

IL-33 mediates cross-talk between immune and stromal cells in localized fibrosis.
Abstract
Dissecting the molecular landscape of fibrotic disease, a major unmet need, will inform the development of novel treatment strategies to target disease progression and identify desperately needed therapeutic targets. Here, we provide a detailed single-cell analysis of the immune landscape in Dupuytren’s disease, a localized fibrotic condition of the hand, and identify a pathogenic signaling circuit between stromal and immune cells. We demonstrate M2 macrophages and mast cells as key cellular sources of tumor necrosis factor (TNF) that promotes myofibroblast development. TNF acts via the inducible TNFR2 receptor and stimulates interleukin-33 (IL-33) secretion by myofibroblasts. In turn, stromal cell IL-33 acts as a potent stimulus for TNF production from immune cells. Targeting this reciprocal signaling pathway represents a novel therapeutic strategy to inhibit the low-grade inflammation in fibrosis and the mechanism that drives chronicity.
INTRODUCTION
Fibrotic diseases represent a major unmet medical need in Western populations (1). Efforts to develop new therapeutics have been hampered by the fact that animal models fail to recapitulate many aspects of fibrotic diseases in humans and a limited access to human tissue, especially at early stages of the disorders (2). Localized fibrotic diseases represent a rich source of readily accessible early-stage tissue (2). Dupuytren’s disease (DD) is a common and progressive fibroproliferative disorder of the palmar and digital fascia of the hand and, in Western populations, affects 12% of those aged 55 years, increasing to 29% of those 75 years and older (3). The initial clinical presentation is the appearance of a firm nodule in the hand, which expands into fibrous collagenous cords that extend into the digits. As the disease progresses, the cords mature, thicken, and contract, leading to permanent flexion deformities of the fingers. Disease progression is highly variable, but approximately 20 to 40% of patients eventually develop some degree of flexion deformity that can impair hand function (4). These flexion contractures can severely limit daily activities, including self-care and employment, reducing health-related quality of life (5). There are no approved treatments for early-stage disease, and there is a high recurrence rate in late-stage disease following standard-of-care therapies, including fasciotomy and collagenase injections.
Chronic low-grade inflammation has been identified as the initiator and driver in the early stages of many fibrotic disorders (6). How inflammation arises in DD remains to be established, but as in other diseases, local production of chemokines is likely to be crucial for the recruitment of immune cells. Common to all fibrotic diseases, the key effector cell in DD is the myofibroblast, which is responsible for the deposition, remodeling, and contraction of the excessive matrix (1) that ultimately compromises hand function. We have previously demonstrated that inhibition of tumor necrosis factor (TNF) down-regulates the myofibroblast phenotype in DD (7), and following a phase 2a dose-ranging, proof-of-efficacy study (8), we are now conducting a phase 2b clinical trial to assess the effects of TNF inhibition by injecting adalimumab into nodules of patients with progressive early-stage disease (9). Although the safety profile of TNF inhibition is well known, it has potential drawbacks, including the reactivation of latent infections such as tuberculosis. TNF is a pleotropic cytokine that acts via two receptors, TNFR1 and TNFR2 (10). While TNFR1 is expressed by all cells, TNFR2 expression is inducible and typically considered to be restricted to cells of the lymphoid lineage (11). Signaling via TNFR1 drives most of the inflammatory responses, while TNFR2 has been associated with immunomodulatory, reparative, and profibrotic responses in murine models of renal and intestinal fibrosis (11–13). However, it is not clear how TNF signals to influence fibroblast behavior in human fibrotic disease, nor are the mechanisms underpinning the localized inflammation that leads to sustained tissue levels of TNF in DD. Understanding these mechanisms would permit a targeted therapeutic strategy and may, in turn, provide a more selective, disease-specific, means of inhibiting TNF-mediated myofibroblast activation.
Because DD develops over many years (14), we hypothesized that persistence of low-grade localized inflammation is due to the local expression of a mediator that perpetuates production of low-level TNF. There is clear evidence that alarmins play a key role in sustaining the inflammatory response in a variety of chronic diseases (15). The alarmin interleukin-33 (IL-33) can function as both a classical cytokine and a nuclear factor–regulating gene transcription (16) and is secreted by activated fibroblasts and myofibroblasts, and elevated IL-33 levels have been found in the serum of patients with liver cirrhosis (17). While typically associated with inflammation of epithelial barriers, we hypothesized that IL-33 may perpetuate the local production of TNF and thereby promote activation of myofibroblasts, leading to and/or sustaining fibrosis.
On the basis of clinical samples from patients with relatively early-stage DD, here, we show that TNF in DD is predominantly secreted by M2 macrophages and mast cells. Our results demonstrate that TNF signals via TNFR2 on stromal cells, triggering low-level production of IL-33. Subsequently, IL-33 signals via ST2 on local immune cells to perpetuate further TNF expression. We suggest that inhibiting TNFR2 may provide a novel therapeutic opportunity for selectively targeting myofibroblast activation, and combination therapy with IL-33 inhibition may offer a comprehensive strategy to restrict further development of disease, requiring less frequent dosing.
RESULTS
The inflammatory landscape in early-stage human fibrosis
Low-grade chronic inflammation is a characteristic of the early stages of many fibrotic diseases (6). To understand more about the inflammatory signaling networks in DD, we systematically examined cytokine levels in freshly disaggregated nodular cells. TNF (mean ± SEM, 50 ± 17.2 pg/ml) and IL-33 (mean ± SEM, 13 ± 4.1 pg/ml) were consistently expressed in nodules, along with high levels of IL-6 (mean ± SEM, 4140 ± 186.1 pg/ml) and transforming growth factor–β (TGF-β1) (mean ± SEM, 244 ± 124.1 pg/ml) (Fig. 1A). Cytokine expression in the disease tissue was not associated with a concomitant increase in circulating levels of proinflammatory cytokines TNF, IL-6, and IL-1β and the chemokine CXCL8 compared to samples from age-matched controls (fig. S1), confirming that inflammation is localized to the diseased nodules. To determine which cytokines were being secreted by the immune cells, we performed detailed phenotyping of the immune cell compartment in Dupuytren’s nodules, which represent early-stage disease (18), using droplet-based single-cell RNA sequencing (RNA-seq) (Fig. 1, B and C, and fig. S2).
Fig. 1. Mast cells and M2 macrophages are the predominant source of TNF in DD.
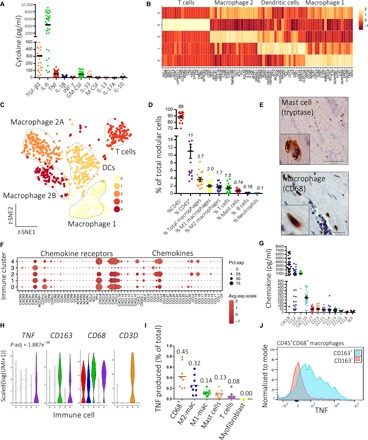
(A) Scatter plot of cytokines secreted by freshly isolated nodular cells in monolayer culture for 24 hours (n = 20 DD patients for TGF-β1 and 40 for all other cytokines; lines represent mean). (B) Heatmap of marker genes of four major immune cell types in single-cell RNA-seq data, displaying average row scaled expression in log(UMI + 1). (C) t-distributed stochastic neighbor embedding (t-SNE) embedding of single-cell RNA-seq of immune cells in Dupuytren’s nodules (n = 12 DD patients, k = 1033 cells). DCs, dendritic cells. (D) Scatter plot of flow cytometry analysis showing percentage of cell types as a proportion of the total population in Dupuytren’s nodules (n = 20 DD patients, line represents mean ± SEM). (E) Representative images of immunohistochemistry for macrophages (CD68) and mast cells (tryptase) in Dupuytren’s nodules. Scale bars, 10 μm. (F) Dot plot mapping genes encoding chemokines and chemokine receptors to immune cell clusters (0 to 4). Expression in scaled(log(UMI + 1)). Pct.exp., percentage of cells in cluster-expressing gene. (G) Scatter plot of chemokines secreted by freshly isolated nodular cells in monolayer culture for 24 hours (n = 40 DD patients; line represents mean). (H) Violin plots showing expression of genes in scaled(log(UMI + 1)) in immune cell clusters. P-adjust denotes adjusted P value (Benjamini-Hochberg correction) for TNF following differential expression analysis (Wilcoxon rank sum test) between macrophage 2B (cluster 4) and all other clusters. (I) Scatter plot of flow cytometry analysis showing TNF expression by nodular cells as a percentage of total TNF produced. Data points are independent DD patients. Lines represent mean (n = 9 DD patients). (J) Representative histograms of flow cytometry analysis showing TNF expression in macrophage populations (n = 5 DD patients).
Immune cells (k = 1033 cells) from nodules were partitioned using unsupervised clustering into five major clusters (Fig. 1, B and C). This revealed a complex immune landscape, with predominant cell types being macrophages, T cells, and dendritic cells. The five immune cell clusters showed three macrophage-rich clusters (CD68high), one T cell cluster (IL7R+CD3D+), and one cluster enriched for dendritic cells (FCER1A+CD1C+CLEC10A+) (Fig. 1, B and C, and fig. S2F). Using flow cytometry, we validated a CD1C+ immune population in nodular cells (fig. S2F). Within the macrophage population, we noted that two clusters (clusters 2 and 4) exhibited a transcriptomic signature suggestive of an M2 macrophage phenotype, with high expression of CD163, CCL18, and STAB1 (fig. S2, G to I). We termed these clusters macrophage 2A and 2B (Fig. 1C). In contrast, one macrophage cluster (cluster 0) displayed a transcriptomic profile of M1 macrophages, with high expression of IL1B, CCR7, and KYNU, and we term this population macrophage 1 (Fig. 1, B and C, and fig. S2, G to I) (19, 20).
To complement this, we undertook broad phenotyping of freshly dissociated cells from nodules using flow cytometry (Fig. 1D and fig. S3, A to C). This showed that most of the cells were CD45− stromal cells (~89%), with a smaller number of CD45+ immune cells (~11%). The major immune cells were macrophages (3.7%), with a similar proportion of classically activated M1 (2.0%) and alternatively activated M2 (1.7%) phenotypes, as identified by CD68+CD163low and CD68+CD163high labeling, respectively. In addition, nodules contained a small number of T cells (1.5%) and mast cells (0.74%) (Fig. 1D and fig. S2J). A number of the rarer immune cells (basophils, B cells, and neutrophils) were not identified in our single-cell RNA-seq dataset, and we identified only a few mast cells (fig. S2J). This may be due to their low overall number in the immune cells sampled or bias introduced during tissue digestion. We confirmed the distribution of macrophages and mast cells in nodules by immunohistochemistry (Fig. 1E and fig. S3D). Our single-cell data revealed a diverse range of chemokines expressed throughout the immune cell clusters (Fig. 1F). In accordance with these findings, freshly disaggregated nodular cells secreted a wide range of chemokines, including high levels of CXCL8 and CCL11, known chemokines for mast cells (21), while elevated CCL2, CCL4, and CXCL8 are consistent with the presence of macrophages in DD tissue (Fig. 1G) (22).
We have previously shown that TNF selectively leads to the differentiation of myofibroblast precursors in DD via the Wnt signaling pathway (7) and the efficacy of inhibiting TNF in a phase 2a clinical trial (12). In contrast, TGF-β1 induced myofibroblast differentiation indiscriminately in DD and control fibroblasts (7). Hence, we sought to define the source of TNF in DD. Our single-cell transcriptomic data showed that TNF was most highly expressed in macrophage 2 and was a significant marker (P = 1.89 × 10−8) of this population (Fig. 1H and figs. S2H and S3E). Using flow cytometry, we confirmed this by showing that resident M2 macrophages (~32%) and mast cells (~13%) were the two highest sources of TNF, together contributing to ~45% of the total TNF produced (Fig. 1, I and J, and fig. S3, F to H). In contrast, stromal cells in DD nodules exhibited far lower mRNA and protein expression of TNF (fig. S3, I and J).
Stromal cell receptors and immune-mediated cytokine synthesis
Next, we sought to identify the receptors through which TNF was signaling. Immunohistochemistry staining confirmed widespread expression of TNFR1 throughout Dupuytren’s nodules (Fig. 2A). Surprisingly, TNFR2, which is usually associated with immune cells, in particular T helper cells (11), was also expressed by most of the cells in the nodules (Fig. 2B). We did not observe TNFR1 and TNFR2 protein expression in Dupuytren’s cords, which represent a relatively acellular later-stage matrix-rich fibrotic structure (fig. S4A). After this, we confirmed our previous findings that TNF is crucial in promoting the differentiation of myofibroblast precursors. We treated myofibroblast precursors, palmar dermal fibroblasts from patients with DD (PF-D), with recombinant TNF (rTNF) and noted an increased expression of key profibrotic genes, COL1A1 and ACTA2, with corresponding increase in smooth muscle actin (α-SMA) protein (fig. S4, B and C). Extending these observations, we found that only myofibroblast precursors, PF-D, and not genetically matched nonpalmar dermal fibroblasts (NPF-D) from the same patients, showed enhanced protein and gene expression of TNFR2 following stimulation with low levels of exogenous TNF, similar to those secreted by freshly disaggregated nodular cells (Fig. 2, C and D). The enhanced TNFR2 expression on TNF stimulation of palmar fibroblasts from patients with DD and by myofibroblasts was confirmed using immunofluorescence staining (fig. S4D). Together, these results demonstrate elevated TNFR2 expression by Dupuytren’s myofibroblasts and their precursors on exposure to low-dose TNF. Last, as transmembrane TNF (tmTNF) has been shown to preferentially bind TNFR2, we measured protein expression of tmTNF in freshly dissagregated nodular cells using flow cytometry (fig. S4E). This demonstrated tmTNF expression by DD nodular cells, with a higher expression by immune cells (CD45+) as compared to stromal cells (CD45−). This finding correlates with the pattern of total TNF expression by nodular cells, which also shows a higher expression by immune cells (fig. S3, I and J).
Fig. 2. Stromal cell receptors and immune-mediated cytokine synthesis.
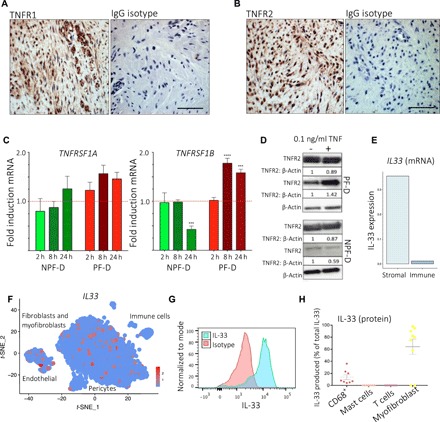
(A and B) Representative images of immunohistochemistry for TNFR1 (A) and TNFR2 (B) expression in Dupuytren’s nodule. Scale bars, 10 μm. IgG is isotype control (rabbit). (C) Bar plots showing TNFRSF1A and TNFRSF1B gene expression by palmar (PF-D) and nonpalmar (NPF-D) dermal fibroblasts from DD patients following stimulation with rTNF (0.1 ng/ml) (n = 3 DD patients, mean ± SEM). (D) Western blot analysis of TNFR1 and TNFR2 protein expression by PF-D and NPF-D from DD patients following stimulation with rTNF (0.1 ng/ml) for 24 hours. (E) Bar plot of IL33 expression (mean normalized counts) in stromal and immune cells in single-cell RNA-seq (n = 6 DD patients, k = 7332 cells). (F) t-SNE embedding of single-cell RNA-seq dataset marking IL33 expression in scaled(log(UMI + 1)). (G) Representative histogram of flow cytometry analysis of freshly isolated nodular cells showing IL-33 expression in CD45− stromal cells with isotype control. (H) Scatter plot of flow cytometry analysis showing IL-33 expression in freshly isolated nodular cells (n = 9 DD patients, mean ± SEM). ***P < 0.001, ****P < 0.0001.
IL-33 is the most recently described member of the IL-1 superfamily and has been implicated in allergy as well as playing a key role in fibrosis in various organs, including the lung, liver, and skin (16, 17, 23, 24). Specifically, IL-33 was shown to induce M2 macrophage polarization in murine models of lung fibrosis (25). However, most data pertaining to the role of IL-33 in fibrosis derive from animal models (2, 17, 23, 24), where it has been shown to induce resident stromal cell activation and increase extracellular matrix secretion (17). The role of IL-33 in human fibrotic diseases remains unclear. Within the single-cell RNA-seq data, we detected low IL33 gene expression in the immune compartment (Fig. 2E), with most of the IL33 expression localized to the fibroblast and myofibroblast clusters, in addition to the endothelial cells (Fig. 2F). To complement this, we used flow cytometry to profile IL-33 protein expression in nodular cells. We found that α-SMA+ myofibroblasts demonstrated strong expression of this cytokine, secreting ~60% of the total IL-33 in nodules (Fig. 2, G and H), and, in contrast to our gene expression data, detected a higher proportion of immune cells expressing IL-33 protein. This apparent discrepancy may relate to transcripts with low expression being below threshold for detection in the droplet-based single-cell RNA-seq. Following this, using immunohistochemistry, we confirmed the expression of IL-33 by Dupuytren’s myofibroblasts along with the expression of ST2, the canonical receptor for IL-33 (fig. S5, A and B). Moreover, immunohistochemistry staining confirmed the expression of IL-33 and ST2 throughout nodule tissue sections (fig. S5, A and B). Notably, flow cytometry and immunofluorescence showed high intracellular staining of IL-33 in stromal cells, while we detected low levels of soluble IL-33 secreted in freshly isolated nodular cells (Fig. 1A). One explanation for this finding may relate to the well-known intracellular localization of IL-33, in addition to its role as a secreted cytokine. Collectively, the protein and mRNA expression data established low expression of IL33 in immune cells (Fig. 2, E and F) and stromal cells as the major source of IL-33 in DD.
Stromal–immune cell cross-talk mediated by IL-33 and TNF
On the basis of accumulating evidence for the role of IL-33 in promoting inflammation in disorders involving epithelial barrier surfaces (16, 26), we hypothesized that it may play a similar role in DD. We investigated whether mast cells and M2 macrophages, the immune cells mainly responsible for TNF production in nodules, were responsive to IL-33 stimulation. Using three separate mast cell lines and human monocyte–derived macrophages, we observed a dose-dependent increase in TNF secretion in response to IL-33 in both mast cells and M2 macrophages primed with TNF (Fig. 3, A and B). In contrast, there was no effect on M0 or M1 macrophages (Fig. 3B). Recombinant human IL-33 (10 pg/ml) led to secretion of TNF (~50 to 100 pg/ml) by immune cells, both levels corresponding to the cytokine concentrations in the supernatant of freshly disaggregated Dupuytren’s nodular cells and also with the optimal concentration for promoting the differentiation of myofibroblast precursor (7). Last, we confirmed that both mast cells and macrophages from Dupuytren’s nodules expressed the ST2 receptor (Fig. 3C).
Fig. 3. Stromal–immune cell crosstalk mediated by IL-33 and TNF.
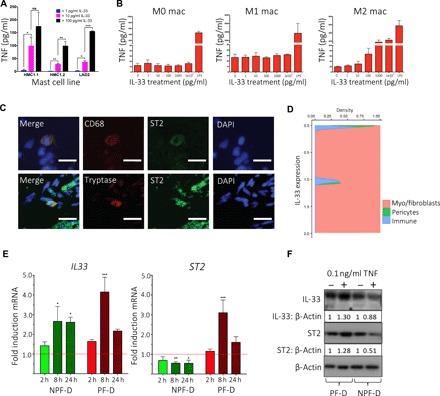
(A) Bar plot of enzyme-linked immunosorbent assay (ELISA) showing TNF secretion in mast cell lines (HMC1.1, HMC1.2, and LAD2) following stimulation with increasing doses of rIL-33 (n = 3, mean ± SEM). (B) Bar plots of ELISA showing TNF secretion by human monocytes and macrophages following stimulation with increasing doses of rIL-33 (n = 3 independent donors, mean ± SEM). (C) Representative confocal images of immunofluorescence for ST2 expression on mast cells (tryptase) and macrophages (CD68) in DD nodules. Scale bars, 10 μm. (D) Conditional density plot of single-cell RNA-seq dataset showing IL33 expression by labeled cell types. Density is Gaussian kernel density estimate (n = 6 DD patients, k = 7332 cells). (E) Bar plots showing IL33 and ST2 gene expression by PF-D and NPF-D dermal fibroblasts from DD patients following stimulation with rTNF at 2, 8, and 24 hours (n = 3 DD patients, mean ± SEM). (F) Western blot analysis showing IL-33 and ST2 protein expression by PF-D and nonpalmar NPF-D dermal fibroblasts from DD patients following stimulation with rTNF(0.1 ng/ml) for 24 hours. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
Reciprocal cytokine signaling is a feature of many chronic inflammatory diseases (27–30), and as IL-33 resulted in the expression of TNF (Fig. 3, A and B), we sought evidence for reciprocal regulation of IL-33 by TNF. As stromal cells are the major source of IL-33 (Fig. 4D), we treated myofibroblast precursors, palmar dermal fibroblasts from Dupuytren’s patients (PF-D), and control NPF-D from genetically matched patients with rTNF (0.1 ng/ml) over 24 hours and measured mRNA and protein expression of IL-33 and ST2 (Fig. 3, E and F). While NPF-D showed a small increase in mRNA for IL-33, the effect was greater in PF-D, and an increase in protein was only observed in the latter (Fig. 3, E and F). We confirmed this finding, with immunofluorescence staining showing enhanced expression of IL-33 by PF-D and DD myofibroblasts (fig. S5C). Both cell types also showed enhanced staining for ST2 on TNF stimulation, a result that was confirmed with quantitative immunofluorescence (fig. S4E).
Fig. 4. IL-33 and TNFR2 inhibition is a novel therapeutic avenue for DD.
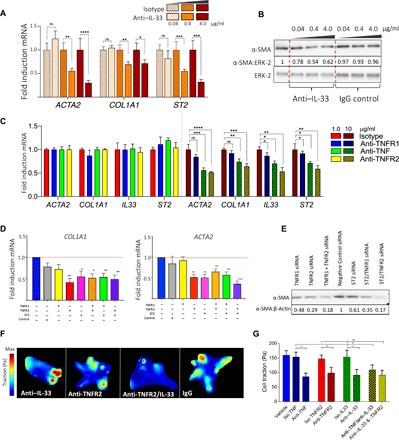
(A) Bar plot showing COL1A1, ACTA2, and ST2 gene expression by DD myofibroblasts treated with neutralizing anti–IL-33 antibody compared to isotype control (n = 8 DD patients, mean ± SEM). (B) Western blot analysis showing α-SMA protein expression by DD myofibroblasts following treatment with neutralizing anti–IL-33 antibody compared to isotype control. (C) Bar plot showing COL1A1, ACTA2, IL33, and ST2 gene expression by DD myofibroblasts following treatment with neutralizing anti-TNFR1, anti-TNFR2, and anti-TNF antibodies (n = 5 DD patients, mean ± SEM). (D) Bar plot showing COL1A1 and ACTA2 gene expression following siRNA-mediated knockdown for TNFRSF1A, TNFRSF1B, ST2, or combination treatment (n = 5 DD patients, mean ± SEM). (E) Western blot analysis of α-SMA protein expression by DD myofibroblasts following siRNA knockdown of TNFRSF1A, TNFRSF1B, and ST2. (F) Heatmaps of cellular traction force (in pascals) in DD myofibroblasts following treatment with indicated neutralizing antibodies. (G) Bar plots showing average traction force following treatment with anti-TNF, anti-TNFR2, anti–IL-33 antibodies, or combination therapy (n = 5 DD patients, mean ± SEM). Gene expression changes (mRNA fold induction) were calculated relative to GAPDH using the ΔΔ cycle threshold (ΔΔCt) method and normalized to isotype control at the corresponding concentration. *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001.
Inhibition of TNF and IL-33 signaling down-regulates key profibrotic genes in myofibroblasts
Next, we investigated the effect of IL-33 inhibition on the expression of key profibrotic genes in myofibroblasts compared to an isotype control at the corresponding concentration. We found that myofibroblasts exhibited reduced expression of α-SMA at both the mRNA and protein level and COL1A1 and ST2 mRNA in a dose-dependent manner (Fig. 4, A and B). Furthermore, treatment of myofibroblasts with a neutralizing TNFR2 antibody, but not anti-TNFR1, resulted in a dose-dependent inhibition of ACTA2, COL1A1, IL33, and ST2 at the mRNA level and α-SMA and ST2 at the protein level (Fig. 4C and fig. S6). The inhibitory effects of ST2 and TNFR2 down-regulation in myofibroblasts were confirmed using small interfering RNA (siRNA) knockdowns (Fig. 4, D and E, and fig. S7).
A key feature of myofibroblast phenotype is contraction of the matrix (31). Therefore, we investigated whether specific TNFR2 and IL-33 blockade could influence myofibroblast function. We seeded freshly isolated Dupuytren’s stromal cells directly onto hydrogels and used traction force microscopy to quantify cellular force production. We found that neutralizing antibodies to TNFR2 and IL-33 inhibited myofibroblast mechanical peak force production and thus contractility (Fig. 4, F and G). This was most significant with selective TNFR2 inhibition in concordance with the down-regulation of key myofibroblast gene and protein markers. Together, our results elucidate selective TNFR2 and IL-33 blockade as a novel therapeutic axis in DD (fig. S8).
DISCUSSION
Currently, there is no approved treatment to control the progression of early-stage DD (7). This mirrors many other fibrotic disorders, including renal and liver fibrosis (32, 33), which, unlike DD, cannot be readily diagnosed at an early stage. DD provides a readily accessible source of human tissue at a relatively early stage of the disease process, and here, we identify novel pathways that may be targeted for attenuating chronic low-grade inflammation. However, the initial trigger of this low-grade inflammation in DD remains unknown. Genetic susceptibility is likely important, as are metabolic factors given the higher prevalence of DD in diabetics.
A commonly used simplified macrophage nomenclature describes classically activated (M1) and alternatively activated (M2) phenotypes based on their cytokine profiles, and the two subtypes play crucial but differing roles in the pathogenesis of fibrosis. M1 macrophages are thought to predominate during the onset of injury, releasing proinflammatory cytokines that unchecked can result in tissue damage (34). As the acute phase of inflammation subsides, the landscape shifts to one that promotes the polarization and recruitment of M2 macrophages (35). Although previous reports have demonstrated that M2 macrophages are important for tissue repair and regeneration, recent studies show these cells to be an important source of profibrotic cytokines and potentially key mediators of the dysregulated wound healing program that characterizes fibrosis (25, 27, 35, 36). Our data report a single-cell census of macrophage phenotypes within a human fibrotic microenvironment and implicate these cells as potentially key mediators of disease through the secretion of TNF. Our paired single-cell protein and gene expression data provide an important taxonomy of immune cell states in human fibrosis.
We have previously shown that DD myofibroblasts and their precursors express higher levels of TNFR2 relative to genetically matched control fibroblasts (7). Our results here expand upon this finding by demonstrating that selective TNFR2 blockade and RNA interference (RNAi) knockdown can down-regulate the myofibroblast phenotype. Antibody-mediated TNFR2 inhibition in myofibroblasts resulted in a significant reduction in both cellular traction force and the expression of profibrotic genes COL1A1 and ACTA2, and this effect was significantly greater than TNFR1 blockade alone and as effective as targeting TNF itself. Selective local inhibition of TNFR2 signaling may provide a highly specific therapeutic strategy in DD that could help to reduce potential adverse effects. In addition, our findings may have potential application in other chronic inflammatory diseases.
Unlike systemic inflammatory disorders such as rheumatoid arthritis, inflammation in DD is restricted to the nodular tissue and persists over decades (37). Previous reports highlight stromal cell–derived IL-33 as a key driver of diverse disease states, including inflammatory bowel disease and cancer (16, 17, 23, 24, 26, 35). Moreover, although recent reports have shown that IL-33 mediates fibrosis in both liver and lung murine models (17, 23, 28), the role of this cytokine has not been well characterized in human fibrotic disease. Here, we show that primary human fibrotic stromal cells are an important source of IL-33, and this cytokine can act directly on local immune cells, including M2 macrophages and mast cells. On exposure to low levels of IL-33, these immune cells secrete TNF at levels capable of promoting fibroblast activation and myofibroblast differentiation. In our single-cell RNA-seq data, endothelial cells exhibited IL33 expression and thus may act to promote sustained inflammation in DD alongside other stromal cell populations. Our findings derived exclusively from the study of human tissue support previous reports based largely on animal models that identified IL-33 as a key profibrotic cytokine (17). Moreover, in accordance with data from a murine model of ulcerative colitis, we found that inhibition of TNF resulted in down-regulation of IL-33 (29). The extracellular functions of IL-33 are mediated via the primary receptor ST2 and the co-receptor IL-1RAcP (16). Our data show that specific blockade of TNFR2 was especially effective in down-regulating expression of both IL-33 and ST2 in myofibroblasts from patients with DD.
While combination therapy has become the standard of care in many cancers and infections, attempts to inhibit multiple proinflammatory cytokines in chronic inflammatory diseases have so far resulted in unacceptably high rates of infective complications (30). Targeting the cross-talk between the immune and resident stromal cells may thus provide a novel therapeutic approach (37). Our data support the key role of resident activated fibroblasts in perpetuating the local low-grade inflammation seen in the early stages of many fibrotic diseases (fig. S8) (1, 35). Our in vitro data show that inhibition of either IL-33 or TNFR2 alone effectively down-regulated the myofibroblast phenotype. However, there appeared to be no benefit to combined blockade of both cytokines. One explanation may be that we only assessed the effects on isolated myofibroblasts and we would speculate that combination therapy may be more effective in vivo in the presence of both immune and stromal cells. Given the lack of an animal model for DD, we have been unable to further validate our findings in an in vivo model.
Our current clinical trial for early-stage DD involves administration of adalimumab every 3 months (9). Potentially, combination therapy based on TNFR2 and IL-33 blockade may improve the efficacy of this treatment regimen, possibly permitting less frequent dosing, and at the same time avoid potential adverse effects associated with global TNF inhibition. While TNF can lead to cell death in some systems, we have previously shown that TNF does not promote apoptosis of DD myofibroblasts (10). It is possible that clinical trials using etanercept may be less effective as it does not readily bind tmTNF, and we found that, at the doses tested, it was less effective than adalimumab or golimumab in vitro (7). It is also possible that long-term treatment with etanercept may be associated with adverse effects via the stimulation of TNFR2. For the future, our data would suggest that selective TNFR2 inhibition, potentially combined with anti–IL-33, may be more efficacious than targeting TNF alone.
In summary, our studies based exclusively on human samples have identified a novel stromal–immune cell interaction that may help to explain the persistent localized low-grade inflammation in DD, a localized fibrotic condition that only occurs in humans, and identified IL-33 as a key signaling molecule and potential therapeutic target. We have also shown that, in DD, TNF signals predominantly via TNFR2 to promote myofibroblast differentiation, and selectively targeting this receptor may offer significant advantages over global TNF blockade.
MATERIALS AND METHODS
DD tissue collection
Dupuytren’s nodular tissue was obtained from patients with DD undergoing dermofasciectomy. Informed written consent was obtained through approval of the regional ethics committee (REC 07/H0706/81), in accordance with the Declaration of Helsinki.
Cell culture
Tissue samples were dissected into small pieces and digested in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with type I collagenase (2 mg/ml; Worthington Biochemical Corporation) + deoxyribonuclease I (0.15 mg/ml; Roche Diagnostics) for up to 2 hours at 37°C. Digested tissue fragments were filtered through a 70-μm cell strainer, and the cell suspension was centrifuged at 1500 rpm for 10 min. Isolated cells were cultured in DMEM with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Lonza, Belgium) and used until passage 2. Human mast cell lines HMB1.1 and HMC1.2 were a gift from J. Butterfield (Mayo Clinic). The cells were grown in Iscove’s basal medium (Gibco) supplemented with 10% defined, iron-supplemented calf serum (HyClone, Utah), 1% penicillin-streptomycin, and 0.01% (v/v) monothioglycerol (Sigma). The human mast cell line LAD2 was maintained in complete StemPro-34 medium (Gibco) supplemented with l-glutamine (2 mM), 1% penicillin-streptomycin, and recombinant human stem cell factor (100 ng/ml; PeproTech, UK). The medium was changed every 3 days. Single-donor platelet phoresis residue packs were purchased from the North London Blood Transfusion Service. Mononuclear cells were isolated by Ficoll-Hypaque centrifugation (specific density, 1.077 g/ml) preceding T cell/monocyte separation in a JE-6 elutriator (Beckman Coulter). Macrophages were derived from elutriated monocytes by culturing the cells with either M1-polarizing conditions [granulocyte-macrophage colony-stimulating factor (GM-CSF), 25 ng/ml; PeproTech] or M2-polarizing conditions [macrophage colony-stimulating factor (M-CSF), 50 ng/ml; PeproTech] in 5% heat-inactivated fetal calf serum (Gibco) RPMI 1640 for 5 days. Cells were stimulated with either IL-33 or TNF (PeproTech, UK) or lipopolysaccharide (Enzo Life Sciences Inc.).
Measurement of mRNA
RNA was extracted using the RNeasy Kit (Qiagen), and complementary DNA (cDNA) was generated using the RNA-Ct Kit (Life Technologies). Gene expression was determined by quantitative polymerase chain reaction (qPCR) on ViiA 7 using Reverse Transcriptase qPCR Mastermix No ROX (RT-QPRT-032XNR, Eurogentec) and Assay-On-Demand premixed TaqMan probe master mixes [ACTA2Hs00426835_g1, Col1 Hs00164004_m1, ST-2 Hs00249384_m1, IL-33 Hs00369211_m1, ST2 Hs00545033_m1, TNFR1 Hs01042313-m1, TNFR2 Hs00961749-m1, and GAPDH (glyceraldehyde phosphate dehydrogenase) 4352665; Life Technologies]. The relative gene expression was calculated using the ΔΔCt method with the GAPDH gene for normalization of RNA levels.
Transfections
For transfection experiments, cells were seeded and, the following day transfected with siRNA-inventoried silencer select reagents, used for IL1RL1 (s17532, Applied Biosystems), and negative control siRNAs 1 (4390843) and 2 (4390846, Applied Biosystems) were used with sequences that do not target any gene product and provide a baseline to compare siRNA-treated samples. All transfections were performed with AMAXA System Nucleofector Kits for Human Dermal Fibroblast (VPD-1001, Lonza, Belgium) and Opti-MEM (31985062, Life Technologies) according to the manufacturer’s instructions. Cells were immediately transferred to a six-well plate with 2 ml of Opti-MEM (31985062, Life Technologies). After 16 hours, the transfection medium was washed three times with phosphate-buffered saline (PBS), before being replaced by DMEM with 10% FBS and 1% penicillin-streptomycin and incubated for another 32 hours.
Antibodies
Antibodies used in immunoblotting were the following: anti–α-SMA (Abcam, ab5694), anti–β-actin (Sigma, A5441), anti-ST2 (Thermo Scientific, PA5-23316), anti-vimentin (Abcam, Ab92547), mouse immunoglobulin G (IgG; R&D Systems, MAB002), anti–TNF-α/TNF-R2 (R&D Systems, MAB2001), anti-TNFR1 (Santa Cruz Biotechnology, sc-8436), TNF-R (New England BioLabs, 37275), extracellular signal–regulated kinase–2 (ERK-2) (Santa Cruz Biotechnology, sc-153), rabbit Ig (PeproTech, 500-P002), and anti–IL-33 (PeproTech, 500-P261).
Immunoblot analysis
Cells were cultured in monolayer and lysed in 1% Triton X-100 lysis buffer [50 mM tris (pH 7.6), 150 mM NaCl, and 2 mM EDTA], supplemented with 500 μM sodium fluoride, 1 mM sodium vanadate, and protease inhibitor mixture I (Sigma-Aldrich), or nuclear proteins were lysed directly in 1× gel sample buffer [50 mM tris (pH 6.8), 10% glycerol, 2% SDS, 0.01% bromophenol blue, and 2.5% β-mercaptoethanol] followed by sonication. Cell extracts (10 μg) were resolved by precast gradient gels (4 to 15%) SDS–polyacrylamide gel electrophoresis (Bio-Rad). Protein transfer to a nitrocellulose membrane was performed with the semidry Trans-Blot Turbo System (Bio-Rad). Polyvinyl difluoride membranes (Millipore) were blocked for 1 hour with blocking buffer [5% (w/v) fat-free milk and 0.1% (v/v) Tween 20 in PBS], followed by 1 to 24 hours of incubation with the antibodies. Horseradish peroxidase–conjugated anti-mouse IgG or anti-rabbit IgG (GE Healthcare NXA931) was used as secondary antibodies at a dilution of 1:2000. Visualization was achieved with chemiluminescence using Enhanced Chemiluminescence Plus (GE Healthcare).
Flow cytometry
After tissue disaggregation into a single-cell suspension, cells were first stained with a panel of fluorescently labeled antibodies to surface antigens, washed with fluorescence-activated cell sorting wash buffer [1% bovine serum albumin (BSA) and 0.01% NaN3 in PBS], and then fixed using Cytofix (Foxp3 Staining Buffer Set, eBioscience, 00-5523-00) for a minimum of 30 min at 4°C. After three washes in perm wash (Foxp3 Staining Buffer, eBioscience), intracellular antigens were stained with another panel of fluorescently labeled antibodies, and cells were then washed with perm wash and analyzed by flow cytometry (BD LSR Fortessa X20) and FlowJo software. BD CompBeads (anti-mouse Igκ, 552843) were used to establish compensation settings. Dead cells were deselected using live/dead stain, added to surface staining panel before cell fixation (Live/Dead Fixable Near-IR Dead Cell Stain Kit, Life Technologies L10119). For intracellular cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml; Calbiochem, D00145019)/ionomycin (1 μg/ml; Calbiochem, 407950) in the presence of brefeldin A (12.5 μg/ml; Sigma, B6542) for 2 hours at 37°C, 5% CO2 before cell staining as outlined above. Antibodies used are as follows: CD45-BV421 (BioLegend, 305032), CD117-BV605 (BioLegend, 313217), FceR1-APC (eBioscience, 17-5899-42), CD3-BV711 (BioLegend, 317328), CD163-PerCp-Cy5.5 (BioLegend, 333608), CD68-PeCy7 (eBioscience, 25-0689-42), α-SMA-AF700 (R&D Systems, 502937), TNF-BV650 (BioLegend, 502937), and IL-33-PECy7 (R&D Systems, IC3625P). We used a CD45+FCER1A+CD117+ gating strategy to identify mast cells in flow cytometry.
Immunohistochemistry
All Dupuytren’s tissue samples were fixed in formalin, longitudinally bisected, and embedded in paraffin wax, and 7-μm sections from the cut surface were processed for immunohistochemistry (18). Sequential sections were stained with mouse monoclonal anti–α-SMA antibody (Sigma, A2547), anti-CD68 antibody (Dako, clone K61), and tryptase (Sigma, 342M-17). Antibodies were detected using a two-stage polymer enhancer system (Sigma). Mouse serum at the same protein concentration as the monoclonal antibody solution was used as a control.
Immunofluorescence and confocal microscopy
Myofibroblasts were grown on poly-l-lysine (Sigma)–coated glass coverslips and then washed with PBS and fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.2% Triton X-100, and blocked with 3% BSA. Then, the cells were stained with the following antibodies: anti–α-SMA (Sigma, A2547), anti-CD68 (Dako, clone K61), anti-tryptase (Sigma, 342M-17), anti-ST2 (Abcam, ab25877), and anti–IL-33 (R&D Systems, AF3625). This was followed by incubation with fluorescent dye–conjugated secondary antibodies (Life Technologies). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich), and F-actin was visualized with Alexa Fluor 488 phalloidin (Life Technologies) and mounted using ProLong Gold Antifade (Life Technologies). Fluorescent images were obtained with a confocal system (Zeiss, LSM 710), and image files were analyzed and processed.
Single-cell RNA-seq
Single-cell suspensions of primary DD cells were derived from freshly resection nodules following surgical excision as previously described. Single cells were isolated and lysed, and subsequently, RNA was reverse-transcribed and converted into cDNA libraries for RNA-seq analysis using a Chromium Controller and a Chromium Single Cell 3′ v2 Reagent kit (Genomics 10×) following the manufacturer’s protocol. Libraries were pooled together for sequencing on a HiSeq 4000 (Illumina) using 96– and 28–base pair reads to a mean depth of >35,000 reads per cell. Alignment of reads to the genome and generation of gene counts per cell were performed by Cell Ranger 1.2 software (Genomics 10×). Quality control was performed on each individual sample to remove poor-quality cells on the basis of the number of genes expressed, number of unique molecular identifiers (UMIs), and percentage of mitochondrial reads mapped. We used a global-scaling normalization procedure calling Seurat’s LogNormalize() function. Briefly, the cell ranger UMI count value for each gene was divided by the sum of the total UMI counts per cell to normalize for differences in library size and then multiplied by a scaling factor that represented the median library complexity (10,000) producing transcript per million (TPM)–like values. We then took the log transform of this procedure for downstream analysis. Following feature selection using highly variable genes and principal components analysis, high-quality cells were clustered using a graph-based routine implemented in Seurat R package (Satija Lab).
Traction force microscopy
Polyacrylamide (PAA) hydrogels for traction force microscopy were prepared as previously described. Briefly, acid-washed glass coverslips (18 mm) were incubated with poly-l-lysine (10 μg/ml in H2O) for 30 min at 4°C and then coated with 0.04 μm of carboxylate-modified red FluoSpheres [1:5000 in double-distilled H2O (ddH2O)]. Polyacrylamide formation was initiated with ammonium persulfate (10% solution in ddH2O, Sigma) and TEMED (tetramethylethylenediamine; Sigma). Polymerized PAA gels were functionalized with sulfo-SANPAH (Invitrogen) and coated with type 1 collagen (200 μg/ml rat tail, Thermo Fisher Scientific). The Young’s modulus of the PAA gels was 2.55 ± 0.5 kPa. Cells were allowed to adhere to the gel for 4 to 6 hours before image acquisition. Images were captured using a 40× Plan Apo objective on a Zeiss LSM 710 confocal microscope, and the experiments were performed at 37°C and 5% CO2 in phenol red–free DMEM (Thermo Fisher Scientific) in a microscope stage incubation chamber. Bead positions were acquired before and after the addition of trypsin, which removed cells from the gel surface. Bead displacement was tracked with an ImageJ PIV plugin (38), and cellular forces were reconstructed using an fourier transform traction cytometry (FTTC) algorithm also implemented in ImageJ (38) Further downstream analysis and visualization were implemented in MATLAB (MathWorks).
Mesoscale discovery
Digested nodular cells were cultured in DMEM with 5% FBS and 1% penicillin-streptomycin. Twenty-four hours later, culture supernatants were harvested, and cytokine and chemokine levels were determined using the Meso Scale Discovery platform using the following kits: U-PLEX Pro-Inflam Combo (K15049K), U-PLEX TGF-β1 (K151XWK), and V-PLEX chemokine panel 1 (K15047D). In addition, a custom plex of IL-33 was used: IL-33 custom-made mesoscale discovery (MSD), IL-33 capture antibody (0.8 μg/ml; R&D DuoSet DY3625), anti–IL-33 detect antibody (400 ng/ml; R&D DuoSet DY3625) plus streptavidin/SULFO-TAG (K15A01-1).
Supplementary Material
Acknowledgments
We thank our clinical collaborators C. Bainbridge, D. Warwick, L. Harry, and D. Davidson for provision of tissue samples from their patients. We thank the Wellcome Trust Centre for Human Genetics for preparation of the single-cell libraries. In addition, we thank the Imaging Centre at the Kennedy Institute of Rheumatology for their support in microscopy. Funding: This work was funded by the Royal College of Surgeons, the Kennedy Institute of Rheumatology Studentship (MSP 13 14 11.), and the Oxford-Celgene Research Fellowship (grant no. AZR00610) Author contributions: J.N., K.S.M., D.I., M. Fritzsche, M. Feldmann, and T.B.L. conceived and designed the study. D.I., T.B.L., L.W., F.M., M.C., H.C.-Y., A.I.E.S., M. Fritzsche, and H.C.-Y. performed and contributed to the analysis of the experiments. J.N., T.L., and K.M. wrote the manuscript with contribution from all co-authors. Competing interests: J.N. and M. Feldmann have interests in 180 Therapeutics, which has exclusively licensed IP from the University of Oxford for the treatment of DD. 180 Therapeutics has filed patent applications based on data presented in the paper. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaay0370/DC1
Fig. S1. DD is a localized inflammatory disorder.
Fig. S2. Single-cell RNA-seq of immune cells from freshly isolated Dupuytren’s nodules.
Fig. S3. Mast cells and M2 macrophages are the predominant sources of TNF in DD.
Fig. S4. TNF induced protein expression and TNF receptor expression in Dupuytren’s cord.
Fig. S5. IL-33 and its receptor ST2 are expressed on Dupuytren’s myofibroblasts.
Fig. S6. No reduction in cell viability following treatment with neutralizing antibodies.
Fig. S7. siRNA knockdown validation for TNFRSF1A, TNFRSF1A, and ST2.
Fig. S8. IL-33 and TNF stromal–immune cell cross-talk in DD.
REFERENCES AND NOTES
- 1.Wynn T. A., Ramalingam T. R., Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nanchahal J., Hinz B., Strategies to overcome the hurdles to treat fibrosis, a major unmet clinical need. Proc. Natl. Acad. Sci. U.S.A. 113, 7291–7293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lanting R., Broekstra D. C., Werker P. M. N., van den Heuvel E. R., A systematic review and meta-analysis on the prevalence of dupuytren disease in the general population of western countries. Plast. Reconstr. Surg. 133, 593–603 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reilly R. M., Stern P. J., Goldfarb C. A., A retrospective review of the management of Dupuytren’s nodules. J. Hand Surg. Am. 30, 1014–1018 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Wilburn J., McKenna S. P., Perry-Hinsley D., Bayat A., The impact of Dupuytren disease on patient activity and quality of life. J. Hand Surg. Am. 38, 1209–1214 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Wick G., Grundtman C., Mayerl C., Wimpissinger T. F., Feichtinger J., Zelger B., Sgonc R., Wolfram D., The immunology of fibrosis. Annu. Rev. Immunol. 31, 107–135 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Verjee L. S., Verhoekx J. S. N., Chan J. K. K., Krausgruber T., Nicolaidou V., Izadi D., Davidson D., Feldmann M., Midwood K. S., Nanchahal J., Unraveling the signaling pathways promoting fibrosis in Dupuytren’s disease reveals TNF as a therapeutic target. Proc. Natl. Acad. Sci. U.S.A. 110, E928–E937 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nanchahal J., Ball C., Davidson D., Williams L., Sones W., McCann F. E., Cabrita M., Swettenham J., Cahoon N. J., Copsey B., Anne Francis E., Taylor P. C., Black J., Barber V. S., Dutton S., Feldmann M., Lamb S. E., Anti-tumour necrosis factor therapy for Dupuytren’s disease: A randomised dose response proof of concept phase 2a clinical trial. EBioMedicine 33, 282–288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nanchahal J., Ball C., Swettenham J., Dutton S., Barber V., Black J., Copsey B., Dritsaki M., Taylor P., Gray A., Feldmann M., Lamb S., Study protocol: A multi-centre, double blind, randomised, placebo-controlled, parallel group, phase II trial (RIDD) to determine the efficacy of intra-nodular injection of anti-TNF to control disease progression in early Dupuytren's disease, with an embedded dose response study. Wellcome Open Res. 2, 37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward-Kavanagh L. K., Lin W. W., Šedý J. R., Ware C. F., The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 44, 1005–1019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faustman D. L., Davis M., TNF receptor 2 and disease: Autoimmunity and regenerative medicine. Front. Immunol. 4, 478 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo G., Morrissey J., McCracken R., Tolley T., Klahr S., Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am. J. Physiol. 277, F766–F772 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Theiss A. L., Simmons J. G., Jobin C., Lund P. K., Tumor necrosis factor (TNF) α increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J. Biol. Chem. 280, 36099–36109 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Shih B., Bayat A., Scientific understanding and clinical management of Dupuytren disease. Nat. Rev. Rheumatol. 6, 715–726 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Chan J. K., Roth J., Oppenheim J. J., Tracey K. J., Vogl T., Feldmann M., Horwood N., Nanchahal J., Alarmins: Awaiting a clinical response. J. Clin. Invest. 122, 2711–2719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liew F. Y., Girard J. P., Turnquist H. R., Interleukin-33 in health and disease. Nat. Rev. Immunol. 16, 676–689 (2016). [DOI] [PubMed] [Google Scholar]
- 17.McHedlidze T., Waldner M., Zopf S., Walker J., Rankin A. L., Schuchmann M., Voehringer D., McKenzie A. N., Neurath M. F., Pflanz S., Wirtz S., Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39, 357–371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verjee L. S., Midwood K., Davidson D., Essex D., Sandison A., Nanchahal J., Myofibroblast distribution in Dupuytren’s cords: Correlation with digital contracture. J. Hand Surg. Am. 34, 1785–1794 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Martinez F. O., Gordon S., The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 6, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray P. J., Allen J. E., Biswas S. K., Fisher E. A., Gilroy D. W., Goerdt S., Gordon S., Hamilton J. A., Ivashkiv L. B., Lawrence T., Locati M., Mantovani A., Martinez F. O., Mege J. L., Mosser D. M., Natoli G., Saeij J. P., Schultze J. L., Shirey K. A., Sica A., Suttles J., Udalova I., van Ginderachter J. A., Vogel S. N., Wynn T. A., Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juremalm M., Nilsson G., Chemokine receptor expression by mast cells. Chem. Immunol. Allergy 87, 130–144 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Griffith J. W., Sokol C. L., Luster A. D., Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 32, 659–702 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Li D., Guabiraba R., Besnard A. G., Komai-Koma M., Jabir M. S., Zhang L., Graham G. J., Kurowska-Stolarska M., Liew F. Y., McSharry C., Xu D., IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin. Immunol. 134, 1422–1432.e11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rankin A. L., Mumm J. B., Murphy E., Turner S., Yu N., McClanahan T. K., Bourne P. A., Pierce R. H., Kastelein R., Pflanz S., IL-33 induces IL-13-dependent cutaneous fibrosis. J. Immunol. 184, 1526–1535 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Kurowska-Stolarska M., Stolarski B., Kewin P., Murphy G., Corrigan C. J., Ying S., Pitman N., Mirchandani A., Rana B., van Rooijen N., Shepherd M., McSharry C., McInnes I. B., Xu D., Liew F. Y., IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 183, 6469–6477 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Schiering C., Krausgruber T., Chomka A., Fröhlich A., Adelmann K., Wohlfert E. A., Pott J., Griseri T., Bollrath J., Hegazy A. N., Harrison O. J., Owens B. M. J., Löhning M., Belkaid Y., Fallon P. G., Powrie F., The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez F. O., Gordon S., Locati M., Mantovani A., Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Kakkar R., Hei H., Dobner S., Lee R. T., Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 287, 6941–6948 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastorelli L., Garg R. R., Hoang S. B., Spina L., Mattioli B., Scarpa M., Fiocchi C., Vecchi M., Pizarro T. T., Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl. Acad. Sci. U.S.A. 107, 8017–8022 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nyboe Andersen N., Pasternak B., Friis-Møller N., Andersson M., Jess T., Association between tumour necrosis factor-α inhibitors and risk of serious infections in people with inflammatory bowel disease: Nationwide Danish cohort study. BMJ 350, h2809 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu F., Lagares D., Choi K. M., Stopfer L., Marinković A., Vrbanac V., Probst C. K., Hiemer S. E., Sisson T. H., Horowitz J. C., Rosas I. O., Fredenburgh L. E., Feghali-Bostwick C., Varelas X., Tager A. M., Tschumperlin D. J., Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L344–L357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boor P., Ostendorf T., Floege J., Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 6, 643–656 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Friedman S. L., Liver fibrosis in 2012: Convergent pathways that cause hepatic fibrosis in NASH. Nat. Rev. Gastroenterol. Hepatol. 10, 71–72 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Duffield J. S., Forbes S. J., Constandinou C. M., Clay S., Partolina M., Vuthoori S., Wu S., Lang R., Iredale J. P., Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 115, 56–65 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wynn T. A., Vannella K. M., Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolic-Paterson D. J., Wang S., Lan H. Y., Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 4, 34–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orr C., Vieira-Sousa E., Boyle D. L., Buch M. H., Buckley C. D., Cañete J. D., Catrina A. I., Choy E. H. S., Emery P., Fearon U., Filer A., Gerlag D., Humby F., Isaacs J. D., Just S. A., Lauwerys B. R., le Goff B., Manzo A., McGarry T., McInnes I. B., Najm A., Pitzalis C., Pratt A., Smith M., Tak P. P., Tas S. W., Thurlings R., Fonseca J. E., Veale D. J., Synovial tissue research: A state-of-the-art review. Nat. Rev. Rheumatol. 13, 463–475 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Martiel J.-L., Leal A., Kurzawa L., Balland M., Wang I., Vignaud T., Tseng Q., Théry M., Measurement of cell traction forces with ImageJ. Methods Cell Biol. 125, 269–287 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaay0370/DC1
Fig. S1. DD is a localized inflammatory disorder.
Fig. S2. Single-cell RNA-seq of immune cells from freshly isolated Dupuytren’s nodules.
Fig. S3. Mast cells and M2 macrophages are the predominant sources of TNF in DD.
Fig. S4. TNF induced protein expression and TNF receptor expression in Dupuytren’s cord.
Fig. S5. IL-33 and its receptor ST2 are expressed on Dupuytren’s myofibroblasts.
Fig. S6. No reduction in cell viability following treatment with neutralizing antibodies.
Fig. S7. siRNA knockdown validation for TNFRSF1A, TNFRSF1A, and ST2.
Fig. S8. IL-33 and TNF stromal–immune cell cross-talk in DD.