
Figure 2. Regulation of the cell cycle arrest and inflammatory SASP in the induction of cellular senescence and its interconnection with apoptosis.
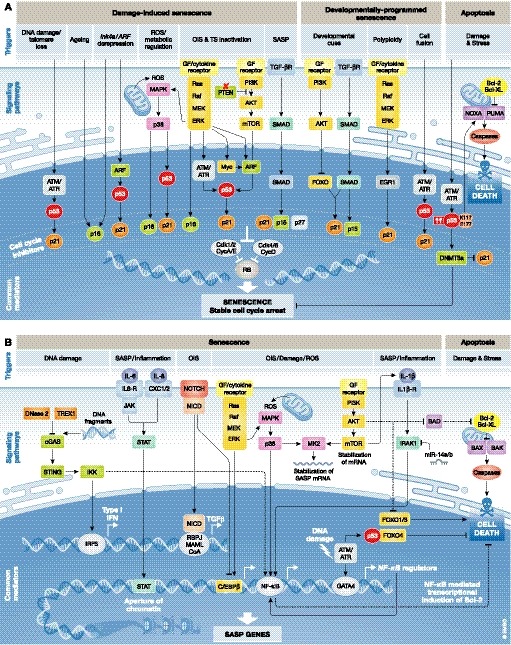
(A) Most senescence‐inducing triggers converge in the activation of the cell cycle inhibitor pathways p53/p21 and/or p16INK 4a. These result in the inhibition of cyclin‐dependent kinase 1 (CDK1), CDK2, CDK4 and CDK6, which prevents the phosphorylation of the retinoblastoma protein (RB), leading to the suppression of S‐phase genes and an ensuing stable cell cycle arrest. DNA‐damaging triggers activate the DNA damage response (DDR) pathway resulting in the activation of p53 and p21. Ageing and epigenetic derepression of the Ink4a/ARF locus also lead to the activation of cell cycle inhibitors p16 and p21. ROS lead to the activation of the MAPK signalling pathway and its downstream effector p38. The aberrant expression of oncogenes or the loss of tumour suppressors leads to p53 activation through the Ras‐Raf‐MEK‐ERK or AKT signalling pathways, and TGFβ, and important factor of the SASP, leads to p15, p21 and p27 upregulation via SMAD signalling. Other triggers such as developmental cues and polyploidy activate the AKT, SMAD and/or Ras‐Raf‐MEK‐ERK pathway for p21 upregulation, while processes such as cell fusion signal through the DDR for p53 activation. In response to damage and different types of stress high levels of p53 with specific post‐translational modifications (such as acetylated K117 and E177) target DNMT3a, a suppressor of p21 and senescence, and trigger the apoptotic programme by upregulating PUMA and NOXA, which in turn activate the caspase cascade leading to cell death. (B) SASP implementation is orchestrated by the activation of the transcription factors NF‐κB and C/EBPβ through upstream signalling pathways. DNA‐damaging agents, ROS and OIS, generally activate the expression of SASP TFs via the AKT and/or the Ras‐Raf‐MEK‐ERK axis. In addition, DNA fragments are also known to trigger the activation of the cGAS/STING signalling, resulting in the activation of the IRF3 TF and subsequent transcription of Type 1 IFN. OIS‐derived SASP is dynamic and can also be orchestrated by NOTCH signalling, a process that restrains the inflammatory secretion by inhibiting C/EBPβ at initial stages, and allows the activation of SASP‐related super enhancers through NF‐κB later on. Accumulating increased levels of TFs reinforce the senescent phenotype through autocrine and paracrine signalling. SASP‐derived inflammatory chemokines such as IL‐6 and IL‐8 promote epigenetic modifications reinforcing the cell cycle arrest through the JAK/STAT cascade, while IL‐1α stimulates the activity of NF‐κB and C/EBPβ promoting a positive feedback loop on the secretion of other cytokines. Finally, senescence promotes survival networks by the regulation anti‐apoptotic pathways. This includes PI3K‐AKT signalling, which can inhibit pro‐apoptotic BAD and FOXO1/3, and phosphorylate caspase‐9; anti‐apoptotic FOXO4, that is present in senescent cells and interacts with p53; and NF‐κB, that may also promote survival responses by transcriptional induction of anti‐apoptotic proteins of the Bcl‐2 family. ATM/ATR, ataxia‐telangiectasia mutated and Rad3‐related homologue; IFN, interferon; OIS, oncogene‐induced senescence; ROS, reactive oxygen species; SASP, senescence‐associated secretory phenotype; TFs, transcription factors; TS, tumour suppressor.