
Figure 1. Classification of structural systems biology studies into six use categories.
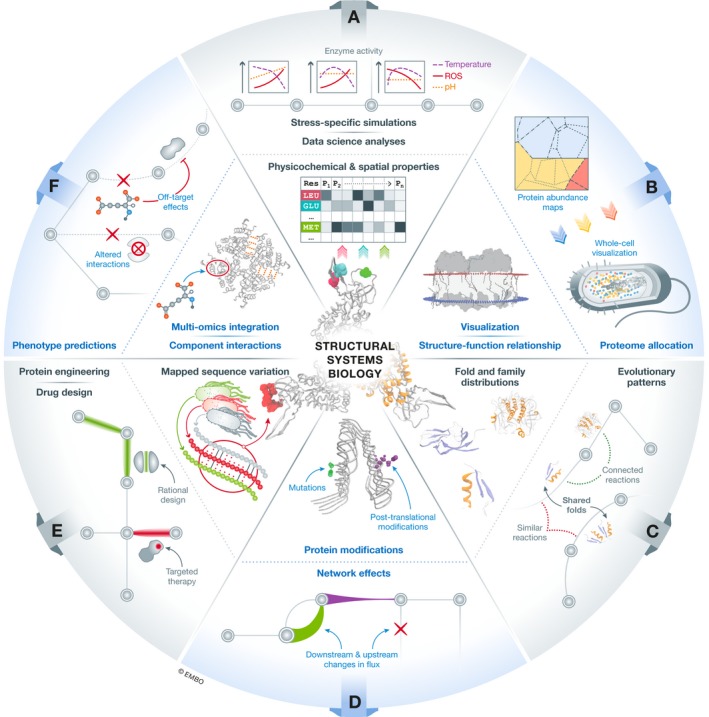
(A) Detailed physicochemical and spatial properties of the structural proteome enable the use of protein structures as an “omics” data source. This additional information has been utilized for downstream data science analyses and advanced metabolic modeling simulations incorporating residue‐level measurements for applications such as stress‐specific simulations. (B) A better understanding of functional assignments of proteins based on their structures leads to improved genome‐scale models, which better predict protein abundances in different conditions. Currently available structural determination techniques lead to visualizations of intricate protein complexes and their place within the cell, leading to potential realistic models of whole cells that reflect these environmental conditions. (C) Classical analyses of protein fold usage in the context of metabolic networks add an additional level of functional understanding to the network. The analysis of how folds are distributed within a network, and between strains or species, answers questions about the patterns we observe in metabolic pathway evolution. (D) In silico molecular modeling tools enable residue‐level predictions of mutations or post‐translational modifications that can then be used to modulate changes within a metabolic network, leading to an understanding of the global effect of small changes upon an entire cell. (E) Large‐scale analyses of sequence variation mapped to structure can uncover how small differences in protein structure potentially lead to metabolic changes within different strains of an organism. Furthermore, these small differences are crucial to finding regions in proteins that may cause undesirable pharmacogenomic interactions and can be crucial in the drug design process. As an extension of this, enzyme engineers can utilize this information to understand where highly variable regions of proteins are, to design more targeted libraries in the engineering cycle. (F) The interactions between proteins and the other components of the cell have only now been characterized at a large scale. Previously, interactions such as protein–ligand binding events have largely been limited to those directly involved in catalysis and studied by enzymatic assays. A large‐scale understanding of all small molecule interactions with proteins inside a cell adds yet another “interactome” to systems biology models, uncovering competitive and non‐competitive interactions that regulate processes alongside the transcriptome. Beyond these, protein–protein, protein–DNA, and gene–metabolome interaction data sets all contribute to our better understanding of the cell, but require a scaffold on which data need to be mapped.