2 | 0.1 |
| 2A-L611Q | 55 | 95 |
| 2A-N614S | -- | -- |
| 2A-N615K | 24 | 49 |
| WT GluN1/WT 2B | 1.0 | 1.0 |
| 2B-W607C | 7.0 | 3.6 |
| 2B-G611V | 28 | 18 |
| 2B-N615I | 17 | 24 |
| 2B-N615K | 21 | 32 |
| 2B-N616K | 50 | 47 |
| 2B-V618G | -- | -- |
| 2B-V620M | 8.6 | 16 |
The values were calculated by Equations 8–10 and indicate the fold difference in synaptic and non-synaptic function for the variants relative to the corresponding WT receptors (set as 1.0).
4. Discussion
The most important finding of this study is that all 12 patient-ascertained de novo variants in the M2 reentrant pore loop region appear to have strong effects on functional properties of the NMDAR. Furthermore, all 12 M2 pore loop variants appear to reduce the extent of Mg2+ block, a dominant feature controlled by residues in this part of the receptor. A previous in vivo study by Chen et al. (P. E. Chen et al., 2009) shows similar results in that a heterogeneous population of NMDAR with modifications to M2 in GluN1 showed a reduced macroscopic sensitivity to Mg2+. This homogeneous functional effect in which all M2 variants diminish Mg2+ block, coupled with the significant depletion of missense variation in the general population in this M2 pore loop, suggests future neurodevelopmental disease-ascertained de novo variants identified in the M2 region are likely to also have strong functional consequences. However, it is important to note that all variants observed here reside on the pore forming side of the reentrant loop (Figure. 1). While it is hard to predict the influences of variants on portions of M2 pore loop that do not face the pore, previous mutagenesis studies suggest these residues also control ion permeation and block (Béhé et al., 1995; Buck, Howitt, & Clements, 2000; Dingledine et al., 1999; Kuner & Schoepfer, 1996; Kupper et al., 1996; Premkumar & Auerbach, 1997; Sharma & Stevens, 1996). Furthermore, these effects suggest changes in NMDAR properties with M2 pore loop variants will alter microcircuit function, and thus likely contribute to patients’ clinical phenotype.
A second important conclusion from this study is that the M2 region can influence a host of receptor properties in addition to Mg2+ block and Ca2+ permeability, including agonist potency, proton sensitivity, open probability, and response time course, consistent with the recent studies (Fedele et al., 2018; K. Marwick, Skehel, Hardingham, & Wyllie, 2015; K. F. Marwick, Hansen, Skehel, Hardingham, & Wyllie, 2019; Mullier et al., 2017). For some variants, multiple functional effects have seemingly opposite actions when viewed simplistically as either enhancing or decreasing NMDAR function. We previously described an approach that combines relative changes in properties to predict whether synaptic and non-synaptic variant receptor will show increased or decreased function (Swanger et al., 2016). The result of the combined analysis of effects suggests that the majority of M2 pore loop variants showed an enhanced NMDAR function (Table 6), and therefore are gain-of-function variants. These data strengthen the rationale for a comprehensive evaluation of NMDA receptor activity to define the impact of genetic variation. In addition, the current data are using receptors expressed with GluN1-1a splice variant and that in different regions of the brain, other GluN1 splice variants predominate and Mg2+ potentiation and hence the magnitude of the gain-of-function effect of M2 mutations may vary between brain regions.
It is important to note that although it can be valuable to view variants as producing an overall gain- or loss-of-function; this binary characterization oversimplifies what could be a myriad of other actions that are important in the context of neurobiology. For example, changes in the NMDAR response time course that also change the synaptic current time course will alter spike timing-dependent plasticity. In addition, reduction in NMDAR Ca2+ permeability could have profound influence on synaptic plasticity (Paoletti et al., 2013), as well as neuronal development and perhaps synaptic pruning (Hickmott & Constantine-Paton, 1997; Zhang, Peterson, & Liu, 2013). Furthermore, a strong reduction in Mg2+ block could lead to selective cell loss through excitotoxic cell death (Choi, 1992), with consequent changes in circuit composition and function. Consistent with this idea, the smaller current responses amplitudes observed for some variants may reflect death of HEK cells that express higher receptor levels, and thus is not necessarily indicative of a change in cell surface expression. For these reasons, it is important to perform a comprehensive analysis of all effects of a putatively clinically relevant variant on NMDAR properties.
5. Conclusions
In summary, this work shows a fairly homogeneous response to genetic variation in an intolerant portion of the NMDA receptor that is known to control critical functions such as Mg2+ block and Ca2+ permeation. On the basis of this work, there is an excellent probability that future variants in this region of GRIN1 or any GRIN2 gene identified in patients will have similar effects, and thus we could potentially group this subset of variants into a likely functional phenotype. Such subdivision of intolerant regions based on functional analysis could stratify patients, and speed diagnosis and application of clinically-tested as well as empirically-derived treatment strategies.
Supplementary Material
Supp. Figure S1 Control recordings assessing the escape of non-tri-heteromeric receptors from ER retention
Supp. Figure S2 The pooled open time duration histograms and amplitude histograms are shown for WT and the GluN2B M2 variants
Supp. Table S2. Statistical analysis for data in Table 3
Supp. Table S3. Statistical analysis for data in Table 4
Supp. Table S4. Statistical analysis for data in Table 5
Supp. Table S5. Summary of patch clamp data with a brief application
Supp. Table S6. Statistical analysis for data in Supp. Table S5
Supp. Table S7. Summary of beta-lac total protein
Supp. Table S8. Statistical analysis for beta-lac total protein
Supp. Table S1. Patients’ clinical and genetic information
Acknowledgements
The authors thank Jing Zhang, Phoung Le, Gil Shaulsky, Kayrn Ding, and Sukhan Kim for technical support.
Funding Information
HY was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) of the National Institutes of Health (NIH) under award number R01HD08237. SFT was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the NIH under award numbers NIH-NINDS R01NS036654 and R24NS092989. LPW was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the NIH under award numbers R01 NS088479.
List of abbreviations
- 7-CKA
7-chlorokynuernic acid
- ABD
agonist binding domain
- ATD
amino-terminal domain
- CTD
carboxy-terminal domain
- EPSC
excitatory postsynaptic current
- HBSS
Hank’s Balanced Salt Solution
- I-V
current-voltage
- MTSEA
2-aminoethyl methanethiosulfonate hydrobromide
- NMDARs
N-methyl-D-aspartate receptors
- TMD
transmembrane domain
- WT
wild type
- β-lac
β-lactamase
Footnotes
Competing interests
SFT is a consultant for Janssen Pharmaceuticals Inc., is PI on a research grant from Janssen to Emory University School of Medicine, is PI on a research grant from Allergan to Emory University School of Medicine, is a member of the SAB for Sage Therapeutics, is co-founder of NeurOp Inc, and receives royalties for software. SFT is co-inventor on Emory-owned Intellectual Property that includes allosteric modulators of NMDA receptor function. HY is PI on a research grant from Sage Therapeutics to Emory University School of Medicine. SP is an employee of AstraZeneca.
References
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, & Mizuno N (1994). Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. Journal of Comparative Neurology, 347(1), 150–160. [DOI] [PubMed] [Google Scholar]
- Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, … Lynch SA (2016). Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia, 57(1), e12–e17. [DOI] [PubMed] [Google Scholar]
- Amengual-Gual M, Sanchez Fernandez I, & Wainwright MS (2018). Novel drugs and early polypharmacotherapy in status epilepticus. Seizure. doi: 10.1016/j.seizure.2018.08.004 [DOI] [PubMed] [Google Scholar]
- Béhé P, Stern P, Wyllie DJ, Nassar M, Schoepfer R, & Colquhoun D (1995). Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proceedings of the Royal Society of London. Series B: Biological Sciences, 262(1364), 205–213. [DOI] [PubMed] [Google Scholar]
- Beck C, Wollmuth LP, Seeburg PH, Sakmann B, & Kuner T (1999). NMDAR channel segments forming the extracellular vestibule inferred from the accessibility of substituted cysteines. Neuron, 22(3), 559–570. [DOI] [PubMed] [Google Scholar]
- Buck D, Howitt S, & Clements JD (2000). NMDA channel gating is influenced by a tryptophan residue in the M2 domain but calcium permeation is not altered. Biophysical Journal, 79(5), 2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, & Sakmann B (1992). Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron, 8(1), 189–198. [DOI] [PubMed] [Google Scholar]
- Burnashev N, & Szepetowski P (2015). NMDA receptor subunit mutations in neurodevelopmental disorders. Current opinion in pharmacology, 20, 73–82. [DOI] [PubMed] [Google Scholar]
- Chen C, & Okayama H (1987). High-efficiency transformation of mammalian cells by plasmid DNA. Molecular and cellular biology, 7(8), 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PE, Errington ML, Kneussel M, Chen G, Annala AJ, Rudhard YH, … Nassar MA (2009). Behavioral deficits and subregion-specific suppression of LTP in mice expressing a population of mutant NMDA receptors throughout the hippocampus. Learning & Memory, 16(10), 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Shieh C, Swanger SA, Tankovic A, Au M, McGuire M, … Traynelis SF (2017). GRIN1 mutation associated with intellectual disability alters NMDA receptor trafficking and function. Journal of human genetics, 62(6), 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1992). Excitotoxic cell death. Journal of neurobiology, 23(9), 1261–1276. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, & Hawkes A (1990). Stochastic properties of ion channel openings and bursts in a membrane patch that contains two channels: evidence concerning the number of channels present when a record containing only single openings is observed. Proc. R. Soc. Lond. B, 240(1299), 453–477. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, & Traynelis SF (1999). The glutamate receptor ion channels. Pharmacol Rev, 51(1), 7–62. [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, … Pientka FK (2010). Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nature genetics, 42(11), 1021. [DOI] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Davis BT, … Parra MC (2015). Enhanced utility of family-centered diagnostic exome sequencing with inheritance model–based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genetics in Medicine, 17(7), 578. [DOI] [PubMed] [Google Scholar]
- Fedele L, Newcombe J, Topf M, Gibb A, Harvey RJ, & Smart TG (2018). Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nature communications, 9(1), 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, & Traynelis SF (2014). Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron, 81(5), 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard M, Hansen KB, Andersen KT, Bräuner-Osborne H, & Traynelis SF (2012). Molecular pharmacology of human NMDA receptors. Neurochemistry international, 61(4), 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickmott PW, & Constantine-Paton M (1997). Experimental down-regulation of the NMDA channel associated with synapse pruning. Journal of neurophysiology, 78(2), 1096–1107. [DOI] [PubMed] [Google Scholar]
- Hu C, Chen W, Myers SJ, Yuan H, & Traynelis SF (2016). Human GRIN2B variants in neurodevelopmental disorders. Journal of pharmacological sciences, 132(2), 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Talos DM, Jackson MC, Park HK, Graham DA, Lechpammer M, … Jensen FE (2015). Developmental expression of N-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb Cortex, 25(2), 482–495. doi: 10.1093/cercor/bht246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jatzke C, Watanabe J, & Wollmuth LP (2002). Voltage and concentration dependence of Ca2+ permeability in recombinant glutamate receptor subtypes. The Journal of physiology, 538(1), 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KS, VanDongen HM, & VanDongen AM (2002). The NMDA receptor M3 segment is a conserved transduction element coupling ligand binding to channel opening. Journal of Neuroscience, 22(6), 2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, & Furukawa H (2014). Crystal structure of a heterotetrameric NMDA receptor ion channel. Science, 344(6187), 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi K, Pahk AJ, Masuko T, Igarashi K, & Williams K (1997). Block and Modulation ofN-Methyl-D-Aspartate Receptors by Polyamines and Protons: Role of Amino Acid Residues in the Transmembrane and Pore-Forming Regions of NR1 and NR2 Subunits. Molecular pharmacology, 52(4), 701–713. [DOI] [PubMed] [Google Scholar]
- Kuner T, & Schoepfer R (1996). Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. Journal of Neuroscience, 16(11), 3549–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner T, Seeburg PH, & Guy HR (2003). A common architecture for K+ channels and ionotropic glutamate receptors? Trends in neurosciences, 26(1), 27–32. [DOI] [PubMed] [Google Scholar]
- Kupper J, Ascher P, & Neyton J (1996). Probing the pore region of recombinant N-methyl-D-aspartate channels using external and internal magnesium block. Proceedings of the National Academy of Sciences, 93(16), 8648–8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam VM, Beerepoot P, Angers S, & Salahpour A (2013). A Novel Assay for Measurement of Membrane-Protein Surface Expression using a β-lactamase Reporter. Traffic, 14(7), 778–784. [DOI] [PubMed] [Google Scholar]
- Law AJ, Weickert CS, Webster MJ, Herman MM, Kleinman JE, & Harrison PJ (2003). Expression of NMDA receptor NR1, NR2A and NR2B subunit mRNAs during development of the human hippocampal formation. European Journal of Neuroscience, 18(5), 1197–1205. [DOI] [PubMed] [Google Scholar]
- Lee C-H, Lü W, Michel JC, Goehring A, Du J, Song X, & Gouaux E (2014). NMDA receptor structures reveal subunit arrangement and pore architecture. Nature, 511(7508), 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JR, Geider K, Helbig KL, Heyne HO, Schütz H, Hentschel J, … Heron D (2016). Delineating the GRIN1 phenotypic spectrum A distinct genetic NMDA receptor encephalopathy. Neurology, 86(23), 2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, … Hörtnagel K (2014). GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol, 75(1), 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, & Jahr CE (1990). Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature, 346(6284), 565. [DOI] [PubMed] [Google Scholar]
- Marwick K, Skehel P, Hardingham G, & Wyllie D (2015). Effect of a GRIN2A de novo mutation associated with epilepsy and intellectual disability on NMDA receptor currents and Mg2+ block in cultured primary cortical neurons. The Lancet, 385, S65. [DOI] [PubMed] [Google Scholar]
- Marwick KF, Hansen KB, Skehel PA, Hardingham GE, & Wyllie DJ (2019). Functional assessment of triheteromeric NMDA receptors containing a human variant associated with epilepsy. The Journal of physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, Howell KB, Cross JH, Kurian MA, & Scheffer IE (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. The Lancet Neurology, 15(3), 304–316. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, & Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron, 12(3), 529–540. [DOI] [PubMed] [Google Scholar]
- Mullier B, Wolff C, Sands ZA, Ghisdal P, Muglia P, Kaminski RM, & André VM (2017). GRIN2B gain of function mutations are sensitive to radiprodil, a negative allosteric modulator of GluN2B-containing NMDA receptors. Neuropharmacology, 123, 322–331. [DOI] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA, McDaniel MJ, Fan LZ, Hu C, … Schulien AJ (2017). Molecular mechanism of disease-associated mutations in the pre-M1 helix of NMDA receptors and potential rescue pharmacology. PLoS Genet, 13(1), e1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, & Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience, 14(6), 383. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J, & Ascher P (1995). Glycine-independent and subunit-specific potentiation of NMDA responses by extracellular Mg2+. Neuron, 15(5), 1109–1120. [DOI] [PubMed] [Google Scholar]
- Platzer K, Yuan H, Schütz H, Winschel A, Chen W, Hu C, … Tang S (2017). GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. Journal of medical genetics, jmedgenet-2016-104509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, & Auerbach A (1997). Stoichiometry of recombinant N-methyl-D-aspartate receptor channels inferred from single-channel current patterns. The Journal of general physiology, 110(5), 485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MC, Romero-Hernandez A, & Furukawa H (2015). A structural biology perspective on NMDA receptor pharmacology and function. Current opinion in structural biology, 33, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, … Monaghan KG (2016). Clinical application of whole-exome sequencing across clinical indications. Genetics in Medicine, 18(7), 696. [DOI] [PubMed] [Google Scholar]
- Sakurada K, Masu M, & Nakanishi S (1993). Alteration of Ca2+ permeability and sensitivity to Mg2+ and channel blockers by a single amino acid substitution in the N-methyl-D-aspartate receptor. Journal of Biological Chemistry, 268(1), 410–415. [PubMed] [Google Scholar]
- Sharma G, & Stevens CF (1996). A mutation that alters magnesium block of N-methyl-D-aspartate receptor channels. Proceedings of the National Academy of Sciences, 93(17), 9259–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, & Gouaux E (2009). X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature, 462(7274), 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto D, Altafaj X, Sindreu C, & Bayés À (2014). Glutamate receptor mutations in psychiatric and neurodevelopmental disorders. Communicative & integrative biology, 7(1), e27887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehlow V, Heyne HO, Vlaskamp DR, Marwick KF, Rudolf G, de Bellescize J, … Callenbach PM (2018). GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain, 142(1), 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, … Zhang J, (2016). Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. The American Journal of Human Genetics, 99(6), 1261–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis J, Silk M, Wang Q, Berkovic SF, Liu L, Ascher DB, … Petrovski S (2017). Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome research, 27(10), 1715–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, … Dingledine R (2010). Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol Rev, 62(3), 405–496. doi: 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Stülpnagel C, Ensslen M, Møller RS, Pal DK, Masnada S, Veggiotti P, … Herberhold T (2017). Epilepsy in patients with GRIN2A alterations: genetics, neurodevelopment, epileptic phenotype and response to anticonvulsive drugs. European Journal of Paediatric Neurology, 21(3), 530–541. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, & Mishina M (1993). Distinct distributions of five N-methyl-D-aspartate receptor channel subunit mRNAs in the forebrain. Journal of Comparative Neurology, 338(3), 377–390. [DOI] [PubMed] [Google Scholar]
- Williams K, Pahk AJ, Kashiwagi K, Masuko T, Nguyen ND, & Igarashi K (1998). The selectivity filter of theN-methyl-D-aspartate receptor: A tryptophan residue controls block and permeation of Mg2+. Molecular pharmacology, 53(5), 933–941. [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, & Sakmann B (1998). Adjacent asparagines in the NR2‐subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg2+. The Journal of physiology, 506(1), 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, & Sakmann B (1998). Different mechanisms of Ca2+ transport in NMDA and Ca2+-permeable AMPA glutamate receptor channels. The Journal of general physiology, 112(5), 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie D, Livesey M, & Hardingham G (2013). Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology, 74, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XiangWei W, Jiang Y, & Yuan H (2018). De novo mutations and rare variants occurring in NMDA receptors. Current opinion in physiology, 2, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XiangWei W, Kannan V, Xu Y, Kosobucki GJ, Schulien AJ, Kusumoto H,… Jiang Y (2019). Heterogeneous clinical and functional features of GRIN2D-related developmental and epileptic encephalopathy. Brain, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavarna T, Al-Dewik N, Al-Mureikhi M, Ali R, Al-Mesaifri F, Mahmoud L, … Nawaz Z (2015). High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Human genetics, 134(9), 967–980. [DOI] [PubMed] [Google Scholar]
- Yi F, Zachariassen LG, Dorsett KN, & Hansen KB (2018). Properties of Triheteromeric N-Methyl-d-Aspartate Receptors Containing Two Distinct GluN1 Isoforms. Molecular pharmacology, 93(5), 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Erreger K, Dravid SM, & Traynelis SF (2005). Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. Journal of Biological Chemistry, 280(33), 29708–29716. [DOI] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Zhang J, Pierson TM, Markello TC, Fajardo KVF, … Boerkoel CF (2014). Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nature communications, 5, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Low C-M, Moody OA, Jenkins A, & Traynelis SF (2015). Ionotropic GABA and Glutamate Receptor Mutations and Human Neurological Diseases. Molecular pharmacology, mol. 115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.-w., Peterson M, & Liu H (2013). Essential role of postsynaptic NMDA receptors in developmental refinement of excitatory synapses. Proceedings of the National Academy of Sciences, 110(3), 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Figure S1 Control recordings assessing the escape of non-tri-heteromeric receptors from ER retention
Supp. Figure S2 The pooled open time duration histograms and amplitude histograms are shown for WT and the GluN2B M2 variants
Supp. Table S2. Statistical analysis for data in Table 3
Supp. Table S3. Statistical analysis for data in Table 4
Supp. Table S4. Statistical analysis for data in Table 5
Supp. Table S5. Summary of patch clamp data with a brief application
Supp. Table S6. Statistical analysis for data in Supp. Table S5
Supp. Table S7. Summary of beta-lac total protein
Supp. Table S8. Statistical analysis for beta-lac total protein
Supp. Table S1. Patients’ clinical and genetic information