

Abstract
Xeroderma pigmentosum (XP) encompasses a group of rare diseases characterized in most cases by malfunction of nucleotide excision repair (NER), which results in an increased sensitivity to UV radiation in affected individuals. Approximately 25–30% of XP patients present with neurological symptoms, such as sensorineural deafness, mental deterioration and ataxia. Although it is known that dysfunctional DNA repair is the primary pathogenesis in XP, growing evidence suggests that mitochondrial pathophysiology may also occur. This appears to be secondary to dysfunctional NER but may contribute to the neurodegenerative process in these patients. The available pharmacological treatments in XP mostly target the dermal manifestations of the disease. In the present review, we outline how current understanding of the pathophysiology of XP could be used to develop novel therapies to counteract the neurological symptoms. Moreover, the coexistence of cancer and neurodegeneration present in XP led us to focus on possible new avenues targeting mitochondrial pathophysiology.
Linked Articles
This article is part of a themed section on Mitochondrial Pharmacology: Featured Mechanisms and Approaches for Therapy Translation. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.22/issuetoc
Abbreviations
- ∆Ψm
mitochondrial membrane potential
- CoQ10
coenzyme Q10
- CS
Cockayne syndrome
- cyPu
8,5‐cyclopurine deoxynucleotides
- ETC
electron transport chain
- mtDNA
mitochondrial DNA
- NER
nucleotide excision repair
- OXPHOS
oxidative phosphorylation
- SYT‐9
synaptotagmin‐9
- TrkB
tropomyosin receptor kinase B
- XP
xeroderma pigmentosum
Introduction
Xeroderma pigmentosum (XP) is an autosomal recessive disorder caused by mutations in genes involved in the DNA repair machinery. XP has an estimated incidence of 2.3 per million live births in Western Europe (Kleijer et al., 2008) but is more common in other geographical regions, including Japan (Hirai et al., 2006). Eight causative proteins have been identified so far (XPA, XPB, XPC, XPD, XPE, XPF, XPG and XPV), allowing XP to be divided into clinically heterogeneous complementation groups (Bootsma and Hoeijmakers, 1991; Bowden et al., 2015). The XPA to XPG proteins are involved in different steps of the nucleotide excision repair (NER) in the presence of DNA damage. Patients with XP variant harbour mutations in the DNA polymerase η, which is involved in DNA synthesis after UV radiation‐related damage (Lehmann et al., 1975; Masutani et al., 1999). The signs and symptoms of patients with XP can broadly be classified into cutaneous and neurological manifestations, although additional symptoms, such as ophthalmological abnormalities and a predisposition to cancers, are well recognized (Bradford et al., 2011; Brooks et al., 2013; Fassihi et al., 2016). A recently published study by Fassihi et al. (2016) has provided detailed clinical and molecular information on the largest analysed cohort of XP patients to date. The study highlighted the clinical heterogeneity of XP even within complementation groups, which is strongly dependent on distinct locations and types of mutations in the causative genes (Fassihi et al., 2016).
Dermatological symptoms and therapeutic strategies
XP patients share the common characteristic of extreme sensitivity to UV radiation. This may manifest with severe skin burning and blistering in infants, but not all patients exhibit this acute abnormal reaction to sunlight (DiGiovanna and Kraemer, 2012; Sethi et al., 2013; Fassihi et al., 2016). Freckling‐like skin changes, however, develop in all patients and eventually progress into atrophy, telangiectasias and intermixed hypo‐ and hyperpigmented areas (Black, 2016). Premalignant lesions, such as actinic keratoses and skin neoplasms in sun‐exposed areas, are observed at an early age and are related to complementation group (DiGiovanna and Kraemer, 2012). The most prevalent skin tumours in XP patients are basal and squamous cell carcinomas, followed by malignant melanomas, with a 10 000‐fold and 2000‐fold increased incidence respectively (Bradford et al., 2011). Interestingly, complementation groups presenting with an abnormal acute sunburn reaction are associated not only with neurodegeneration but also with a lower prevalence of skin cancer due to early diagnosis and initiation of sun protection (Sethi et al., 2013; Fassihi et al., 2016).
In the absence of specific treatments that target the underlying DNA‐repair dysfunction, the multidisciplinary clinical management of XP patients mainly focuses on strict UV protection and treatment of malignancies (Tamura et al., 2014). The former encompasses the reduction of exposure to sunlight using UV‐protective long‐sleeved clothing, filters on windows in buildings and cars and sunscreen lotions with the highest possible protective filters (Moriwaki et al., 2017). Regular skin cancer screening is essential to detect early malignancies, which are treated in accordance with guidelines used for non‐XP patients (Naik et al., 2013). First‐line treatment is surgical excision, but case reports on conservative approaches with topical application of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5003 5% (Malhotra et al., 2008; Yang et al., 2015) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4789 (Lambert and Lambert, 2015) have demonstrated favourable results. One prospective randomized controlled trial suggested a reduced frequency of actinic keratoses and basal cell carcinomas using a liposome preparation containing the bacterial DNA repair enzyme T4N5 endonuclease (Yarosh et al., 2001). However, subsequent studies were terminated due to lack of efficacy.
Neurological symptoms and lack of causative treatment
The prevalence of neurodegeneration varies across and even within the complementation groups and is most commonly associated with XPA and XPD, followed by XPB, XPG and XPF (Anttinen et al., 2008; Niedernhofer et al., 2011; Karass et al., 2015; Fassihi et al., 2016). Overall, in Europe and North America, approximately 25–30% of XP patients are affected by neurological impairment of variable severity. In affected patients, the progressive cerebral and cerebellar degeneration with frequent involvement of the peripheral nervous system results in a wide range of symptoms including (i) progressive cognitive impairment, (ii) sensorineural hearing loss, (iii) ataxia, (iv) pyramidal and (v) extrapyramidal tract signs and (vi) areflexia (Rass et al., 2007; Niedernhofer, 2008; Lehmann et al., 2011; Fassihi et al., 2016). The mean age of death of affected patients has been reported as 29 years, compared to 37 years in patients without neurodegeneration (Bradford et al., 2011).
To date, there is no effective treatment for the neurological manifestations of XP, and symptoms are managed with supportive measures. Exposure to UV‐B radiation is crucial in cutaneous carcinogenesis in XP, however, the aetiology of the neurological symptoms is poorly understood. Recently, it has been found that NER is required for repair not only of UV radiation damage but also of some endogenous DNA lesions due to generation of reactive species (see Brooks, 2017). These lesions are generated by the reaction of hydroxyl radicals with DNA, forming 8,5‐cyclopurine deoxynucleotides (cyPu).
Tomas Lindahl (Kuraoka et al., 2000) and Jay Robbins (Brooks et al., 2000) groups reported that cyPu are exclusive substrates for NER, suggesting that mutations in this specific DNA repair process contribute to the neurological symptoms in XP (see Brooks, 2017). An improved understanding of the pathophysiology of neurological dysfunction, which will be discussed later in the review, seems crucial for the development of causative treatment.
Related disorders
The most closely related NER disorder to XP is Cockayne syndrome (CS). CS‐A is caused by mutations of the ERCC8 gene, while CS‐B patients harbour mutations in the ERCC6 gene (Spivak, 2004). The CS‐A and CS‐B proteins are required for a sub‐branch of NER (transcription‐coupled‐NER) that rapidly repairs damage in the transcribed strand of actively transcribed genes (Kamenisch et al., 2010). They also have a role in transcription and neuronal differentiation (Wang et al., 2014). CS has a severe developmental and neurological phenotype, which overlaps with the relatively milder neurological phenotype of XP (Kraemer et al., 2007). Neurological manifestations include progressive spasticity, peripheral neuropathy, ataxia, weakness and dementia. Underlying these impairments is both a failure of brain development and progressive neuronal loss. Although patients are photosensitive, CS is not associated with an increased risk of skin malignancies (Rapin et al., 2006). Life expectancy is markedly reduced in all patients but differs according to clinical subtype (Rapin et al., 2006). XP‐CS complex refers to a rare neurodegenerative disorder that combines clinical characteristics of XP and CS. Patients present with growth retardation and neurodevelopmental decline while at the same time suffering from the cutaneous manifestations observed in XP (Natale and Raquer, 2017). Although CS and XP have different genetic defects, they share cellular hypersensitivity to UV radiation and defective NER, which will be further discussed below.
Other related disorders that share some clinical and molecular features with XP include the following: (i) ataxia telangiectasia characterized by a similar neurological phenotype and the occurrence of cancer; (ii) ataxia with oculomotor apraxia type 1 (iii) and 2 (AOA1; AOA2; Clements et al., 2004); and (iv) spinocerebellar ataxia with axonal neuropathy (SCAN1; El‐Khamisy et al., 2005; Gilmore, 2014) (v) and Riddle syndrome (Stewart et al., 2009), sharing some neurological feature such as ataxia and with underlying DNA repair defects. Mitochondrial dysfunction is a common pathophysiological feature of all these disorders (Le Ber et al., 2007; Scheibye‐Knudsen et al., 2013), and although the cause of cancer in XP is molecularly understood, the pathophysiology causing neurodegeneration is still a matter of debate (Table 1).
Table 1.
XP and related disorders
| Disease | Gene | Protein function | Defective pathway | Clinical features |
|---|---|---|---|---|
| Xeroderma pigmentosum |
XPA XPB/ERCC3 XPC XPD/ERCC2 XPE/DDB2 XPF/ERCC4 XPG/ERCC5 XPV/POLH |
Damage verification Helicase Damage recognition Helicase Damage recognition Nuclease Nuclease Polymerase |
NER NER NER NER NER NER NER Translesion synthesis |
40% of the patients show extreme sensitivity to sunlight and sunburn reaction, while 60% do not show any sunburn reaction. 20/30% of the patients show neurological abnormalities: neuronal degeneration resulting in deafness, ataxia, areflexia, microcephaly and intellectual deficiency and impaired eye sight. XPC, XPE and XPV do not show signs of neurological abnormalities. (Lehmann et al., 2011; Fassihi et al., 2016) |
| Cockayne syndrome |
CSA/ERCC8 CSB/ERCC6 |
Damage recognition and Ubiquitination Damage recognition |
TC‐NER (transcription‐coupled NER) | Microcephaly, ataxia, failure to thrive and delayed development. Increased sensitivity to sunlight (photosensitivity), and in some cases, even a small amount of sun exposure can cause a sunburn or blistering of the skin. Hearing loss, vision loss, severe tooth decay, bone abnormalities, abnormal thermoregulation in hands and feet and liver dysfunction.(Rapin et al., 2006; Wilson et al., 2016) |
| Ataxia telangiectasia | ATM | Damage‐activated protein kinase | DSB (DNA double strand‐break) | Ataxia, chorea, myoclonus neuropathy. Slurred speech and oculomotor apraxia. Small clusters of enlarged blood vessels called telangiectases, which occur in the eyes and on the surface of the skin, are also characteristic of this condition.High amounts of a protein called α‐fetoprotein (AFP) in the blood. |
| Ataxia with oculomotor apraxia type 1 | – | DNA‐adenylate hydrolase | SSB (DNA single‐strand Break) | Ataxia, oculomotor apraxia and peripheral vision. (Clements et al., 2004) |
| Ataxia with oculomotor apraxia type 2 | – | DNA–RNA helicase | SSB | Ataxia, oculomotor apraxia and peripheral vision. High amounts of a protein AFP in blood. (Clements et al., 2004) |
| Spinocerebellar ataxia with axonal neuropathy | – | Tyrosyl phosphodieaterase involved in SSB repair | SSB | Spinocerebellar ataxia with axonal neuropathy.El‐Khamisy et al., 2005) |
| RIDDLE syndrome | – | Ubiquitination | DSB | Microencephaly, facial dysmorphism, telangeiectasia, pulmonary fibrosis, learning difficulties and ataxia. (Stewart et al., 2009) |
Pathophysiology
Oxidative damage in XP
Oxidative stress and cumulative oxidative DNA damage in neurons are the primary causes of neurodegeneration (Hayashi, 2009; Niedernhofer et al., 2011). Neurons have a high metabolic load and are thus sensitive to alterations in energy metabolism (Rothe et al., 1993). High oxygen consumption leads to greater generation of ROS (Hayashi, 2009). Endogenous genotoxic processes, such as defective oxidative cellular metabolism and ROS generation, can alter cell integrity as well as result in many different types of oxidative DNA damage. Most of this damage, such as single‐strand breaks and oxidized purines and pyrimidines, is repaired by processes such as base excision repair that are not deficient in XP. However, as described above, certain types of oxidative damage such as cyclopurines can only be repaired by NER and so are thought to accumulate in XP (Brooks et al., 2000; Kraemer et al., 2007; Brooks, 2008). This unrepaired oxidative DNA damage accumulates over time in terminally differentiated post‐mitotic cells such as neurons and has deleterious effects on transcription and apoptosis regulation, resulting in neurodegeneration.
Silencing the genes that produce the NER proteins, CSA, CSB, XPA and XPC, alters redox homeostasis by increasing ROS levels, affecting oxidative phosphorylation (OXPHOS) and cell energy metabolism through oxidative damage to the electron transport chain (ETC) subunits and membrane phospholipids (Parlanti et al., 2015; Brennan‐Minnella et al., 2016). This leads to a further increase in oxidative stress (Kowaltowski and Vercesi, 1999). XPC down‐regulation also resulted in an increase in oxidative nuclear and mitochondrial DNA (mtDNA) damage, impairing OXPHOS (Pascucci et al., 2011). However, mtDNA lacks NER, and damage is corrected primarily by base excision repair (Wilson and Bohr, 2007; Boesch et al., 2011).
The absence of NER proteins from mitochondria suggests that mitochondrial abnormalities are secondary to nuclear disruptions and the resultant defective signalling pathways (Fang et al., 2014). In addition, the clinical heterogeneity of XP indicates that there are pathological processes occurring beyond the inefficient repair of helix‐distorting DNA lesions. Therefore, non‐DNA repair‐related oxidative stress could be involved in the pathogenesis of cancer and neurodegeneration in XP. It may be involved in many different aspects, causing, and being caused by, many interconnected pathogenic processes, the direction of which is difficult to determine.
Mitochondrial dysfunction in XP
Mitochondrial pathophysiology is strictly linked to oxidative stress, as free radicals are normally produced during respiration. Energy production is driven by the activity of the ETC within the mitochondria, which generates a proton gradient across the mitochondrial membranes, called the mitochondrial membrane potential (∆Ψm). The maintenance of ∆Ψm is necessary for functional ETC complexes and normal OXPHOS (Droge, 2002). Changes in ∆Ψm, such as hyperpolarization or depolarization, are considered pathological because they underlie defects within the ETC. The health of mitochondria is, in fact, pivotal to cellular physiology, and in particular, OXPHOS is critical for cell survival and fundamental for aerobic cell life (Chretien and Rustin, 2003).
The role of mitochondria and oxidative stress in ageing, neurodegeneration and cancer is well established (DiMauro and Schon, 2003; Plun‐Favreau et al., 2010). ROS are generated in normal cell metabolism with important roles in cell signalling for metabolism and growth (Jezek and Hlavata, 2005; Valko et al., 2007) and are therefore tightly regulated. Increased ROS levels are associated with altered energy states in the ETC (Jezek and Hlavata, 2005). ETC dysfunction allows more electron leakage and increases ROS production, which is detrimental to the cell (Koopman et al., 2010). ROS can also induce apoptosis directly via death‐receptor activation and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1624 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619 (Kulms et al., 2002), but ROS‐induced oxidative stress is likely the most significant contributor to cell death (Chretien and Rustin, 2003). Moreover, when ROS generation is not efficiently counteracted by the endogenous antioxidant systems, it increases and subsequently leads to deleterious effects on DNA, lipids and proteins (Cooke et al., 2003; Hayashi, 2009).
Antioxidants, such as glutathione and coenzyme Q10 (CoQ10), and detoxification enzymes, such as catalase, glutathione peroxidase and SOD, neutralize ROS and represent the primary protection against oxidative stress (Barrientos et al., 2009).
Mechanisms of mitochondrial dysfunction in XP are still a matter for debate. High and prolonged levels of ROS generation have been reported in XP‐A, XP‐D (Arbault et al., 2004; Arczewska et al., 2013; Parlanti et al., 2015) and XP‐C patient cells (Frechet et al., 2008). Additionally, XP‐patient cells show remarkably low levels of antioxidants (Nishigori et al., 1989; Vuillaume et al., 1992).
Mitochondrial dysfunction in CS‐B patients
Although the mitochondrial defect has likewise been considered secondary in CS models, it has been demonstrated that the CS‐B protein localizes to the mitochondria, suggesting a potential role of this protein in mtDNA repair (Arnold et al., 2012). CS‐B‐deficient cells showed an increased mitochondrial content, increasing the ∆Ψm and free radicals, and increased oxygen consumption (Osenbroch et al., 2009; Cleaver et al., 2014). However, these changes did not seem to be related to an increased mitochondrial biogenesis as the transcription factors PGC‐1α, TFAM and ERRα (the mitochondrial transcription factors related to mitochondrial biogenesis) were not altered in CS‐B‐deficient cells. As the amount of mitochondria is dependent on biogenesis and degradation, Scheibye‐Knudsen et al. (2012) investigated a probable inhibition of autophagy. Interestingly, they found a decreased co‐localization of LC3, P62 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4789 in response to stress in CS‐B‐deficient cells, resulting in autophagy inhibition thereby explaining the mitochondrial phenotype. The authors were also able to reverse the phenotype by treating these cells with http://www.guidetopharmacology.org/GRAC/LigandTextSearchForward?searchWildcard=rapamycin&order=rank&submitWildcard=Do+wildcard+search, stimulating autophagy (this will be discussed further below in the text). Moreover, rapamycin seems to be neuroprotective and could potentially attenuate the neurological symptoms in this disease (Bove et al., 2011; Dello et al., 2013). This is compatible with the finding that XPA‐deficient cells harbour impaired autophagy, leading to increased mitochondrial content, which could contribute to the neurodegenerative phenotype observed in these patients (Fang et al., 2014).
Potential pharmacological approaches
Antioxidant therapy with CoQ10
The available pharmacological therapy for neurological symptoms in XP patients is limited to symptomatic treatment. As it has been demonstrated that oxidative stress increases and mitochondrial efficiency decreases with age (Bohr et al., 1998; Muller et al., 2007), CoQ10 was investigated as a potential therapeutic option. However, these changes cannot be explained by alterations in CoQ10 levels as these appear to be stable over time in both control and disease populations (Duncan and Heales, 2005). Preliminary data from our XP cohort of patients (XPA, XPD, XPF and XPG) with variable neurological phenotype showed a trend towards a decreased level of CoQ10 concentrations with age in mononuclear cells (MNCs) from XP patients (Giunti, personal communication), although the lower levels were still within the normal range. This may suggest a possible decline along with age though not with the severity of the phenotype. This differs with data from Tanaka et al. (1998), reporting a pathologically low CoQ10 level in plasma that correlated with disease progression (Tanaka et al., 1998). However, the neurological phenotype, in Tanaka et al., was severe and the age of the patients was within a range of 3 to 25 years, which appears notably younger than that in our cohort (mean: 34 years, range 5–46 years). For all this, we can explain the difference in the results achieved by the two studies. Additionally, the CoQ10 levels were measured in MNCs and plasma using two different assays.
Interestingly, a decline of CoQ10 with age was not observed in XP plasma samples of all complementation groups. However, by measuring the CoQ10 concentration in fibroblasts from two different complementation groups, XPC (prone to cancer) and XPD (severe neuropathology), we found that levels in XPC fibroblasts were similar to controls, while XPD fibroblasts had a significantly lower concentration. This raises the possibility that CoQ10 supplementation may be beneficial in XP complementation groups prone to neurodegeneration. Although treatment of CoQ10 deficiency and ETC disorders with CoQ10 supplementation is difficult owing to the insolubility of CoQ10 (Hargreaves, 2014), the above‐mentioned non‐randomized study suggested that an oral dose of 0.9–1.5 mg·kg−1 daily improves daily activity in a subset of XP patients (Tanaka et al., 1998). As information about complementation groups was not provided by Tanaka et al., it is not clear whether this subgroup consisted primarily of patients with neurological involvement.
A trial of CoQ10 (180 mg·day−1) in one XPF patient from our cohort was initiated due to constant fatigue but did not have a beneficial effect on this symptom or the Scale for the Assessment and Rating of Ataxia rating scale over the course of 3 years (Giunti, personal communication). Randomized controlled clinical trials are needed to evaluate the efficacy of CoQ10 supplementation in XP.
Autophagy stimulation therapy with rapamycin
An emerging therapy to counteract neurodegeneration is the up‐regulation of autophagy. This is a physiological process responsible for the removal of misfolded protein aggregates and cellular organelles helping to maintain cellular homeostasis and integrity (Mizushima and Komatsu, 2011). Autophagy is a dynamic recycling system that seems to be down‐regulated in neurodegeneration in general and in particular in CS and XPA (Scheibye‐Knudsen et al., 2012; Fang et al., 2014). Rapamycin is used to activate autophagy through the selective inhibition of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2109, and although this can inactivate cell proliferation and survival, it does not affect neurons, as rapamycin showed to be beneficial in neurodegeneration. For example, in Alzheimer's disease (AD), the most common neurodegenerative disorder, the stimulation of autophagy through rapamycin was associated with up‐regulation of synapsin I, synapthopysin and postsynaptic protein 95 (Anttinen et al., 2008; Singh et al., 2017). These proteins are down‐regulated in AD and crucial for the maintenance of synaptic integrity. Moreover, oxidative stress, a marker for AD, was also attenuated. Above all, rapamicyin is currently in phase II clinical trials for analogous but different neurodegenerative disorders such as amyotrophic lateral sclerosis and Huntington disease.
Neurite development therapy with amitriptyline
Another possible strategy is the use of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=200, a tricyclic antidepressant, which is licenced for the treatment of depression and neuropathic pain. As the neuropathophysiology of CS‐B is characterized by abnormal neuronal development (unlike XP neurons that undergo normal development but degenerate later in life), Wang et al. (2016) attempted to rectify this by using amitriptyline to promote neurite development in cellular models of CS‐B. Further to this, they demonstrated that by up‐regulating the usually inhibited cascade involving synaptotagmin‐9 (SYT‐9), neurite proliferation was restored (Wang et al., 2016). Moreover, amitriptyline was one of the pharmacological agents that up‐regulated the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1818 and increased neurite growth (Wang et al., 2016). One of the key mediators of aberrant neuronal development in CS‐B is SYT‐9, which is down‐regulated in knock down CS‐B neurons impeding the formation of neurites (Wang et al., 2016). The SYT family is a group of proteins which regulates membrane trafficking and fusion (Dean et al., 2012). In particular SYT‐1, ‐2 and ‐9 are calcium sensors on synaptic vesicles and play a major role in synaptic vesicles membrane fusion events (Yoshihara and Montana, 2004). By up‐regulating SYT‐9 in CS‐B models, neurite proliferation was recovered. This was corroborated by pharmacological experiments using amitriptyline, which effectively up‐regulates TrkB (Wang et al., 2016). This effect is also mimicked by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4872, which is unstable in cultures and degrades quickly compared to amitriptyline. However, in addition to the beneficial effect of this compound on neurite growth, amitriptyline appears to induce mitochondrial fragmentation in neuronal models of Parkinson's disease (Lee et al., 2015). This effect would need to be carefully weighed against possible benefits and could possibly be counteracted by the addition of antioxidants in the therapeutic regime.
Conclusions
In conclusion, although no pharmacological therapies for neurological symptoms in XP are yet available, we have, here, discussed possible avenues that are being investigated to ameliorate these symptoms. We highlighted the possible role for antioxidant therapy with CoQ10 in attenuating the oxidative stress generated by mitochondrial dysfunction, which occurs secondarily to NER deficiency. Furthermore, we raised the possibility of re‐activating the autophagic machinery that is down‐regulated in CS and XPA, with rapamycin, and finally, to restore synaptic contacts by triggering neurite growth with amitriptyline (Figure 1).
Figure 1.
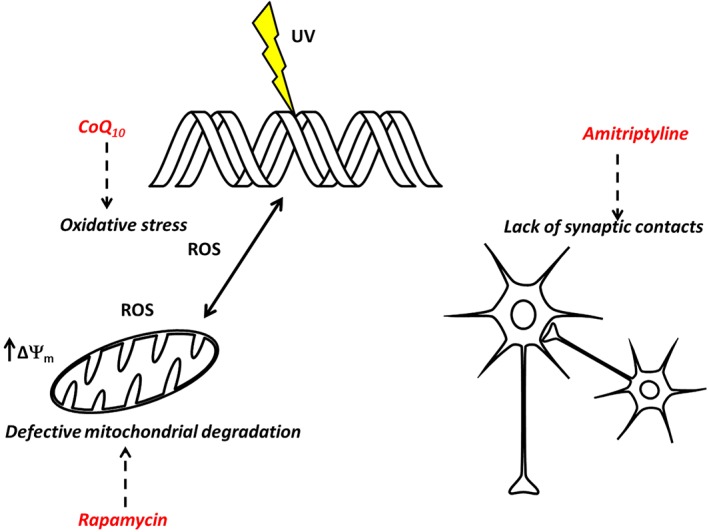
Therapeutic targets. DNA damage elicits an oxidative stress reaction with a positive feedback which contributes to an even more extensive damage of the DNA. Oxidative stress generated by mitochondria induces hyperpolarization and defective degradation (CoQ10 and rapamycin could counteract these effects). At the same time in neurons, synaptic contacts are lost (amitriptyline increases synaptic clefts).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Conflict of interest
The authors declare no conflicts of interest.
Abeti, R. , Zeitlberger, A. , Peelo, C. , Fassihi, H. , Sarkany, R. P. E. , Lehmann, A. R. , and Giunti, P. (2019) Xeroderma pigmentosum: overview of pharmacology and novel therapeutic strategies for neurological symptoms. British Journal of Pharmacology, 176, 4293–4301. 10.1111/bph.14557.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttinen A, Koulu L, Nikoskelainen E, Portin R, Kurki T, Erkinjuntti M et al (2008). Neurological symptoms and natural course of xeroderma pigmentosum. Brain 131: 1979–1989. [DOI] [PubMed] [Google Scholar]
- Arbault S, Sojic N, Bruce D, Amatore C, Sarasin A, Vuillaume M (2004). Oxidative stress in cancer prone xeroderma pigmentosum fibroblasts. Real‐time and single cell monitoring of superoxide and nitric oxide production with microelectrodes. Carcinogenesis 25: 509–515. [DOI] [PubMed] [Google Scholar]
- Arczewska KD, Tomazella GG, Lindvall JM, Kassahun H, Maglioni S, Torgovnick A et al (2013). Active transcriptomic and proteomic reprogramming in the C. elegans nucleotide excision repair mutant XPA‐1. Nucleic Acids Res 41: 5368–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Smidansky ED, Moustafa IM, Cameron CE (2012). Human mitochondrial RNA polymerase: structure‐function, mechanism and inhibition. Biochim Biophys Acta 1819: 948–960. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Fontanesi F, Diaz F (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet Chapter 19, Unit19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JO (2016). Xeroderma pigmentosum. Head Neck Pathol 10: 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesch P, Weber‐Lotfi F, Ibrahim N, Tarasenko V, Cosset A, Paulus F et al (2011). DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim Biophys Acta 1813: 186–200. [DOI] [PubMed] [Google Scholar]
- Bootsma D, Hoeijmakers JH (1991). The genetic basis of xeroderma pigmentosum. Ann Genet 34: 143–150. [PubMed] [Google Scholar]
- Bowden NA, Beveridge NJ, Ashton KA, Baines KJ, Scott RJ (2015). Understanding xeroderma pigmentosum complementation groups using gene expression profiling after UV‐light exposure. Int J Mol Sci 16: 15985–15996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J et al (2011). Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow‐up characterises the role of DNA repair. J Med Genet 48: 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan‐Minnella AM, Arron ST, Chou KM, Cunningham E, Cleaver JE (2016). Sources and consequences of oxidative damage from mitochondria and neurotransmitter signaling. Environ Mol Mutagen 57: 322–330. [DOI] [PubMed] [Google Scholar]
- Bohr V, Anson RM, Mazur S, Dianov G (1998). Oxidative DNA damage processing and changes with aging. Toxicol Lett 102‐103: 47–52. [DOI] [PubMed] [Google Scholar]
- Brooks PJ (2008). The 8,5′‐cyclopurine‐2′‐deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair (Amst) 7: 1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ (2017). The cyclopurine deoxynucleosides: DNA repair, biological effects, mechanistic insights, and unanswered questions. Free Radic Biol Med 107: 90–100. [DOI] [PubMed] [Google Scholar]
- Brooks BP, Thompson AH, Bishop RJ, Clayton JA, Chan CC, Tsilou ET et al (2013). Ocular manifestations of xeroderma pigmentosum: long‐term follow‐up highlights the role of DNA repair in protection from sun damage. Ophthalmology 120: 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL et al (2000). The oxidative DNA lesion 8,5′‐(S)‐cyclo‐2′‐deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem 275: 22355–22362. [DOI] [PubMed] [Google Scholar]
- Bove J, Martinez‐Vicente M, Vila M (2011). Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 12: 437–452. [DOI] [PubMed] [Google Scholar]
- Chretien D, Rustin P (2003). Mitochondrial oxidative phosphorylation: pitfalls and tips in measuring and interpreting enzyme activities. J Inherit Metab Dis 26: 189–198. [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Brennan‐Minnella AM, Swanson RA, Fong KW, Chen J, Chou KM et al (2014). Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc Natl Acad Sci U SA 111: 13487–13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P et al (2004). The ataxia‐oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst) 3: 1493–1502. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003). Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17: 1195–1214. [DOI] [PubMed] [Google Scholar]
- Dello RC, Lisi L, Feinstein DL, Navarra P (2013). mTOR kinase, a key player in the regulation of glial functions: relevance for the therapy of multiple sclerosis. Glia 61: 301–311. [DOI] [PubMed] [Google Scholar]
- Dean C, Dunning FM, Liu H, Bomba‐Warczak E, Martens H, Bharat V et al (2012). Axonal and dendritic synaptotagmin isoforms revealed by a pHluorin‐syt functional screen. Mol Biol Cell 23: 1715–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna JJ, Kraemer KH (2012). Shining a light on xeroderma pigmentosum. J Invest Dermatol 132: 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA (2003). Mitochondrial respiratory‐chain diseases. N Engl J Med 348: 2656–2668. [DOI] [PubMed] [Google Scholar]
- Droge W (2002). Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95. [DOI] [PubMed] [Google Scholar]
- Duncan AJ, Heales SJ (2005). Nitric oxide and neurological disorders. Mol Aspects Med 26: 67–96. [DOI] [PubMed] [Google Scholar]
- El‐Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR et al (2005). Defective DNA single‐strand break repair in spinocerebellar ataxia with axonal neuropathy‐1. Nature 434: 108–113. [DOI] [PubMed] [Google Scholar]
- Fang EF, Scheibye‐Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H et al (2014). Defective mitophagy in XPA via PARP‐1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157: 882–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S et al (2016). Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A 113: E1236–E1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frechet M, Warrick E, Vioux C, Chevallier O, Spatz A, Benhamou S et al (2008). Overexpression of matrix metalloproteinase 1 in dermal fibroblasts from DNA repair‐deficient/cancer‐prone xeroderma pigmentosum group C patients. Oncogene 27: 5223–5232. [DOI] [PubMed] [Google Scholar]
- Gilmore EC (2014). DNA repair abnormalities leading to ataxia: shared neurological phenotypes and risk factors. Neurogenetics 15: 217–228. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves IP (2014). Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol 49: 105–111. [DOI] [PubMed] [Google Scholar]
- Hayashi M (2009). Oxidative stress in developmental brain disorders. Neuropathology 29: 1–8. [DOI] [PubMed] [Google Scholar]
- Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, Macphee DG et al (2006). Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res 601: 171–178. [DOI] [PubMed] [Google Scholar]
- Jezek P, Hlavata L (2005). Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol 37: 2478–2503. [DOI] [PubMed] [Google Scholar]
- Kamenisch Y, Fousteri M, Knoch J, von Thaler AK, Fehrenbacher B, Kato H et al (2010). Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med 207: 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karass M, Naguib MM, Elawabdeh N, Cundiff CA, Thomason J, Steelman CK et al (2015). Xeroderma pigmentosa: three new cases with an in depth review of the genetic and clinical characteristics of the disease. Fetal Pediatr Pathol 34: 120–127. [DOI] [PubMed] [Google Scholar]
- Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A et al (2008). Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 7: 744–750. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA et al (2010). Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal 12: 1431–1470. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Vercesi AE (1999). Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med 26: 463–471. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ (2007). Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype‐phenotype relationship. Neuroscience 145: 1388–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulms D, Zeise E, Poppelmann B, Schwarz T (2002). DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation‐induced apoptosis in an essential and independent way. Oncogene 21: 5844–5851. [DOI] [PubMed] [Google Scholar]
- Kuraoka, I. Bender, A. Romieu, J. Cadet, R.D. Wood, T. (2000). Removal of oxygen free‐radical‐induced 5′,8‐purine cyclodeoxynucleosides from DNA by the nucleotide excision‐repair pathway in human cells. Proc Natl Acad Sci USA, 3832–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert WC, Lambert MW (2015). Development of effective skin cancer treatment and prevention in xeroderma pigmentosum. Photochem Photobiol 91: 475–483. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Dubourg O, Benoist JF, Jardel C, Mochel F, Koenig M et al (2007). Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology 68: 295–297. [DOI] [PubMed] [Google Scholar]
- Lee MY, Hong S, Kim N, Shin KS, Kang SJ (2015). Tricyclic antidepressants amitriptyline and desipramine induced neurotoxicity associated with Parkinson's disease. Mol Cells 38: 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, Kirk‐Bell S, Arlett CF, Paterson MC, Lohman PH, de Weerd‐Kastelein EA et al (1975). Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV‐irradiation. Proc Natl Acad Sci U S A 72: 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, McGibbon D, Stefanini M (2011). Xeroderma pigmentosum. Orphanet J Rare Dis 6: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra AK, Gupta S, Khaitan BK, Verma KK (2008). Multiple basal cell carcinomas in xeroderma pigmentosum treated with imiquimod 5% cream. Pediatr Dermatol 25: 488–491. [DOI] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M et al (1999). The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399: 700–704. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147: 728–741. [DOI] [PubMed] [Google Scholar]
- Moriwaki S, Kanda F, Hayashi M, Yamashita D, Sakai Y, Nishigori C (2017). Xeroderma pigmentosum clinical practice guidelines. J Dermatol 44: 1087–1096. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A et al (2007). Denervation‐induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293: R1159–R1168. [DOI] [PubMed] [Google Scholar]
- Naik SM, Shenoy AM, Nanjundappa A, Halkud R, Chavan P, Sidappa K et al (2013). Cutaneous malignancies in xeroderma pigmentosum: earlier management improves survival. Indian J Otolaryngol Head Neck Surg 65: 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale V, Raquer H (2017). Xeroderma pigmentosum‐Cockayne syndrome complex. Orphanet J Rare Dis 12: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ (2008). Tissue‐specific accelerated aging in nucleotide excision repair deficiency. Mech Ageing Dev 129: 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Bohr VA, Sander M, Kraemer KH (2011). Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: molecules to patients. Mech Ageing Dev 132: 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigori C, Miyachi Y, Imamura S, Takebe H (1989). Reduced superoxide dismutase activity in xeroderma pigmentosum fibroblasts. J Invest Dermatol 93: 506–510. [DOI] [PubMed] [Google Scholar]
- Osenbroch PØ, Auk‐Emblem P, Halsne R, Strand J, Forstrøm RJ, van der Pluijm I et al (2009). Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J 276: 2811–2821. [DOI] [PubMed] [Google Scholar]
- Parlanti E, Pietraforte D, Iorio E, Visentin S, De Nuccio C, Zijno A et al (2015). An altered redox balance and increased genetic instability characterize primary fibroblasts derived from xeroderma pigmentosum group A patients. Mutat Res 782: 34–43. [DOI] [PubMed] [Google Scholar]
- Pascucci B, D'Errico M, Parlanti E, Giovannini S, Dogliotti E (2011). Role of nucleotide excision repair proteins in oxidative DNA damage repair: an updating. Biochemistry (Mosc) 76: 4–15. [DOI] [PubMed] [Google Scholar]
- Plun‐Favreau H, Lewis PA, Hardy J, Martins LM, Wood NW (2010). Cancer and neurodegeneration: between the devil and the deep blue sea. PLoS Genet 6: e1001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC (2007). Defective DNA repair and neurodegenerative disease. Cell 130: 991–1004. [DOI] [PubMed] [Google Scholar]
- Rapin I, Weidenheim K, Lindenbaum Y, Rosenbaum P, Merchant SN, Krishna S et al (2006). Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J Child Neurol 21: 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M, Werner D, Thielmann HW (1993). Enhanced expression of mitochondrial genes in xeroderma pigmentosum fibroblast strains from various complementation groups. J Cancer Res Clin Oncol 119: 675–684. [DOI] [PubMed] [Google Scholar]
- Scheibye‐Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK et al (2012). Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med 209: 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye‐Knudsen M, Scheibye‐Alsing K, Canugovi C, Croteau DL, Bohr VA (2013). A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging (Albany NY) 5: 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi M, Lehmann AR, Fawcett H, Stefanini M, Jaspers N, Mullard K et al (2013). Patients with xeroderma pigmentosum complementation groups C, E and V do not have abnormal sunburn reactions. Br J Dermatol 169: 1279–1287. [DOI] [PubMed] [Google Scholar]
- Singh AK, Kashyap MP, Tripathi VK, Singh S, Garg G, Rizvi SI (2017). Neuroprotection through rapamycin‐induced activation of autophagy and PI3K/Akt1/mTOR/CREB signaling against amyloid‐beta‐induced oxidative stress, synaptic/neurotransmission dysfunction, and neurodegeneration in adult rats. Mol Neurobiol 54: 5815–5828. [DOI] [PubMed] [Google Scholar]
- Spivak G (2004). The many faces of Cockayne syndrome. Proc Natl Acad Sci U S A 101: 15273–15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al‐Hakim AK, Kolas NK, Miller ES (2009). The RIDDLE syndrome protein mediates a ubiquitin‐dependent signaling cascade at sites of DNA damage. Cell 136: 420–434. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44–84. [DOI] [PubMed] [Google Scholar]
- Vuillaume M, Daya‐Grosjean L, Vincens P, Pennetier JL, Tarroux P, Baret A et al (1992). Striking differences in cellular catalase activity between two DNA repair‐deficient diseases: xeroderma pigmentosum and trichothiodystrophy. Carcinogenesis 13: 321–328. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jones‐Tabah J, Chakravarty P, Stewart A, Muotri A, Laposa RR et al (2016). Pharmacological bypass of Cockayne syndrome B function in neuronal differentiation. Cell Rep 14: 2554–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chakravarty P, Ranes M, Kelly G, Brooks PJ, Neilan E et al (2014). Dysregulation of gene expression as a cause of Cockayne syndrome neurological disease. Proc Natl Acad Sci U S A 111: 14454–14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BT, Stark Z, Sutton RE, Danda S, Ekbote AV, Elsayed SM et al (2016). The Cockayne Syndrome Natural History (CoSyNH) study: clinical findings in 102 individuals and recommendations for care. Genet Med 18: 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura D, DiGiovanna JJ, Khan SG, Kraemer KH (2014). Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Photodermatol Photoimmunol Photomed 30: 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Nagai T, Okada S (1998). Serum concentration of coenzyme Q in xeroderma pigmentosum. Rinsho Shinkeigaku 38: 57–59. [PubMed] [Google Scholar]
- Wilson DM III, Bohr VA (2007). The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst) 6: 544–559. [DOI] [PubMed] [Google Scholar]
- Yang JQ, Chen XY, Engle MY, Wang JY (2015). Multiple facial basal cell carcinomas in xeroderma pigmentosum treated with topical imiquimod 5% cream. Dermatol Ther 28: 243–247. [DOI] [PubMed] [Google Scholar]
- Yarosh D, Klein J, O'Connor A, Hawk J, Rafal E, Wolf P (2001). Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet 357: 926–929. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Montana ES (2004). The synaptotagmins: calcium sensors for vesicular trafficking. Neuroscientist 10: 566–574. [DOI] [PubMed] [Google Scholar]