

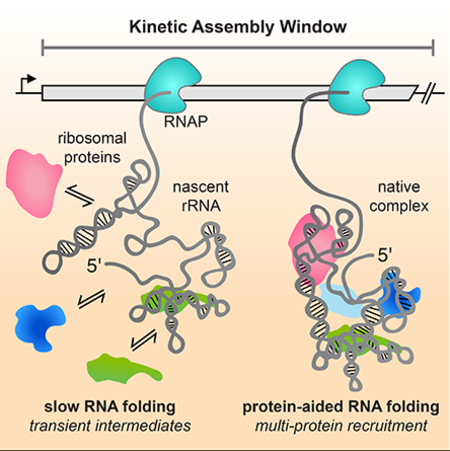
Summary
The synthesis of new ribosomes begins during transcription of the rRNA and is widely assumed to follow an orderly 5′ to 3′ gradient. To visualize co-transcriptional assembly of ribosomal protein-RNA complexes in real time, we developed a single-molecule platform that simultaneously monitors transcription and protein association with the elongating transcript. Unexpectedly, the early assembly protein uS4 binds newly made pre-16S rRNA only transiently, likely due to non-native folding of the rRNA during transcription. Stable uS4 binding became more probable only in the presence of additional ribosomal proteins that bind upstream and downstream of protein uS4 by allowing productive assembly intermediates to form earlier. We propose that dynamic sampling of elongating RNA by multiple proteins overcomes heterogeneous RNA folding, preventing assembly bottlenecks and initiating assembly within the transcription time window. This may be a common feature of transcription-coupled RNP assembly.
Keywords: Ribonucleoprotein Assembly, RNP, Co-transcriptional RNP assembly, Transcription, 30S ribosome, 16S RNA, Ribosome biogenesis, Single-molecule fluorescence, Single-molecule PIFE, Colocalization single-molecule spectroscopy
Graphical Abstract
INTRODUCTION
A hallmark of ribonucleoprotein (RNP) assembly is that it begins during synthesis of an RNA, when the RNA begins to fold as soon as it emerges from the transcription elongation complex (TEC). As a result, RNA binding proteins can begin to interact with local domains of RNA structure long before the entire RNA is transcribed. Properties of transcription, such as polymerase speed and pausing, have been shown to influence the functions of bacterial riboswitches and the assembly of ribosomes and splicing complexes (Aslanzadeh et al., 2018; Chauvier et al., 2016; Herzel et al., 2017; Lewicki et al., 1993; Perdrizet et al., 2012; Saldi et al., 2018). RNP assembly during transcription is often assumed to be simpler because the 5’ to 3’ direction favors a smaller number of assembly intermediates. Studies on riboswitches, however, showed that metastable RNA structures can exchange on the same time scale as transcription, creating a window for ligand binding and regulation (Steinert et al., 2017; Widom et al., 2018). Similarly, the mechanism of co-transcriptional RNP assembly may be more complex than previously thought.
An early example of transcription-coupled assembly is the biosynthesis of ribosomal subunits (Davis and Williamson, 2017). Electron micrographs of E. coli chromatin revealed that the elongating rRNA is compacted into nodular structures corresponding to RNA-protein complexes that form prior to rRNA processing (French and Miller, 1989). That assembly is coupled to transcription in E. coli is supported by the observations that faster transcription and mutations in nusA and nusB transcription elongation factors lead to defective subunit assembly in vivo (Bubunenko et al., 2013; Lewicki et al., 1993).
Pioneering work by the Nomura lab established a hierarchical order of ribosomal protein binding to the mature 16S rRNA in which subsets of proteins must bind first before the next proteins can add to the complex (Held et al., 1973, 1974; Mizushima and Nomura, 1970; Traub and Nomura, 1968, 1969). Although the assembly hierarchy implies a preferred order of protein addition, kinetics studies demonstrated that assembly can begin at multiple places along the RNA and follow heterogeneous pathways to reach the native subunit (Adilakshmi et al., 2008; Bunner et al., 2010; Talkington et al., 2005). In the cell, assembly is expected to follow the 5′ to 3′ direction of rRNA synthesis. This directionality may be encoded in the rRNA, since the 5’ domain is rich in RNA tertiary interactions and folds more rapidly than the central and 3’ domains (Adilakshmi et al., 2008; Powers et al., 1993).
We used the primary assembly protein uS4 (hereafter referred to as S4) to study the early steps of 30S assembly during transcription. S4 nucleates 30S assembly, and tightly binds to a 5-way helix junction (5WJ) formed by a long-range interaction between the beginning and end of the 16S 5’ domain (Gerstner et al., 2001; Nowotny and Nierhaus, 2002; Sapag et al., 1990; Vartikar and Draper, 1989). S4 binding induces conformational changes within the 16S rRNA (Kim et al., 2014) that are crucial for stable incorporation of protein S16 and other 30S proteins (Abeysirigunawardena et al., 2017; Powers et al., 1993; Stern et al., 1986, 1989). Single molecule FRET experiments suggested a multi-step binding mechanism for S4, in which heterogeneous and unstable S4 encounter complexes (~0.1 s) convert into non-native intermediates and long-lived, native complexes (>1 min; Kim et al., 2014).
To determine whether transcription alters the path for protein S4 recruitment and the nucleation of 30S assembly, we developed a new single-molecule method for simultaneously measuring transcription and protein binding in real time, called single-molecule Co-localization Co-transcriptional Assembly (smCoCoA). Surprisingly, we find that S4 alone binds poorly during transcription of the pre-16S RNA and can only form long-lived native complexes when other ribosomal proteins (RPs) are present. Our results suggest that recruitment of multiple proteins in parallel are required to promote 30S assembly on biologically relevant timescales. We propose that heterogeneous RNA folding during transcription amplifies the need for RNA binding proteins to smooth the assembly landscape and ensure the timely establishment of the proper RNA-protein interactions.
RESULTS
Synchronized transcription of full-length single pre-rRNAs
To understand how ribosome assembly occurs during transcription of the pre-16S rRNA, we designed a single molecule platform to monitor transcription and protein association in real time. The experimental design required that we time two independent points of transcription to signify how much of the RNA was transcribed and to report on the rate of transcription. To mark the beginning of transcription, we synchronized transcription elongation by generating stalled transcription elongation complexes (TECs) that could be restarted simultaneously by the addition of NTPs to the slide chamber.
Stable TECs can be initiated on DNA templates containing sequences that lack one of the ribonucleotides (Landick et al., 1996). To this end, we designed a DNA template containing the entire sequence for the precursor 16S rRNA from the E. coli rrnB operon proceeded by a T7 promoter and a 43 nt initial transcribed sequence lacking cytidines to allow initial transcription in the absence of CTP (Figure 1). This construct was based on a plasmid used to express functional 16S RNA by T7 RNAP in E. coli cells (Lewicki et al., 1993). Stalled TECs assembled on our DNA template in the absence of CTP were stable, with homogenous pausing at the first guanosine in the DNA template and slow mis-incorporation after 2 minutes (Figure S1). These stalled TECs restarted RNA synthesis to generate full length RNA when all four NTPs are added to the reaction mixture (Figure 1).
Figure 1.

Pre-rRNA transcription from single immobilized TECs. A) DNA template contains a T7 promoter and C-less cassette before the P2 transcription start site of the rrnB operon. See also Table S1. SA5 sequence is complementary to fluorescently labeled oligomer (SA5_aadU3; see Key Resources Table) used for labeling the transcribed RNA. Cy3 is attached to the template strand just upstream of the terminator. B) Single-round transcription assay demonstrating that full length pre-16S RNA can be generated from stalled TECs stalled in the absence of CTP. 32P-labeled products were resolved by 6% PAGE; top and bottom regions containing major bands are shown. Lanes were cropped from the same gel and are shown at the same contrast. C) Quantitation of gel in B. D) Stalled TECs (–CTP) are immobilized on passivated microscope slides through a biotinylated oligomer complementary to the 5′ end of the transcript, and then restarted with injection of NTPs to the slide chamber. RNA is detected using a Cy5-oligomer complementary to 3’ end of the transcript (SA5_aadU3). E) Representative microscope images showing that localization of Cy5-oligomer depends on NTP addition. Stalled Cy3-TECs imaged prior to NTP addition (left); Cy5-oligomer was added to the slide chamber in the absence (middle) or presence of NTPs (right) before imaging. F) Overlay of a different region illustrating colocalization between immobilized Cy3-labeled TECs and full-length RNA after NTPs and a Cy5-labeled oligomer (SA5_aadU3) were added to the slide chamber. White arrows highlight several instances of co-localization indicating that transcription has occurred at these sites. G) Quantification of 10 fields of view; mean ± s.d.
For smCoCoA experiments, stalled TECs were immobilized on the slide surface by annealing a biotinylated DNA oligomer to the 5′ end of the transcript. A similar design was used to tether stalled TECs to surfaces or beads for single molecule RNA folding experiments (Duss et al., 2018; Frieda and Block, 2012). The stalled TECs were directly visualized using a Cy3 fluorophore attached to the DNA template immediately upstream of the transcription terminator (Figure S1). Cy3-labeled TECs only accumulated on the slide surface in the presence of the DNA tether, indicating that immobilization is specific (Figure S1). To test whether immobilized stalled TECs were active, NTPs and a Cy5-labeled DNA oligomer complementary to the 3’ end of the full-length 16S RNA were added to the slide chamber to restart transcription and fluorescently label the resultant transcripts. Spots of Cy5 intensity accumulated only in the presence of NTPs and could be mapped to the locations of stalled Cy3-labeled TECs at the start of the experiment (Figure 1).
Real-time fluorescent signature of RNA synthesis
Simultaneous injection of Cy5-labeled S4 marked the time of NTP addition owing to an increase in the Cy5 background (Figure S2; see Methods). Shortly after NTP addition, the Cy3 fluorescence of TECs began to slowly increase (Figure 2 and Figure S2). This increase in fluorescence intensity was shown to correspond to movement of the fluorophore-labeled template in the TIR illumination field during transcription in similar experiments on tethered TECs (Duss et al., 2018; Yin et al., 1994). Therefore, we attributed this slow increase in Cy3 fluorescence signal to active elongation.
Figure 2.

PIFE signature for transcription speed. A) Single molecule fluorescence signature reports the time of RNA synthesis by immobilized TECs. Transcription pulls the Cy3-labeled DNA template toward the slide during transcription, causing an increase in Cy3 fluorescence as it moves into the TIR field. When T7 RNAP reaches the Cy3 dye in the DNA template, PIFE causes a brief increase in Cy3 intensity followed by a loss of Cy3 signal indicating termination and release of the DNA template or photobleaching. B) Representative single-molecule trajectory showing the fluorescent transcription signature and measurement of transcription time. Transcription restart is marked by NTP injection (defined in Methods and Supplemental Figure S2A and S2B) and end of pre-16S RNA transcription is indicated by PIFE. C) Single PIFE peaks are fit to a Gaussian function to define the peak center which is taken to be the RNA transcription end point. D) Histograms of transcription times for DNA templates generating RNAs of different lengths as indicated. Number of molecules for each histogram: N(234 nts) = 82; N(576 nts) = 103; N(731 nts) = 91; N(1564 nts) = 110; N(1918 nts) = 119. E) Transcription time in different NTP concentrations for the 16S5WJ RNA (234 nts). Number of molecules in each histogram: N(500 μM) = 77; N(100 μM) = 51; N(50 μM) = 79; N(20 μM) = 121. F) Average transcription rate (± s.d.) from histograms in E.
The slow increase in fluorescence was almost always (>95% of active TECs) followed by a brief 3 to 4-fold spike in Cy3 intensity (Figure 2 and Figure S2). We attributed this strong increase in Cy3 intensity to protein induced fluorescence enhancement (PIFE) when T7 RNAP approaches the end of the DNA template. PIFE is a phenomenon which occurs over 1 nm distances and that has been observed for several helicases and DNA binding proteins (Hwang and Myong, 2014; Hwang et al., 2011). Because Cy3 was attached to a thymidine in the template strand immediately upstream of the terminator, the PIFE effect occurs when Cy3 traverses through the RNAP active site during transcription of the terminator hairpin. Therefore, the PIFE signal pinpoints the moment when the full RNA has been transcribed. Following the PIFE peak, a plateau in Cy3 intensity was often followed by the loss of Cy3 signal, corresponding to termination and dissociation of the Cy3-DNA template (See Methods; Figure 2).
Examination of single TECs exhibiting both a slow increase in fluorescence signal followed by a PIFE peak (henceforth referred to as a transcription signature) revealed that single PIFE peaks could be well fit to a single gaussian (Figure 2C) consistent with processive movement of RNAP along the DNA template. Importantly, we found that ~90% Cy3-labeled TECs exhibiting a transcription signature colocalized with Cy5-labeled oligomer after transcription. Additionally, the transcription time, which is defined as the interval between the restart of transcription (NTP injection) and full-length RNA synthesis (PIFE peak center), varied proportionally with DNA length and NTP concentration (Figure 2 and Figure S2). Importantly, under near physiological conditions, the T7 transcription rates measured in this assay (60 – 85 nt/s at 1 mM NTPs) corresponded well with the rates of 16S transcription by E. coli RNAP in E. coli (65 nts/s; (Dennis et al., 2009). Therefore, the transcription signature in our assay can be used to monitor transcription of full-length pre-16S RNA at rates comparable to those in the cell.
Protein S4 binds unstably to pre-16S RNAs during transcription
Protein S4 is required for the addition of many other RPs and is thought to be one of the first RPs to bind the 16S rRNA during transcription. To visualize when S4 is first able to recognize the pre-16S rRNA during transcription, Cy5-S4 was injected into the slide chamber together with NTPs to simultaneously monitor transcription and S4 binding (Figure 3A). Binding events were detected by co-localization of the Cy5 signal with active Cy3-labeled TECs. Although S4 forms very stable complexes in ensemble binding experiments, binding events were surprisingly transient and dynamic during transcription. Many complexes dissociated after ~0.5 s, with rare longer lived binding events (>1 s) occurring after transcription was completed (Figure 3B). S4 did not significantly interact with the stalled TECs in the absence of NTPs or with TECs that did not exhibit a transcription signature (Figure S3A and S3B), indicating that S4 binding depends on elongation of the rRNA.
Figure 3.

Transient binding of protein S4 to pre-16S rRNA during transcription. A) In co-transcriptional experiments, stalled TECs (left) are restarted in the presence of Cy5-labeled S4 and co-localization of Cy5-S4 with Cy3-TEC is monitored during (middle) and after (right) transcription. B) For post-transcriptional binding experiments, unlabeled stalled TECs were immobilized on the surface (left), and transcription was restarted by addition of NTPs for 2 min. Free NTPs were washed away and a Cy3-SA5_aadU3 oligomer complementary to the 3’ end of the pre-16S RNA was added (middle). RNAs were allowed to fold for 30 min before Cy5-S4 was added to the slide chamber during imaging (right). C) Rastergram of S4 binding (black bars) during and just after transcription; each horizontal line represents a single TEC (N = 54). The lengths of horizontal bars indicate the lifetimes of individual complexes. On the x-axis, t = 0 represents the time of transcription restart marked by the flow of Cy5-S4 and NTPs into the slide chamber. Green circles represent the center of the PIFE peak marking the end of RNA transcription. D) Rastergram of S4 binding (black bars) 30 min post-transcription (N = 54). On the x-axis, t = 0 represents the time when Cy5-S4 is added to the slide chamber. E) Probability density histogram of S4 binding lifetimes 30 min after transcription (post-txn; Nobs = 497 on 104 molecules) and during and shortly following transcription (co-txn; Nobs = 1879 on 103 molecules). Fits represent maximum likelihood fitting to a function with three exponential terms. Error bars represent the variance in a binomial distribution. F) S4 binding probabilities as a function of time after transcription restart (x = 0), as in C,D. Binding events were clustered according to the lifetime τ of the complex: Transient binding (left axis), τ < 1 s (black line and grey circles); productive binding (right axis), τ > 10 s (red line and squares) and τ > 50 s (red dotted line and diamonds). Linear and exponential fits are shown to visualize the trends and do not represent physical models of the data. See also Table S2 for fit parameters.
To examine if these short-lived interactions are common among RPs, we used smCoCoA to study co-transcriptional binding of another primary RP, uS7 (S7), which binds the 16S 3′ domain (Figure S4A and B). Protein S7 also bound the pre-16S RNA transiently during and following transcription, with multi-phasic kinetics similar to that of S4 (Figure S4C and D). These data and previous experiments on protein S15 (Duss et al., 2018) suggested that short-lived interactions may be a general feature of ribosome assembly in which primary ribosomal proteins dynamically sample the nascent pre-rRNA.
S4 binding becomes more stable with time after transcription
To determine whether the lack of stable S4 binding was due to poor folding of the RNA during transcription or to some other feature of the assay, we allowed the pre-16S rRNA to fold before adding protein S4. In this experiment, stalled TECs were restarted in the presence of a Cy3-oligomer complementary to the full-length rRNA (Figure 3C). After a 30 min folding delay, Cy5-S4 was added, and we observed S4 association with the Cy3-labeled pre-rRNA in real time as before. We observed many more long-lived binding events after transcription, showing that S4 is able to form specific complexes with the pre-16S rRNA if the RNA has an opportunity to refold (Figure 3D).
Maximum likelihood analysis of S4 binding events for both post-transcriptional and co-transcriptional experiments revealed three distinct lifetimes for interactions between S4 and pre-16S rRNA (Figure 3E and Table S2). The shortest events lasted only a few frames, which could correspond to either specific or nonspecific RNA binding activity of S4. Like many RNA binding proteins, S4 is known to interact nonspecifically with RNA in addition to forming long-lived complexes with its proper binding site (Bellur and Woodson, 2009; Sapag et al., 1990). To evaluate the likelihood of nonspecific binding, we performed smCoCoA experiments with a transcript containing only the first 100 nucleotides of the 16S rRNA, which lacks the binding site for S4. We observed only transient interactions between S4 and the 16S100bp RNA (Figure S3C), with a characteristic lifetime τnonspecific = 0.5 ± 0.01 s, similar to the shortest binding lifetime to the pre-16S rRNA (Table S2).
To further evaluate the probability of non-specific binding, we measured S4 binding post-transcription with a transcript containing only the 16S central and 3′ domains. S4 binding to the truncated 16S was also mainly transient, with only a few longer-lived complexes (Figure S3 and Table S2) similar to the distribution for the full-length RNA during transcription. We interpret this to mean that the 5WJ does not fold properly during transcription, causing S4 to bind transiently with many sites along the pre-rRNA.
Previous single-molecule FRET experiments established that S4 forms a stable native complex (t1/2 > 30 min) when its binding site is well folded, but forms less stable complexes (< 30 s) in lower [Mg2+] when the RNA is less folded (Kim et al., 2014). The intermediate (τ2) and long-lived binding events (τ3) were much more common on transcripts containing the 5WJ and after 30 min folding time (post-txn; Figure 3D) than during transcription (co-txn; Figure 3C). For example, the intermediate events were 10-fold less probable in co-transcriptional experiments relative to post-transcriptional experiments ( versus). When the post-transcription folding time was varied from 5 to 30 min, the number of transient S4 binding events progressively decreased while the numbers of intermediate and long-lived complexes increased (Figure 3F). The time-dependence of this conversion was remarkably similar to the folding time of the 16S 5′ domain at 25 °C (Adilakshmi et al., 2005). In total, these results suggested that the pre-16S rRNA tends not to initially form structures that are competent to stably recruit protein S4 during transcription.
Transcription speed does not significantly influence S4 binding dynamics
We next considered whether long pre-16S transcripts misfold because transcription is too fast. Because our assay monitors the start and end of transcription for each TEC, we can directly evaluate how S4 binding compares to transcription speed. By varying the concentration of NTPs, we obtained a large range of T7 transcription rates from 1 nt/s to 200 nt/s (Figure 2E and F). This allowed us to compare how transcription rate (independent of temperature and polymerase identity) influences rRNA folding and S4 binding activity. In the absence of other factors, there was no correlation between transcription rate and S4 binding lifetimes within this range (Figure S3G) suggesting that the rate of synthesis is not positively or negatively influencing the binding behavior of S4.
S4 binds stably to a minimal 5WJ RNA during transcription
To understand why S4 was unable to stably bind newly transcribed pre-16S rRNA, we examined the binding of S4 to shorter 16S transcripts. A minimal 5WJ RNA was previously shown to form a stable complex with S4 in the absence of other factors (Bellur and Woodson, 2009; Mayerle et al., 2011). SmCoCoA experiments on the minimal 5WJ RNA revealed three types of S4 complexes, as observed for the pre-16S RNA (Figure 4 and Figure S5). In general, all S4 complexes were more stable for the 5WJ RNA compared to the pre-16S RNA (Figure 4E and Table S2). The longest binding lifetime for the 5WJ RNA ( compared to) approached those observed previously on refolded 16S 5’ domain RNA (Kim et al., 2014). The specific complexes were also more common for the 5WJ than pre-16S RNA (~3-fold higher and) suggesting that S4 is capable of stably binding to short transcripts that are more likely to fold correctly.
Figure 4.

Stable S4 binding with a minimal 16S 5WJ RNA during transcription. A) – D) Single-molecule traces highlighting long-lived S4 binding events for each RNA shown at (left); only the Cy5 intensity is shown. Transcription restart (solid black line) and transcription end (dotted black line) were determined as in Figure 2. See Supplemental Figure S5 for additional traces and rastergram analysis for each experiment. E) Probability density histogram of S4 lifetimes on each transcript. Lines represent maximum likelihood fitting to a function with three exponential terms; values are given in Table S2. Error bars in the probability density histogram represent the variance in a binomial distribution. Numbers of observations and molecules: Nobs (16S5WJ) = 1593 on 142 molecules, Nobs (16S5WJ+5’L) = 1049 on 106 molecules, Nobs (16S5’domain) = 640 on 82 molecules.
To test this possibility, we began systematically building on the 5WJ RNA to deduce which parts of the RNA interfere with the formation of stable complexes. First, we extended the 5’ end of the 5WJ RNA to include the entire rrnB 5’ leader (Figure 4 and Figure S5). Surprisingly, the leader had no effect on S4 binding, suggesting that extension of the 5’ end of 16S RNA does not interfere with folding of the 5WJ and S4 recognition. We next examined S4 binding to an RNA containing only the 5’ domain of the 16S rRNA (16S5’domain), which also forms stable complexes with S4. We confirmed that RNAs containing the extensions for smCoCoA experiments fold natively and bind Cy5-S4 by EMSA (Figure S5G, H). Analysis of S4 binding lifetimes revealed that S4 binds similarly to the 5’ domain when compared with the 5WJ RNA suggesting that sequences between the 5′ and 3′ halves of the 5WJ do not significantly hamper 5WJ folding and S4 binding (Figure 4 and Figure S5). In combination with the observation that S4 can interact transiently with 16S sequences downstream of the 5WJ (Figure S3D – F), we concluded that regions downstream of the 5WJ may sequester or otherwise interfere with stable association of S4.
Ribosomal proteins enhance S4 binding to pre-16S RNAs
Ribosomal protein binding influences the folding and conformational dynamics of the 16S RNA, which in turn allows other RPs to add to the complex. Therefore, we wondered if additional proteins that bind to the 5’ domain would influence the binding of S4 on pre-16S RNA. To test this, we performed smCoCoA experiments in the presence of 20 nM S17, 20 nM S20, and 50 nM S16, which is sufficient to fully assemble the 16S 5′ domain (Ramaswamy and Woodson, 2009). Strikingly, the fraction of intermediate S4 complexes increased ( vs.), showing that S4 is more likely to interact productively with the pre-16S rRNA when S17, S20, and S16 are also present (Figure 5). However, the longest-lived events were still three times less frequent (a3) as compared to longest-lived events 30 min post-transcription (Table S2). Therefore, intermediate S4 were more likely to occur in the presence of S16, S17, and S20, but the most stable complexes were still rare.
Figure 5.

Ribosomal proteins (RPs) increase stable co-transcriptional S4 recruitment. A, B) E. coli 30S assembly map (A) and 3D structure (B) highlighting the RPs that assemble on the 5’ domain (Held et al., 1974). 30S ribbon, (Vila-Sanjurjo et al., 2003). C) Rastergram of S4 binding to pre-16S RNA in the presence of 20 μM S17, 20 μM S20, and 50 μM S16. PIFE center from the corresponding Cy3-DNA trace is indicated with green circles to signify the end of RNA transcription. Traces are synchronized such that t = 0 marks restart of transcription. D) Rastergram of S4 binding to pre-16S RNA in the presence of 20 μM S17, 20 μM S20, 20 μM S8, 50 μM S16, 100 μM S5, and 100 μM S12, as in C. E) Probability density histogram of S4 lifetimes in the presence of RPs, as in Figure 4. Number of observations and molecules: Nobs (S4 only) = 1789 on 103 molecules; Nobs (+S16, S17, S20) = 3401 on 176 molecules, Nobs (+S16, S17, S20, S8, S5, S12) = 1789 on 112 molecules.
Downstream ribosomal proteins help recruit S4 to pre-16S transcripts
Since the presence of S17, S20, and S16 did not completely restore stable incorporation of S4, we carried out experiments with all 7 proteins (S4, S5, S8, S12, S16, S17, S20) that contribute to assembly of the 16S 5’ domain (Figure 5A; Held et al., 1974). The additional proteins, S5, S8 and S12, bind near S4 in the mature 30S ribosome and have the potential to stabilize S4 complexes (Figure 5B; Calidas and Culver, 2011; Vila-Sanjurjo et al., 2003).
The presence of all 7 proteins restored stable S4 binding to the pre-rRNA during transcription; the longest lifetime and amplitude were equal to the values for S4 binding to the 5WJ RNA alone (Figure 5C, D). The fraction of longer-lived complexes also dramatically increased in the presence of all 5’ domain proteins (Figure S6). This result suggests that the full set of 5’ domain proteins are required for efficient and stable incorporation of S4 into newly made pre-30S complexes. Remarkably, this group includes proteins, such as S8, whose binding site lies downstream of the binding site for S4. This enhancement of S4 binding was specific for proteins that bind near S4, because neither protein S9 (50 nM), which interacts with the 16S 3′ domain, nor MS2-MBP (50 nM) altered the S4 binding kinetics (Figure S6).
We considered whether the other RPs simply exclude non-specific binding of S4 to other sites in the nascent 16S rRNA. Transient S4 binding events, however, are still very common in the presence of additional RPs (Figure 5). Furthermore, the probability of specific binding correlates with the combinations of proteins used rather than total protein concentration. Finally, transient binding events were relatively less frequent when the 16S was given more time to fold, suggesting that the complexes formed reflect the underlying structure of the rRNA and not merely its accessibility (Figure 3). In total, these results show that proteins which bind the same domain as S4 increase the probability of the longer-lived S4 binding and the stability (lifetimes) of the complexes.
5’ domain proteins promote earlier S4 association relative to transcription
A consequence of more frequent stable binding is that specific S4 recruitment should occur earlier during transcription of the pre-16S rRNA. To assess whether other 5′ domain proteins hasten the productive addition of S4, we examined the delay between the restart of transcription and the first short-lived (τ > 1 s) S4 binding event on each nascent transcript. In the absence of other proteins, these productive binding events were broadly distributed, occurring from ~10 s to 200 s after transcription restart (median = 95.8 s; Figure 6). This is consistent with the idea that heterogeneous co-transcriptional folding of the pre-16S delays the recruitment of S4, because productive S4 binding often occurred long after the 5WJ was synthesized. Furthermore, a significant fraction of molecules (26%) did not experience a productive binding event within the 5 min duration of the experiment.
Figure 6.

Ribosomal proteins increase the number of metastable S4 binding events during transcription. A) Boxplot of the delay from transcription restart until the first specific S4 binding event for each TEC (> 1 s); brown, S4 only (N = 44; median = 95.4 s); orange, S4 + S16, S17, S20 (N = 107; median = 32.6 s); red, S4 + S16, S17, S20, S8, S5, S12 (N = 149; median = 29.2 s). B) Cumulative probability plot of the specific binding delay as in A. The transcription coordinate (top) indicates the average transcription time ± s.d. between the three experiments (ttranscription = 24.5 ± 9.4 s; average = black vertical bar and s.d. = grey box).
Upon addition of the 5’ domain proteins, the delay until the first specific binding event decreased dramatically, with a narrower distribution of binding times (median16,17,20 = 34.6 and median16,17,20,5,8,12 = 29.2; Figure 6A). Nearly all of the initial specific S4 binding events occurred within the time frame for pre-16S transcription (Figure 6B). Importantly, proteins S17, S20 and S16 that bind upstream of S4 significantly enhanced the probability of S4 specific binding (τ1, τ2), whereas proteins S5, S12, and S8 further narrowed the delay before first specific binding event (Figure 6A and 6B). Thus, during transcription the timely recruitment of S4 depends on the presence of other 30S proteins. By contrast, S4 can bind independently of other proteins when the pre-rRNA is allowed to fold after transcription.
DISCUSSION
Dynamic sampling of rRNA by protein S4 during transcription
Real-time observation of protein binding during transcription using single-molecule Colocalization of Co-transcriptional Assembly (smCoCoA) provides a new view of how the assembly of non-coding RNPs is coupled to 5′ to 3′ folding of the nascent transcript. 5′ to 3′ folding of the RNA during transcription has the potential to simplify the assembly process by allowing proteins to capture and solidify RNA structural domains before 5′ residues have a chance to misfold. In reality, we observe a more complex behavior in which the initial RNA structures are not competent to stably recruit protein S4. We find that transient co-transcriptional RNA-protein interactions make the system more sensitive to other proteins that bind the same transcript, intensifying the cooperativity of RNP assembly.
An important observation is that during transcription, protein S4 binds dynamically with nascent pre-16S RNAs, producing many transient complexes (~0.5) and only a few short-lived (~ 5 s) and long-lived complexes (~ 20 – 90 s). This behavior cannot be due to inherently poor binding under our assay conditions, because S4 frequently forms stable complexes with short transcripts containing only the 16S 5WJ, or when the pre-16S is given time to fold before S4 is added. Instead, dynamic binding must arise from variable folding of the pre-16S rRNA during transcription.
Based on previous smFRET and ensemble experiments, we interpret the rare long-lived complexes to represent native interactions between S4 and the 5WJ, which require protein-induced changes in the conformational dynamics of the rRNA (Kim et al., 2014). The short-lived complexes likely represent unstable intermediates in which S4 recognizes the 5WJ, but does not convert to the native conformation required for long-lived binding. Similar short-lived binding was also observed in smFRET experiments in low [Mg2+], which hinders RNA folding, supporting the idea that the stability of the S4 complex depends on the structure of the RNA (Kim et al., 2014). The probability of stable S4 association is strongly influenced by the context of its 5WJ binding site: S4 can form long-lived complexes with the 16S 5′ domain transcripts, but not the full pre-16S which contains residues downstream of the 5WJ.
In the presence of other RPs, short-lived (~ 5 s) S4 complexes occur much earlier with respect to transcription, appearing soon after the S4 binding site is synthesized. This effect was initially surprising, because S4 binds the native 16S rRNA in the absence of other proteins during 30S reconstitution (Held et al., 1973; Mizushima and Nomura, 1970; Schaup et al., 1971). Footprinting and three-color smFRET experiments, however, showed that the 5′ domain proteins communicate with each other through a complex network of conformational switches in the rRNA (Abeysirigunawardena et al., 2017; Ramaswamy and Woodson, 2009). This explains how other 5’ domain proteins may influence S4 binding dynamics by stabilizing rRNA conformations that are productive for assembly. This cooperativity presumably requires the 5′ domain proteins to reside on the RNA simultaneously (Abeysirigunawardena et al., 2017), although we cannot exclude the possibility that the RNA retains some native structure after a protein dissociates. The effect of other proteins on S4 binding is specific because it depends on particular protein combinations (Figure 4). Finally, S5, S8 and S12 increase the numbers of the longest-lived complexes, suggesting that the more protein-RNA interactions that are formed at a time, the greater chance of committing a nascent transcript to stable assembly within a given time window.
Implications for 30S ribosome assembly during transcription
We propose a new model for ribosome assembly in which initial protein binding during transcription provides a series of “kinetic windows” for coupled RNA folding and protein association. In this model, primary assembly proteins dynamically sample the nascent RNA, forming initial complexes with the RNA as soon as their binding site is transcribed. These initial complexes can have different fates depending on the folding pathway of the RNA: they may either dissociate, or evolve into a more stable, native-like complex which may depend on interactions with more distant parts of the RNA chain. Other proteins may accelerate the evolution into a stable complex by switching the conformational states of other RNA domains, as suggested by three-color smFRET experiments (Abeysirigunawardena et al., 2017), or by pre-emptively altering the RNA folding pathway. What we observe is that multiple protein-RNA interactions are needed to achieve stable binding within 1–2 minutes. Because these protein-RNA interactions are coupled to elongation of the RNA, the process of transcription defines the kinetic window for the nucleation of pre-30S assembly.
An implication of this model is that assembly of large RNPs such as the ribosome can occur through short-lived RNA-protein complexes, which may not contain all of the molecular interactions present in the mature RNP. The lifetime of the short-lived S4 complexes (1 – 8 s) is likely relevant for assembly since transcription of the pre-16S RNA occurs in ~ 24 s in cells (Dennis et al., 2009) and the 5’ domain is transcribed in 7 – 9 s. Because S4 binds rapidly (kon ~ 108 M−1s−1; (this study and Adilakshmi et al., 2008), its average occupancy is similar during and after transcription (8% in 5 nM S4), although co-transcriptional binding is unstable. If the other proteins bind independently and similarly to S4, co-binding of the core 5′ domain would still take ~1 hr at these concentrations. However, cooperative binding by other proteins raises the S4 occupancy (14% in 5 nM S4), shortening the average assembly time to ~1 min, similar to the rate of assembly in E. coli. Thus, transient interactions can contribute to productive assembly of multi-protein complexes if protein association is rapid and there is a source of binding cooperativity.
A process based on transient intermediates may be advantageous because it prevents the accumulation of trapped S4-RNA complexes that are not competent for further assembly. In addition, random order binding opens up multiple paths for cooperative protein recruitment, whereas a deterministic binding order would be vulnerable to fluctuations in the pools of available ribosomal proteins. In this way, the native S4-RNA complex may not be required to nucleate assembly in the thermodynamic sense, but may instead act as a pawl to promote forward assembly and prevent disassembly once a quorum of native RNA-protein interactions has been reached. This kinetic control model for co-transcriptional protein association likely applies to other domains of the 30S ribosome, which can assemble independently with a subset of primary RPs. For instance, both primary proteins S7 and S15 also exhibit two phase binding behavior for binding to rRNAs (Figure S4; Batey and Williamson, 1996a, 1996b; Duss et al., 2018; Orr et al., 1998), suggesting that is a general phenomenon.
Linking assembly to transcription speed
For this work, we transcribed the pre-16S rRNA using T7 RNAP, which is faster and more processive than E. coli RNAP. This choice was fortuitous in that the rate of T7 transcription in our experiments at 25 °C (~ 80 nt/s) is very similar to the estimated rate of transcription on the 16S coding region in E. coli at 37 °C (~ 65 nt/s; (Dennis et al., 2009). Our kinetic assembly model may explain why T7 RNAP could be used to express functional ribosomal subunits in E. coli cells at 25 °C but not at 37°C (Lewicki et al., 1993). A transcription speed of 65 nt/s provides an average assembly window of ~ 24 s, whereas a rate of ~ 400 nts/s at 37 °C would shorten this window to ~ 4 – 5 seconds. This is on comparable to the short-lived S4 binding intermediates in our experiment, and perhaps insufficient for consolidating the 5′ domain prior to processing of the pre-rRNA by RNase III. Conversely, extending the transcription window by reducing NTP concentrations did not improve S4 binding kinetics in our experiments. Although T7 RNAP mimics the speed of RNA synthesis, T7 RNAP is less prone to pausing than E. coli RNAP (Zhang and Landick, 2016). However, it is unclear if pausing is important because the nus antitermination complex normally suppresses pausing on rrn genes in E. coli (Condon et al., 1995; Vogel and Jensen, 1995). Future experiments will be needed to determine whether variations in transcription speed or other features are important for ribosome assembly.
Application to studying other RNPs assembled during transcription
We anticipate that our smCoCoA platform will be useful for studying transcription-coupled assembly of other RNPs. One advantage of this approach is that the PIFE signal accurately reports the time when the polymerase reaches a defined point on the template, allowing the transcription time to be determined for individual TECs. Site-specific dye incorporation at other positions in the DNA template would allow transcription speeds to be correlated with protein binding in different regions of the template. Another advantage of this method is that it can be applied to any RNA, including large, highly structured RNAs such as pre-ribosomal RNA or long non-coding RNAs.
Dynamic sampling and synergistic recruitment of proteins to RNAs is likely a recurrent theme in biology. For example, assembly of a nuclear export complex for the HIV Rev-response element (RRE) involves dynamic binding of Rev proteins which induce a conformational change that accelerates recruitment of additional Rev proteins (Bai et al., 2014). Spliceosomes also assemble co-transcriptionally by dynamic and reversible association of the spliceosomal small nuclear RNPs on a pre-mRNA (Herzel et al., 2017; Hoskins et al., 2011, 2016; Larson and Hoskins, 2017; Shcherbakova et al., 2013). Finally, the assembly of many other RNPs, such as the initial stages of eukaryotic ribosome assembly, likely have an underlying kinetic contribution (Hunziker et al., 2019; Zhang et al., 2016).
It is often thought that RNP assembly proceeds via a stepwise assembly mechanism for protein association during RNA transcription, in that sequential protein binding occurs as soon as the binding sites are synthesized. However, our data demonstrate that initial binding of S4 to nascent RNA is unproductive, and that the likelihood of forming stable, native complexes depends on the context of the RNA binding site as well as the presence of other ribosomal proteins. This suggests that a sequential binding model is too simple for RNP assembly and rather an all-or-none kinetic model for global assembly may more accurately reflect the complexity of RNA/protein cooperativity that is central to RNP assembly.
STAR★ METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sarah A. Woodson (swoodson@jhu.edu). All unique and stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
All ribosomal proteins were purified from Escherichia coli BL21(DE3) cells as described below. DNA templates containing the precursor 16S ribosomal RNA sequence was derived from the rrnB operon from the Escherichia coli K-12 strain.
METHOD DETAILS
Protein purification and fluorescent labeling
Unlabeled ribosomal proteins S4(C32S,S189C), S7(S54C), S16, S17, S20, S8, S9, S5, and S12 were expressed and purified as previously described (Abeysirigunawardena and Woodson, 2015; Culver and Noller, 1999, 2000). T7 RNAP was recombinantly expressed in E. coli BL21(DE3) cells as previously described (Davanloo et al., 1984) and natively purified on a P11 phosphocellulose column followed by a Blue Dextran-Sepharose column using a protocol developed for SP6 polymerase (Butler and Chamberlin, 1982). His-tagged MS2-MBP was overexpressed in BL21(DE3) cells and purified on a HisTrap column (GE Healthcare) as described previously (Sharma et al. 2018). For fluorescent labeling of ribosomal proteins S4 and S7, S4:C32S,S189C and S7:S54C were labeled with maleimide-Cy5 dye (GE Healthcare) as in (Kim et al., 2014). Briefly, S4:C32S,S189C or S7:S54C were incubated with a six-fold excess of dye in 80 mM K-HEPES pH 7.6, 1 M KCl, 1 mM TCEP, 3 M urea at 20 °C and unreacted dye was removed by cation exchange followed by dialysis against 80 mM K-HEPEs pH 7.6, 1 M KCl, 6 mM 2-mercaptoethanol.
Single-round bulk transcription assays
Radiolabeled stalled TECs were assembled in 20 μL reactions: 50 nM DNA template, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 50 nM T7 enzyme, 200 uM GTP, 200 uM ATP, 50 uM UTP, 1U/μL RNasin Plus (Thermo), 20 μCi 32P-α-ATP. Stalling reactions were incubated at RT for 2 mins and then heparin was added to 1 mg/mL. Immediately following stalling, reactions were diluted to 30 μL with restart mixture: 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 2 mM NTPs (GTP, ATP, UTP, CTP), 2 U RNasin Plus. Aliquots (3 μL) were taken from the reaction mixture, quenched in stop buffer (95% formamide, 25 mM EDTA), and placed on ice. Samples were loaded onto a pre-run 6% denaturing sequencing gel and run at 55 W for 4 – 5 hours at RT. Sequencing gels were transferred to filter paper, dried, exposed to a phosphorimaging screen overnight, and imaged using a phosphorimager (GE Typhoon). Gels were quantified using FIJI (Schindelin et al., 2012).
DNA template construction and fluorescent labeling
Primers were purchased from Integrated DNA technologies, Inc. (IDT). A reverse primer (Terminator_REV_aadU46; see Key Resources Table) containing an internal amino-alkyl modified nucleotide (IDT; see Table S1) was fluorescently labeled with Cy3-NHS mono reactive dye (GE Healthcare) as follows: Cy3 dye was dissolved in 33 μL of DMSO and added to a reaction mixture containing 5 nmols of modified primer adjusted to 100 μL final volume with 100 mM sodium bicarbonate pH 8.5. Reactions were incubated overnight at room temperature and then free dye was eliminated using a Chroma TE-10 spin column (Takara Bio) followed by ethanol precipitation. Fluorescent DNA templates for transcription were generated from the pUC19-p17S plasmid by PCR using Q5 high fidelity polymerase (NEB). Following PCR, fluorescently labeled DNA templates were separated on 1% agarose, gel purified using a Wizard SV gel clean up system (Promega), and ethanol precipitated.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli DH5a | NEB | C2987I |
| Escherichia coli BL21(DE3) | NEB | C2527I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| T7 RNA polymerase | This study; Purified as in Davanloo et al., 1984 and Butler and Chamberlin, 1982. | N/A |
| S4 | This study | N/A |
| S5 | Purified in Woodson Lab by Arthur Korman as in Culver and Noller, 1999. | N/A |
| S7 | Purified in Woodson Lab by Hui-Ting Lee as in Culver and Noller, 1999. | N/A |
| S8 | Purified by in Woodson Lab by Megan Mayerle as in Culver and Noller, 1999. | N/A |
| S9 | Purified by in Woodson Lab by Megan Mayerle as in Culver and Noller, 1999. | N/A |
| S12 | Purified in Woodson Lab by Megan Mayerle as in Culver and Noller, 1999. | N/A |
| S16 | Purified in Woodson Lab by Sanjaya Abeysirigunawardena as in Abeysirigunawardena et al., 2017. | N/A |
| S17 | Purified in Woodson Lab by Sanjaya Abeysirigunawardena as in Abeysirigunawardena et al., 2017. | N/A |
| S20 | Purified in Woodson Lab by Sanjaya Abeysirigunawardena as in Abeysirigunawardena et al., 2017. | N/A |
| MS2 coat protein fused to MBP (MS2-MBP) | Purified in Woodson Lab by Indra Mani Sharma as in Sharma et al., 2018 | N/A |
| NTPs (ATP, GTP, CTP, UTP) | ThermoFisher Scientific | Cat#R0481 |
| α–32P ATP 3000 Ci/mmol | Perkin Elmer | BLU003H250UC |
| Cy5 maleimide mono reactive dye pack | GE Healthcare | Cat#PA25031 |
| Cy3 mono reactive NHS-ester dye pack | GE Healthcare | Cat#PA23001 |
| Cy5 mono reactive NHS-ester dye pack | GE Healthcare | Cat#PA25001 |
| DMSO | ThermoFisher Scientific | Cat#D12345 |
| Biotinylated Bovine Serum Albumin | Sigma | Cat#A8549–10MG |
| Tween-20 | Sigma | Cat#9005–64–5 |
| Streptavidin | Prozyme | Cat#SA10 |
| Glucose Oxidase | Sigma | Cat#G2133 |
| Catalase | Sigma | Cat#C9322 |
| TROLOX | Sigma | Cat# 648471 |
| Glucose | Sigma | Cat#G8270 |
| RNasin plus | Promega | Cat# N2611 |
| Heparin Sodium Salt | Sigma | Cat#H3393 |
| Critical Commercial Assays | ||
| Wizard SV Gel and PCR Clean-Up System | Promega | Cat#A9281 |
| Oligonucleotides | ||
| SA5_aadU35: 5’ – CCTGTGTCCTGTGTGTCCTGTCCAAAGTGTGTCG /iAmMC6T/CC – 3’ | Integrated DNA Technologies, Inc. | N/A |
| Terminator_REV_aadU46: 5’ – CAAAAAACCCCTCAAGACCCGTTTAGAGGCCCCAAGGGGTTATGC /iAmMC6T/AG – 3’ | Integrated DNA Technologies, Inc. | N/A |
| T7Pro_Tether_FOR: 5’ – GATCCTAATACGACTCACTATAGGGTGAGTGAGAGATGGATGGGTAGAGAGTTAGTAGTA – 3’ | Integrated DNA Technologies, Inc. | N/A |
| Tether_T3_33nts_3’BIO: 5’ – CTAACTCTCTACCCATCCATCTCTCACTCACCC /3BIO/ – 3’ | Integrated DNA Technologies, Inc. | N/A |
| Recombinant DNA | ||
| Sequences for linear DNA templates listed in Table S2 | This study | N/A |
| Plasmids for unlabeled r-proteins | Gift from G. Culver Culver and Noller, 1999 | N/A |
| pET24-S4:C32S,S189C | Kim et al., 2014 | N/A |
| pET24-S7:S54C | Gift from Hui-Ting Lee | N/A |
| pAR1219-T7RNAP | Davanloo et al., 1984 | N/A |
| pUC19-p17S | This study | N/A |
| Software and Algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| PyMol | Schrödinger, LLC (Version 2.2) | https://pymol.org/2/ |
| FIJI | Schindelin et al., 2012 | https://fiji.sc/ |
| Imscroll | Friedman and Gelles, 2015 | https://github.com/gelles-brandeis/CoSMoS_Analysis |
Co-transcriptional colocalization single-molecule experiments
Single-molecule experiments were carried out on a custom-built prism-type total internal reflection fluorescence (prism-TIRF) microscope. Green (532 nm) and Red (640 nm) lasers were used to excited Cy3 and Cy5 fluorophores, respectively. Single-molecules were imaged on a EMCCD camera (Andor) utilizing custom software developed in the Ha Lab implemented in IDL (smCamera, Ha Lab). DDS-Tween20 passivated quartz slides were prepared and assembled as described previously (Hua et al., 2014). Flow channels were created by adhering a tip to both holes in the slide channel using epoxy: one tip end served as a reservoir and the other tip served to hold tubing connected to a syringe.
Immediately before immobilization, stalled TECs were assembled with the same conditions as in the single-round transcription assays. Briefly, stalling reactions were assembled at RT as follows: 100 nM Cy3-labeled DNA template, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 50 nM T7 enzyme, 200 uM GTP, 200 uM ATP, 50 uM UTP, 2 U RNasin Plus, 100 nM biotinylated tether oligomer (Tether_T3_33nts_3’BIO; see Key Resources Table). Reactions were incubated at RT for 1.5 minutes and then diluted in transcription buffer (40 mM Tris-HCl pH 7.5, 20 mM MgCl2) for immobilization. Dilution was determined empirically for each DNA template, starting at 100-fold dilution, by examining the extent of immobilization on the microscope. Typically, 20-fold dilution of the stalled TEC was sufficient for well-separated spot density. Immobilized stalled TECs were stable and efficiently restarted for up to an hour after immobilization.
Following immobilization, stalled TECs were washed with imaging buffer (40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 150 mM KCl, 1% w/v glucose, 165 U/mL glucose oxidase, 2170 U/mL catalase, 4 mM Trolox, 2 U RNasin Plus) and imaged to check spot density. Since pre-16S DNAs are ~2 kb long, Cy3 intensity was very low at the beginning of the experiment. A restart imaging mixture (100 μL) was assembled immediately before use: 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 150 mM KCl, 5 nM Cy5-S4, 1 mM ATP, 1 mM GTP, 1 mM CTP, 1 mM UTP, 1% w/v glucose, 165 U/mL glucose oxidase, 2170 U/mL catalase, 4 mM Trolox, 2 U RNasin Plus. (NTPs were 1 mM each unless specified in the figure legend).
During imaging, flow of the restart imaging mixture was established briefly (~ 2 s) to simultaneously inject NTPs and Cy5-S4 into the slide chamber. Restart of transcription was marked in each experiment by the increase in background signal of the Cy5-channel indicative of flow of Cy5-S4 to the slide chamber, similar to previous experiments using free Cy5 dye (Hua et al., 2018). An alternating laser excitation scheme was used to image green and red channels to prevent photobleaching of Cy5-S4. Frames were taken every 100 ms for a total of ~ 3000 frames (5 minutes).
Post-transcriptional in situ RNA labeling
Where mentioned, RNAs were post-transcriptionally labeled on the slide with a fluorescently-labeled oligomer complementary to the 3’ extension on the RNA located just upstream of the terminator. Following immobilization of stalled TECs, transcription was restarted in the same manner as above with the addition of 100 nM fluorescently-labeled oligomer and incubated for 5 minutes without imaging. The slide chamber was then thoroughly washed (100 μL; 3 times) with wash buffer (40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 150 mM KCl, 2 U RNasin Plus) followed by imaging buffer (100 μl).
Single-molecule colocalization analyses
Single-molecule experiments were analyzed using the Imscroll software implemented in MATLAB that was developed in the Gelles lab (Friedman and Gelles, 2015). First, Cy3-labeled DNA molecules were automatically selected as areas of interest (AOIs). For Cy3-labeled pre-16S DNAs, spots were selected ~ 50–100 frames following flow of NTPs in order to account for low fluorescence signal before the restart of transcription. The fluorescence intensity in each AOI was integrated at each frame and plotted as a single-molecule time trace. The position of each AOI was then translated to the Cy5 channel using a mapping function, generated as previously described (Friedman and Gelles, 2015). The fluorescence intensity of corresponding locations in the Cy5 channel was then integrated over the duration of the movie.
Determining transcription window and transcription rate
Transcription restart was indicated as the start of flow and was marked in the single-molecule trajectories as the first frame following the increase in the background signal of Cy5 corresponding to injection of Cy5-S4 (Supplemental Figure S2B). The start of flow overlapped with the PIFE peak of for 16S1–100 (RNA is 80 nts after restart; ~ 1 s transcription) indicating that TECs are restarted as soon as NTPs are added to the slide chamber (Supplemental Figure S2B). The start of flow was nearly identical in all traces and therefore, a single timepoint was used as transcription restart for all molecules in a single experiment.
To assess the end of RNA transcription prior to termination, each Cy3 trace was inspected manually for a transcription signature. A transcription signature is defined as a slow increase in fluorescence starting after flow followed by a sharp increase in fluorescence (PIFE). PIFE was almost always followed shortly after by a loss of Cy3 fluorescence indicating either photobleaching or termination and dissociation of the Cy3 labeled DNA (Figure 2B and Supplemental Figure S2). Traces were filtered first for a transcription signature, then for a PIFE peak that could be well approximated by a single Gaussian (Figure 2B; Supplemental Figure S2C). Traces exhibiting multiple PIFE peaks were not analyzed further, because these spots likely contain multiple nascent RNAs tethered to the same DNA template.
The exact timepoint for the end of RNA transcription was marked by center of the PIFE peak approximated as the time frame with the highest Cy3 intensity (Figure 2C). Based on the single nanometer distance dependence of PIFE, the highest Cy3 intensity likely corresponds to RNAP positioned closest to the Cy3 fluorophore located on the DNA template. The transcription window (Figure 2B) was then calculated as the difference between the end of RNA transcription (marked by PIFE) and the restart of transcription (marked by Cy5-S4 flow). Transcription rate was determined by dividing the length of the transcribed RNA by the transcription window. Transcription rate varied with both RNA length and NTP concentration (Figure 2D and 2E).
Single-molecule dwell time analyses
S4 binding dynamics were analyzed on the subset of AOIs that exhibited an appropriate transcription signature (typically 20 – 30% of the DNAs present on the surface). Binding intervals were generated as previously described (Friedman and Gelles, 2015). S4 lifetimes lasting only a single frame (0.2 s) were indistinguishable from nonspecific S4 binding with the surface in the absence of RNA and were excluded from analysis. Global kinetic fitting of the unbinned data was achieved using maximum likelihood methods for single (16S100bp) and triple exponential kinetic binding behavior as in Equation 1 and 2, respectively, where x is the total time duration of the movie; tm is the minimum resolvable time interval in the experiment; tm is the maximum time interval; τ, τ1, τ2, τ3, represent characteristic lifetimes; and a1, and a2 are the amplitudes associated with the fitted lifetimes.
| Eq. 1 |
| Eq. 2 |
Errors in fitted parameters was estimated by bootstrapping the data to obtain a 95% confidence interval as previously described (Friedman and Gelles, 2015). Histograms were generated in MATLAB (the Mathworks) by unequal binning of the data to minimize empty bins and visualize the data with the maximum likelihood fits. Error bars in the histogram represent the variance in a binomial distribution described by Equation 3, where N is the number of observations and P is the event probability.
| Eq.3 |
S4 binding occupancy (O) per transcript was determined by calculating the number of frames with Cy5 colocalization divided by the total number of frames and the number of molecules in the dataset. By assuming that all RPs have the same binding behavior and that each protein binds independently, S4’s binding occupancy can then be used to estimate the probability density, P(n), of S4 binding events that occur on a complex also containing n additional RPs as follows:
| Eq. 4 |
where kon was approximated as the binding occupancy divided by the time interval of observation (200 s) and the concentration of S4 (5 nM). This apparent kon was similar to what was reported previously for S4 (Adilakshmi et al., 2008).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of individual experiments such as number of molecules analyzed and number of observations are as detailed in the manuscript text, figure legends, and figures.
DATA AND SOFTWARE AVAILABILITY
Custom-written MATLAB scripts used for dwell time analysis of transcription traces will be provided upon request to the Lead Contact, Sarah A. Woodson (swoodson@jhu.edu).
Supplementary Material
Figure S1. Preparing Stalled TECs. Related to Figure 1. A) Stalled TECs were assembled on pre-16S DNA as described for single-round transcription reactions (Methods). Heparin was added to prevent re-initiation and stalled complexes were incubated at room temperature for the indicated time points. Slow mis-incorporation (> 3 min) occurs at nt 41 resulting in stalling at the next G in the DNA template. Asterisk indicates shorter products produced by another T7 RNAP loaded and stalled behind the T7 RNAP stalled at nt 41 on the same template. Multiple polymerases were also observed in smCoCoA experiments and could be distinguished from templates with only a single polymerase (see Figure S2C). Samples were quenched in stop buffer and run on a 6% sequencing gel in 1X TBE for 4 hours at 55 W. Gel image shown is cropped from the full gel image. B) Microscope images of single TECs. TECs can only be immobilized in the presence of a biotinylated tether oligomer that is complementary to the C-less cassette of the nascent RNA. C) Quantification of 10 fields of view for images in B. D) DNA templates used in this study; see Table S1 for sequences. PIFE occurs when the RNAP encounters the Cy3 dye on the DNA template. Therefore, the location of the Cy3 dye corresponds to the length of RNA transcribed when PIFE occurs.
Figure S2. Characterizing PIFE and the fluorescence transcription signature. Related to Figure 2. A) Aligned single-molecule time trajectories for Cy3-DNA and Cy5-S4 illustrating the correlation between transcription restart by the increase in Cy5 background signal upon simultaneous Cy5-S4 and NTP injection. B) Aligned single-molecule time trajectories for transcription of a short RNA (80 nts) that is transcribed in ~ 1 s at 1 mM NTPs. Since injection takes 1 – 2 seconds, coincidence of the PIFE peak with the increase in Cy5 background indicates that transcription restart occurs within the timeframe of NTP injection. C) Expansion of Cy3 time traces illustrating multiple separate PIFE peaks for some TECs. Multiple PIFE peaks became more frequent with high polymerase concentration and were interpreted as multiple RNAPs loaded onto the same DNA template due to the length of the initial transcribed sequence, which can accommodate up to three enzymes. Only data from TECs exhibiting single PIFE peaks were included in analysis. D) Example traces of PIFE on Cy3-labeled pre-16S DNAs at 1 mM NTPs. The dotted line indicates the restart of transcription. E) Example traces of PIFE at 1 mM NTPs (top) and 100 μM NTPs (bottom) demonstrating that the PIFE peak occurs at later times with lower NTP concentration, indicating slower transcription. Traces are aligned such that t = 0 represents transcription restart.
Figure S3. S4 binds primarily transiently with RNAs that lack the 5WJ. Related to Figure 3. A) Example trace of S4 nonspecific binding to stalled TECs before NTP addition and transcription restart. Only short (0.2 – 0.4 s) binding events are observed in the absence of NTPs suggests S4 binds specifically to elongated RNA. B) Example trace of a stalled TEC that does not exhibit a transcription signature indicating that it is not restarted upon NTP injection (black line). S4 does not associate significantly with TECs that are not restarted, suggesting S4 specifically interacts with elongated RNAs. C) Example traces of co-transcriptional binding of S4 to 16S100bp RNA that does not contain the 5WJ. S4 only associates transiently with the 16S100bp RNA indicating that longer lived binding events are specific for the 5WJ. D) Secondary structures comparing RNA constructs lacking the S4 binding site used to distinguish nonspecific binding behavior. E) Rastergram of S4 binding events on a 5’ truncated pre-16S RNA occurring 30 minutes after transcription. Compared to S4 binding to full-length pre-16S RNA post-transcriptionally (Figure 3D), S4 binds primarily transiently with few long-lived interactions. F) Probability density plot comparing S4 lifetime distributions of binding to RNAs with (pre-16S RNA, black dotted lines) and without (16S100bp; blue and pre-16S5’truncated; magenta) the 5WJ binding site. Kinetic parameters for maximum likelihood fitting of the unbinned data are given in Table S2. G) Transcription rate does not positively or negatively influence S4 binding dynamics in the absence of other factors. Related to Figure 3. Scatterplot of S4 binding lifetimes from three experiments at 100 μM, 1 mM, and 2 mM NTP concentrations (N = 103 molecules; Nobs = 1879) plotted against the corresponding pre-16S transcription rate. The distribution of transcription rates in a single experiment were narrow owing to synchronized restart and processivity of T7 (see histograms in Figure 2D) explaining why the points are not evenly distributed along the x-axis.
Figure S4. Ribosomal protein S7 associates primarily transiently with pre-16S RNA co-transcriptionally. Related to Figure 3. A) 30S assembly map highlighting that S4 and S7 are both primary proteins that recognize structures within the 5’ and 3’ domains, respectively. B) Depiction of the 16S rRNA secondary structure illustrating the binding sites of S4 and S7. C) Example traces for S7 binding during and immediately following transcription of the pre-16S RNA. S7 binds primarily transiently to nascent transcripts with longer lived events occurring after transcription of the pre-16S has completed. D) Probability density plot of S7 and S4 binding lifetimes. Unbinned lifetimes were analyzed by maximum likelihood analysis as described in the methods (Nmol = 108 transcripts; Nobs = 1054 lifetimes). S7 lifetime distribution was fit to an multi-exponential function with three kinetic parameters (τ1 = 0.9 ± 0.04, τ2 = 9.1 ± 1.5, a= 0.87 ± 0.02). S4 fitting parameters are described in Table S2.
Figure S5. S4 binding to 16S fragments during transcription. Related to Figure 4. A) Example traces for S4 binding to 16S5WJ+5’L. Black line indicates transcription restart (Cy5-S4 and NTP injection) and dotted lines indicate transcription end (PIFE). B) Example traces for S4 binding to 16S5’ domain as in B. C) Rastergram (N = 54 mol) for S4 binding to 16S5WJ. D) Rastergram (N = 54 mol) for S4 binding to 16S5WJ+5’L. E) Rastergram (N = 54 mol) for S4 binding to 16S5’ domain. F) Native gel analysis for Cy5-S4 binding to refolded 5’ domain RNAs. Briefly, 32P-labeled RNAs (1 nM) was incubated in reconstitution buffer (40 mM Tris-HCl pH 7.5, 330 mM KCl, 0 – 20 mM MgCl2, ± 100 or 400 nM Cy5-S4) for 10 min at 37°C. Reactions were run on a native gel (6%) in 1X THEM at 15W at 4°C as previously described (Adilakshmi et al., 2005). (left) Mg2+ titration for folding of the 16S21–556 RNA. Native Cy5-S4/RNA complexes form similarly to previous work (Adilakshmi et al., 2005). (right) Mg2+ titration for folding of the 16S5’domain RNA is similar to 16S21–556. G) EMSA with Cy5-S4 and 5’ domain RNA (16S21–556; left) EMSA with Cy5-S4 and 5’ domain RNA (16S5’domains; right).
Figure S6. Increased likelihood of longer-lived S4 binding events are dependent on the presence of specific ribosomal proteins. Related to Figures 5 and 6. A) Crystal structures of the 30S subunit with highlighted combinations of ribosomal proteins. B) Probability density plot of S4 lifetime distribution in the presence of ribosomal protein S9 (50 nM; blue) and a generic RNA binding protein, MS2-MBP (50 nM; orange) compared to in the absence of additional proteins (black dotted line). Fitting parameters for multi-exponential fits are given in Table S2. C) Analysis of the fraction of S4 binding events for transient interactions (< 1 s) and long-lived interactions (> 10 s and > 50 s) in the presence of different proteins. Number of molecules for each experiment are as follows; S4 only (N = 103), S4 + MS2-MBP (N = 103), S4 + S9 (N = 30), S4 + S16, S17, S20 (N = 112), and S4 + S16, S17, S20, S8, S5, S12 (N = 176).
Table S1. DNA templates used in this study. Related to Figure 1.
Table S2. Kinetic parameters for maximum likelihood fitting of the S4 lifetime data. Related to Figures 3 – 5.
Figure 7.

Model for ribosome assembly during transcription. Primary ribosomal proteins (RPs) can form multiple types of complexes with the nascent pre-rRNA depending on its folding pattern; transient sampling of the elongating RNA dominates until the RNA forms a structure that can form a metastable (1–5 s) protein complex. These metastable complexes hasten stable recruitment of other primary RPs like S4, and contribute to the recruitment of the secondary and tertiary RPs. Once a sufficient number of metastable interactions have accumulated within an assembly domain, they consolidate into native, long-lived complexes that commit the transcript for assembly. This commitment must presumably occur within the time frame of transcription in order to generate stable precursor 30S subunits that can undergo late, post-transcriptionally assembly. Otherwise, the metastable complexes fall apart leaving the rRNA vulnerable to turnover preventing sequestration of RPs on incompetent transcripts.
ACKNOWLEDGEMENT
We thank Dr. Tucker Carrocci, Dr. Joshua Larson, and all the members of the Woodson lab for helpful discussions. We also acknowledge Dr. Sanjaya Abeysirigunawardena, Dr. Hui-Ting Lee, Arthur Korman, Dr. Indra Mani Sharma, and Dr. Megan Mayerle for purifying proteins. This project is funded by the National Institutes of Health [R01 GM60819 to S.A.W and F32 GM125234 to M.L.R.]
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information includes DNA templates used in this study, 6 figures, and 2 tables, and can be found with this article online at https://doi.org/10.1016/j.cell.2019.11.007.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abeysirigunawardena SC, and Woodson SA (2015). Differential effects of ribosomal proteins and Mg 2+ions on a conformational switch during 30S ribosome 5′-domain assembly. RNA 21, 1859–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeysirigunawardena SC, Kim H, Lai J, Ragunathan K, Rappé MC, Luthey-Schulten Z, Ha T, and Woodson SA (2017). Evolution of protein-coupled RNA dynamics during hierarchical assembly of ribosomal complexes. Nat Commun 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adilakshmi T, Bellur DL, and Woodson SA (2008). Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 455, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanzadeh V, Huang Y, Sanguinetti G, and Beggs JD (2018). Transcription rate strongly affects splicing fidelity and cotranscriptionality in budding yeast. Genome Res 28, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Tambe A, Zhou K, and Doudna JA (2014). RNA-guided assembly of Rev-RRE nuclear export complexes. Elife 3, e03656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey RT, and Williamson JR (1996a). Interaction of the Bacillus stearothermophilusRibosomal Protein S15 with 16 S rRNA: I. Defining the Minimal RNA Site. J Mol Biol 261, 536–549. [DOI] [PubMed] [Google Scholar]
- Batey RT, and Williamson JR (1996b). Interaction of the Bacillus stearothermophilusRibosomal Protein S15 with 16 S rRNA: II. Specificity Determinants of RNA-Protein Recognition. J Mol Biol 261, 550–567. [DOI] [PubMed] [Google Scholar]
- Bellur DL, and Woodson SA (2009). A minimized rRNA-binding site for ribosomal protein S4 and its implications for 30S assembly. Proc National Acad Sci 37, 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubunenko M, Court DL, Refaii A, Saxena S, Korepanov A, Friedman DI, Gottesman ME, and Alix J (2013). Nus transcription elongation factors and RNase III modulate small ribosome subunit biogenesis in Escherichia coli. Mol Microbiol 87, 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunner AE, Beck AH, and Williamson JR (2010). Kinetic cooperativity in Escherichia coli 30S ribosomal subunit reconstitution reveals additional complexity in the assembly landscape. Proc National Acad Sci 107, 5417–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler E, and Chamberlin M (1982). Bacteriophage SP6-specific RNA polymerase. I. Isolation and characterization of the enzyme. J Biological Chem 257, 5772–5778. [PubMed] [Google Scholar]
- Calidas D, and Culver GM (2011). Interdependencies govern multidomain architecture in ribosomal small subunit assembly. RNA 17, 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvier A, eacute d eacute ric Picard-Jean F, Berger-Dancause J-C, egrave ne Bastet L, Naghdi M, eacute A, Turcotte P, Perreault J, and Lafontaine DA (2016). Transcriptional pausing at the translation start site operates as a critical checkpoint for riboswitch regulation. Nat Commun 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon C, Squires C, and Squires C (1995). Control of rRNA transcription in Escherichia coli. Microbiol Rev 59, 623–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver G, and Noller H (1999). Efficient reconstitution of functional Escherichia coli 30S ribosomal subunits from a complete set of recombinant small subunit ribosomal proteins. RNA 5, 832 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver G, and Noller H (2000). In vitro reconstitution of 30S ribosomal subunits using complete set of recombinant proteins. Methods in Enzymology 318, 446 460. [DOI] [PubMed] [Google Scholar]
- Davanloo P, Rosenberg A, Dunn J, and Studier F (1984). Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA 81, 2035–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JH, and Williamson JR (2017). Structure and dynamics of bacterial ribosome biogenesis. J Mol Biol 372, 20160181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis P, Ehrenberg M, Fange D, and Bremer H (2009). Varying Rate of RNA Chain Elongation during rrn Transcription in Escherichia coli. J Bacteriol 191, 3740 3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duss O, Stepanyuk GA, Grot A, O’Leary SE, Puglisi JD, and Williamson JR (2018). Real-time assembly of ribonucleoprotein complexes on nascent RNA transcripts. Nat Commun 9, 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S, and Miller O (1989). Transcription mapping of the Escherichia coli chromosome by electron microscopy. Journal of Bacteriology 171, 4207 4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieda KL, and Block SM (2012). Direct observation of cotranscriptional folding in an adenine riboswitch. Science 338, 397 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LJ, and Gelles J (2015). Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods 86, 27 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner RB, Pak Y, and Draper DE (2001). Recognition of 16S rRNA by Ribosomal Protein S4 from Bacillus stearothermophilus. Biochemistry-Us 40, 7165–7173. [DOI] [PubMed] [Google Scholar]
- Held W, Mizushima S, and Nomura M (1973). Reconstitution of Escherichia coli 30 S ribosomal subunits from purified molecular components. J Biol Chem 248, 5720 5730. [PubMed] [Google Scholar]
- Held W, Ballou B, Mizushima S, and Nomura M (1974). Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J Biol Chem 249, 3103–3111. [PubMed] [Google Scholar]
- Herzel L, Ottoz DS, Alpert T, and Neugebauer KM (2017). Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat Rev Mol Cell Bio 18, 637 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, and Moore MJ (2011). Ordered and dynamic assembly of single spliceosomes. Science 331, 1289 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Rodgers ML, Friedman LJ, Gelles J, and Moore MJ (2016). Single molecule analysis reveals reversible and irreversible steps during spliceosome activation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua B, Han K, Zhou R, Kim H, Shi X, Abeysirigunawardena SC, Jain A, Singh D, Aggarwal V, Woodson SA, et al. (2014). An improved surface passivation method for single-molecule studies. Nat Methods 11, 1233–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua B, Panja S, Wang Y, Woodson S, and Ha T (2018). Mimicking Co-Transcriptional RNA Folding Using a Superhelicase. J Am Chem Soc 140, 100067–100070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker M, Barandun J, Buzovetsky O, Steckler C, Molina H, and Klinge S (2019). Conformational switches control early maturation of the eukaryotic small ribosomal subunit. Elife 8, e45185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Abeysirigunawarden SC, Chen K, Mayerle M, Ragunathan K, Luthey-Schulten Z, Ha T, and Woodson SA (2014). Protein-guided RNA dynamics during early ribosome assembly. Nature 506, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, and Hoskins AA (2017). Dynamics and consequences of spliceosome E complex formation. Elife 6, e27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewicki B, Margus T, Remme J, and Nierhaus K (1993). Coupling of rRNA transcription and ribosomal assembly in vivo. Formation of active ribosomal subunits in Escherichia coli requires transcription of rRNA genes by host RNA polymerase which cannot be replaced by bacteriophage T7 RNA polymerase. J Mol Biol 231, 581 593. [DOI] [PubMed] [Google Scholar]
- Mayerle M, Bellur DL, and Woodson SA (2011). Slow Formation of Stable Complexes during Coincubation of Minimal rRNA and Ribosomal Protein S4. J Mol Biol 412, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, and Nomura M (1970). Assembly Mapping of 30S Ribosomal Proteins from E. coli. Nature 226, 1214. [DOI] [PubMed] [Google Scholar]
- Nowotny V, and Nierhaus KH (2002). Assembly of the 30S subunit from Escherichia coli ribosomes occurs via two assembly domains which are initiated by S4 and S7. Biochemistry 27, 7051–7055. [DOI] [PubMed] [Google Scholar]
- Orr JW, Hagerman PJ, and Williamson JR (1998). Protein and Mg2+-induced conformational changes in the S15 binding site of 16 s ribosomal RNA11Edited by D. E. Draper. J Mol Biol 275, 453–464. [DOI] [PubMed] [Google Scholar]
- Perdrizet GA, Artsimovitch I, Furman R, Sosnick TR, and Pan T (2012). Transcriptional pausing coordinates folding of the aptamer domain and the expression platform of a riboswitch. Proc Natl Acad Sci USA 109, 3323–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, Daubresse G, and Noller H (1993). Dynamics of in vitro assembly of 16 S rRNA into 30 S ribosomal subunits. J Mol Biol 232, 362–374. [DOI] [PubMed] [Google Scholar]
- Ramaswamy P, and Woodson SA (2009). S16 throws a conformational switch during assembly of 30S 5′ domain. Nat Struct Mol Biol 16, nsmb.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldi T, Fong N, and Bentley DL (2018). Transcription elongation rate affects nascent histone pre-mRNA folding and 3’ end processing. Genes Dev 32, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapag A, Vartikar JV, and Draper DE (1990). Dissection of the 16S rRNA binding site for ribosomal protein S4. Biochim Biophys Acta 1050, 34–37. [DOI] [PubMed] [Google Scholar]
- Schaup H, Green M, and Kurland C (1971). Molecular interactions of ribosomal components. Mol Gen Genet 112, 1–8. [DOI] [PubMed] [Google Scholar]
- Shcherbakova I, Hoskins AA, Friedman LJ, Serebrov V, Corrêa IR, Xu M-Q, Gelles J, and Moore MJ (2013). Alternative spliceosome assembly pathways revealed by single-molecule fluorescence microscopy. Cell Reports 5, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert H, Sochor F, Wacker A, Buck J, Helmling C, Hiller F, Keyhani S, Noeske J, Grimm S, Rudolph MM, et al. (2017). Pausing guides RNA folding to populate transiently stable RNA structures for riboswitch-based transcription regulation. Elife 6, e21297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Wilson RC, and Noller HF (1986). Localization of the binding site for protein S4 on 16 S ribosomal RNA by chemical and enzymatic probing and primer extension. J Mol Biol 192, 101–110. [DOI] [PubMed] [Google Scholar]
- Stern S, Powers T, Changchien L, and Noller H (1989). RNA-protein interactions in 30S ribosomal subunits: folding and function of 16S rRNA. Science 244, 783–790. [DOI] [PubMed] [Google Scholar]
- Talkington MW, Siuzdak G, and Williamson JR (2005). An assembly landscape for the 30S ribosomal subunit. RNA 438, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub P, and Nomura M (1968). Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc Natl Acad Sci USA 59, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub P, and Nomura M (1969). Studies on the Assembly of Ribosomes in vitro. Cold Spring Harb Sym 34, 63–67. [DOI] [PubMed] [Google Scholar]
- Vartikar JV, and Draper DE (1989). S4–16 S ribosomal RNA complex Binding constant measurements and specific recognition of a 460-nucleotide region. J Mol Biol 209, 221–234. [DOI] [PubMed] [Google Scholar]
- Vila-Sanjurjo A, Ridgeway WK, Seymaner V, Zhang W, Santoso S, Yu K, and Cate JH (2003). X-ray crystal structures of the WT and a hyper-accurate ribosome from Escherichia coli. Proc Natl Acad Sci USA 100, 8682–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel U, and Jensen K (1995). Effects of the antiterminator BoxA on transcription elongation kinetics and ppGpp inhibition of transcription elongation in Escherichia coli. J Biol Chem 270, 18335–18340. [DOI] [PubMed] [Google Scholar]
- Widom JR, Nedialkov YA, Rai V, Hayes RL, Brooks CL, Artsimovitch I, and Walter NG (2018). Ligand Modulates Cross-Coupling between Riboswitch Folding and Transcriptional Pausing. Mol Cell 72, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Landick R, and Gelles J (1994). Tethered particle motion method for studying transcript elongation by a single RNA polymerase molecule. Biophys J 67, 2468–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Landick R (2016). A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure. Trends Biochem Sci 41, 293 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wu C, Cai G, Chen S, and Ye K (2016). Stepwise and dynamic assembly of the earliest precursors of small ribosomal subunits in yeast. Genes Dev 30, 718–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, and Reed R (2003). Purification of Functional RNA‐Protein Complexes using MS2 MBP. Curr Protoc Mol Biol 63, 27.3.1–27.3.7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Preparing Stalled TECs. Related to Figure 1. A) Stalled TECs were assembled on pre-16S DNA as described for single-round transcription reactions (Methods). Heparin was added to prevent re-initiation and stalled complexes were incubated at room temperature for the indicated time points. Slow mis-incorporation (> 3 min) occurs at nt 41 resulting in stalling at the next G in the DNA template. Asterisk indicates shorter products produced by another T7 RNAP loaded and stalled behind the T7 RNAP stalled at nt 41 on the same template. Multiple polymerases were also observed in smCoCoA experiments and could be distinguished from templates with only a single polymerase (see Figure S2C). Samples were quenched in stop buffer and run on a 6% sequencing gel in 1X TBE for 4 hours at 55 W. Gel image shown is cropped from the full gel image. B) Microscope images of single TECs. TECs can only be immobilized in the presence of a biotinylated tether oligomer that is complementary to the C-less cassette of the nascent RNA. C) Quantification of 10 fields of view for images in B. D) DNA templates used in this study; see Table S1 for sequences. PIFE occurs when the RNAP encounters the Cy3 dye on the DNA template. Therefore, the location of the Cy3 dye corresponds to the length of RNA transcribed when PIFE occurs.
Figure S2. Characterizing PIFE and the fluorescence transcription signature. Related to Figure 2. A) Aligned single-molecule time trajectories for Cy3-DNA and Cy5-S4 illustrating the correlation between transcription restart by the increase in Cy5 background signal upon simultaneous Cy5-S4 and NTP injection. B) Aligned single-molecule time trajectories for transcription of a short RNA (80 nts) that is transcribed in ~ 1 s at 1 mM NTPs. Since injection takes 1 – 2 seconds, coincidence of the PIFE peak with the increase in Cy5 background indicates that transcription restart occurs within the timeframe of NTP injection. C) Expansion of Cy3 time traces illustrating multiple separate PIFE peaks for some TECs. Multiple PIFE peaks became more frequent with high polymerase concentration and were interpreted as multiple RNAPs loaded onto the same DNA template due to the length of the initial transcribed sequence, which can accommodate up to three enzymes. Only data from TECs exhibiting single PIFE peaks were included in analysis. D) Example traces of PIFE on Cy3-labeled pre-16S DNAs at 1 mM NTPs. The dotted line indicates the restart of transcription. E) Example traces of PIFE at 1 mM NTPs (top) and 100 μM NTPs (bottom) demonstrating that the PIFE peak occurs at later times with lower NTP concentration, indicating slower transcription. Traces are aligned such that t = 0 represents transcription restart.
Figure S3. S4 binds primarily transiently with RNAs that lack the 5WJ. Related to Figure 3. A) Example trace of S4 nonspecific binding to stalled TECs before NTP addition and transcription restart. Only short (0.2 – 0.4 s) binding events are observed in the absence of NTPs suggests S4 binds specifically to elongated RNA. B) Example trace of a stalled TEC that does not exhibit a transcription signature indicating that it is not restarted upon NTP injection (black line). S4 does not associate significantly with TECs that are not restarted, suggesting S4 specifically interacts with elongated RNAs. C) Example traces of co-transcriptional binding of S4 to 16S100bp RNA that does not contain the 5WJ. S4 only associates transiently with the 16S100bp RNA indicating that longer lived binding events are specific for the 5WJ. D) Secondary structures comparing RNA constructs lacking the S4 binding site used to distinguish nonspecific binding behavior. E) Rastergram of S4 binding events on a 5’ truncated pre-16S RNA occurring 30 minutes after transcription. Compared to S4 binding to full-length pre-16S RNA post-transcriptionally (Figure 3D), S4 binds primarily transiently with few long-lived interactions. F) Probability density plot comparing S4 lifetime distributions of binding to RNAs with (pre-16S RNA, black dotted lines) and without (16S100bp; blue and pre-16S5’truncated; magenta) the 5WJ binding site. Kinetic parameters for maximum likelihood fitting of the unbinned data are given in Table S2. G) Transcription rate does not positively or negatively influence S4 binding dynamics in the absence of other factors. Related to Figure 3. Scatterplot of S4 binding lifetimes from three experiments at 100 μM, 1 mM, and 2 mM NTP concentrations (N = 103 molecules; Nobs = 1879) plotted against the corresponding pre-16S transcription rate. The distribution of transcription rates in a single experiment were narrow owing to synchronized restart and processivity of T7 (see histograms in Figure 2D) explaining why the points are not evenly distributed along the x-axis.
Figure S4. Ribosomal protein S7 associates primarily transiently with pre-16S RNA co-transcriptionally. Related to Figure 3. A) 30S assembly map highlighting that S4 and S7 are both primary proteins that recognize structures within the 5’ and 3’ domains, respectively. B) Depiction of the 16S rRNA secondary structure illustrating the binding sites of S4 and S7. C) Example traces for S7 binding during and immediately following transcription of the pre-16S RNA. S7 binds primarily transiently to nascent transcripts with longer lived events occurring after transcription of the pre-16S has completed. D) Probability density plot of S7 and S4 binding lifetimes. Unbinned lifetimes were analyzed by maximum likelihood analysis as described in the methods (Nmol = 108 transcripts; Nobs = 1054 lifetimes). S7 lifetime distribution was fit to an multi-exponential function with three kinetic parameters (τ1 = 0.9 ± 0.04, τ2 = 9.1 ± 1.5, a= 0.87 ± 0.02). S4 fitting parameters are described in Table S2.
Figure S5. S4 binding to 16S fragments during transcription. Related to Figure 4. A) Example traces for S4 binding to 16S5WJ+5’L. Black line indicates transcription restart (Cy5-S4 and NTP injection) and dotted lines indicate transcription end (PIFE). B) Example traces for S4 binding to 16S5’ domain as in B. C) Rastergram (N = 54 mol) for S4 binding to 16S5WJ. D) Rastergram (N = 54 mol) for S4 binding to 16S5WJ+5’L. E) Rastergram (N = 54 mol) for S4 binding to 16S5’ domain. F) Native gel analysis for Cy5-S4 binding to refolded 5’ domain RNAs. Briefly, 32P-labeled RNAs (1 nM) was incubated in reconstitution buffer (40 mM Tris-HCl pH 7.5, 330 mM KCl, 0 – 20 mM MgCl2, ± 100 or 400 nM Cy5-S4) for 10 min at 37°C. Reactions were run on a native gel (6%) in 1X THEM at 15W at 4°C as previously described (Adilakshmi et al., 2005). (left) Mg2+ titration for folding of the 16S21–556 RNA. Native Cy5-S4/RNA complexes form similarly to previous work (Adilakshmi et al., 2005). (right) Mg2+ titration for folding of the 16S5’domain RNA is similar to 16S21–556. G) EMSA with Cy5-S4 and 5’ domain RNA (16S21–556; left) EMSA with Cy5-S4 and 5’ domain RNA (16S5’domains; right).
Figure S6. Increased likelihood of longer-lived S4 binding events are dependent on the presence of specific ribosomal proteins. Related to Figures 5 and 6. A) Crystal structures of the 30S subunit with highlighted combinations of ribosomal proteins. B) Probability density plot of S4 lifetime distribution in the presence of ribosomal protein S9 (50 nM; blue) and a generic RNA binding protein, MS2-MBP (50 nM; orange) compared to in the absence of additional proteins (black dotted line). Fitting parameters for multi-exponential fits are given in Table S2. C) Analysis of the fraction of S4 binding events for transient interactions (< 1 s) and long-lived interactions (> 10 s and > 50 s) in the presence of different proteins. Number of molecules for each experiment are as follows; S4 only (N = 103), S4 + MS2-MBP (N = 103), S4 + S9 (N = 30), S4 + S16, S17, S20 (N = 112), and S4 + S16, S17, S20, S8, S5, S12 (N = 176).
Table S1. DNA templates used in this study. Related to Figure 1.
Table S2. Kinetic parameters for maximum likelihood fitting of the S4 lifetime data. Related to Figures 3 – 5.
Data Availability Statement
Custom-written MATLAB scripts used for dwell time analysis of transcription traces will be provided upon request to the Lead Contact, Sarah A. Woodson (swoodson@jhu.edu).