

Summary
Intermediary metabolism in cancer cells is regulated by diverse cell-autonomous processes including signal transduction and gene expression patterns arising from specific oncogenotypes and cell lineages. Although it is well established that metabolic reprogramming is a hallmark of cancer, we lack a full view of the diversity of metabolic programs in cancer cells and an unbiased assessment of the associations between metabolic pathway preferences and other cell-autonomous processes. Here we quantified metabolic features, mostly from 13C enrichment of molecules from central carbon metabolism, in over 80 non-small cell lung cancer (NSCLC) cell lines cultured under identical conditions. Because these cell lines were extensively annotated for oncogenotype, gene expression, protein expression and therapeutic sensitivity, the resulting database enables the user to uncover new relationships between metabolism and these orthogonal processes.
Keywords: Non-small cell lung cancer, cancer metabolism, cell lines, 13C stable isotope labeling, glucose, glutamine, oncogenotypes, gene expression, protein expression, therapeutic sensitivity
eTOC Blurb
Metabolic reprogramming influences therapeutic sensitivity in cancer, but the scope of metabolic diversity among cancer cells is unknown. Chen et al. characterized metabolic phenotypes in over 80 non-small cell lung cancer cell lines, then used genomics, transcriptomics, proteomics and therapeutic sensitivities to uncover relationships between metabolism and orthogonal processes.
Introduction
Malignant cells reprogram metabolism to support cancer progression. Metabolic reprogramming is a hallmark of cancer and targeting metabolism is a potential therapeutic strategy (Hanahan and Weinberg, 2011; Tennant et al., 2010). This concept dates to the 1920s, when Otto Warburg observed rapid glucose uptake and lactate secretion (the Warburg effect) in tumors (Koppenol et al., 2011). Copious lactate secretion in the presence of oxygen was interpreted as evidence that suppressed mitochondrial metabolism is a uniform, required component of malignancy (Warburg, 1956). This paradigm was reinforced by studies in the 1980s-2000s demonstrating that oncogenes (c-Myc, Ras, Akt, and others) is stimulate glycolysis (Elstrom et al., 2004; Flier et al., 1987; Shim et al., 1997). It is now recognized that the tricarboxylic acid (TCA) cycle and other aspects of mitochondrial metabolism also contribute to cancer cell proliferation by providing energy and biosynthetic precursors (Cheng et al., 2011; DeBerardinis et al., 2007; Weinberg et al., 2010). An emerging theme is that tumor cells acquire heterogeneous metabolic phenotypes to withstand complex challenges during cancer progression (Boroughs and DeBerardinis, 2015).
Cancer metabolism is regulated in part by signaling and transcriptional networks activated by mutations in oncogenes and tumor suppressors, resulting in heterogeneous, cell-autonomous phenotypes among genetically diverse cancer cells (DeBerardinis and Chandel, 2016). Consistent with this idea, cultured cancer cells display variable rates of nutrient uptake (Jain et al., 2012). However, neither the breadth of metabolic diversity in cancer cells, nor the complement of mechanisms that induce metabolic reprogramming, are known. We set out to characterize cell-autonomous metabolic heterogeneity in cell lines derived from a particular tumor type. We chose non-small cell lung cancer (NSCLC) because: a) NSCLC is the foremost cause of cancer deaths worldwide, indicating the need for new therapies; b) cell lines covering the molecular diversity of NSCLC are available; c) recurrently-mutated genes in NSCLC, including KRAS, EGFR, PIK3CA, TP53, KEAP1 and PTEN, regulate metabolism; and d) access to orthogonal data sets would facilitate correlating metabolism with oncogenotypes, gene and protein expression and other features to understand mechanisms by which metabolic reprogramming occurs.
Results
Experimental design and metabolic analyses.
Over 80 NSCLC cell lines were analyzed for nutrient utilization, nutrient addiction, cell growth and isotope labeling after culture with [U-13C]glucose or [U-13C]glutamine (Table S1), and examined relationships between these activities and orthogonal molecular and therapeutic sensitivity data (Figure 1A). Multiple replicates cultured on different days were used to derive metabolic parameters. Consumption of glucose and glutamine and secretion of lactate and glutamate were measured to estimate rates of glycolysis and glutamine catabolism. Each cell line was assessed for proliferation in complete medium and survival in media lacking glucose or glutamine. Cells were also subjected to two complementary isotope labeling experiments to assess the extent to which glucose and glutamine supply carbon to metabolite pools. One experiment used medium with [U-13C]glucose and unlabeled glutamine, and the other used [U-13C]glutamine and unlabeled glucose.
Figure 1. Experimental design and diversity of nutrient utilization in NSCLC cell lines.
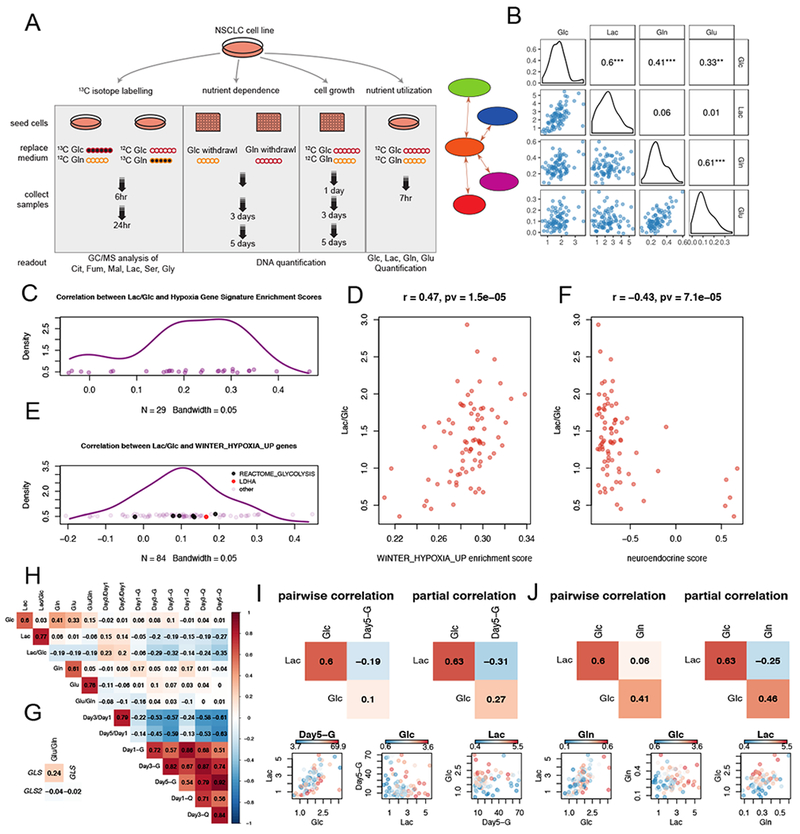
(A). Schematic of design for metabolic profiling and association analyses. See also Table S1.
(B). Scatter plots and density plots for nutrient utilization features, with Pearson correlation coefficients provided. ***, p<=0.001; **, p<=0.01; *, p<=0.05.
(C). Kernel density estimation of correlation coefficient distribution from the pairwise correlations between Lac/Glc and ssGSEA scores from 29 hypoxia-related genesets. The hypoxia related genesets were selected from C2-CGP gene sets in MSigDB with the criterion that the geneset name contained “HYPOXIA” but not “DN” (short for “Down”).
(D). Scatterplot showing the positive correlation between Lac/Glc and ssGSEA scores derived for “WINTER_HYPOXIA_UP” signature.
(E). Distribution from the pairwise correlation coefficients between Lac/Glc and 84 genes from the “WINTER_HYPOXIA_UP” gene set. Glycolytic genes (ALDOA, PGK1, TPI1, PGAM1, GAPDH and PFKFB4) from the “REACTOME_GLYCOLYSIS” geneset and LDHA are indicated by black and red spots.
(F). Scatterplot showing the negative correlation between Lac/Glc and neuroendocrine scores.
(G). Pearson correlation between glutaminolytic rate (Glu/Gln) and expression of GLS and GLS2.
(H). Correlation heatmap revealing pairwise Pearson correlations amongst metabolic features from nutrient utilization, cell growth and nutrient dependence.
(I and J). Comparisons of partial correlations and pairwise correlations for Lac, Glc and Day5-G (I) or Lac, Glc and Gln (J).
Abbreviations: Lac, lactate; Glc, glucose; Gln, glutamine; Glu, glutamate.
Metabolites were extracted after 6 and 24 hours and analyzed by gas chromatography–mass spectrometry (GC-MS) to obtain mass isotopologue distributions (MID) for citrate (Cit), fumarate (Fum), malate (Mal), lactate (Lac), serine (Ser) and glycine (Gly). Other than nutrient deprivation assays, the nutrient milieu was consistent across all these experiments and in experiments to characterize gene/protein expression and therapeutic sensitivity, making it possible to cross-correlate the data. All metabolic features are available through a web application at http://Ice.biohpc.swmed.edu/nsclc_cell_metabolism/ designed to allow users to explore the data.
Nutrient consumption, secretion and dependence.
Nutrient consumption/secretion rates varied 6-7-fold for glucose consumption and 15-fold for lactate secretion (Table S1). These rates are normalized to protein content, and were not affected by further corrections to account for differences in cell proliferation rates (Figure S1A; see STAR Methods for explanation of these corrections). As expected, glucose consumption correlated with lactate secretion and glutamine consumption correlated with glutamate secretion (Figure 1B). On average, cells secreted 1.35 moles of lactate per mole of glucose consumed (standard deviation 0.5 moles), and 0.4 moles of glutamate per mole of glutamine consumed (standard deviation 0.2 moles), emphasizing that cultured cancer cells process both glucose and glutamine at rates exceeding their need to retain carbon from these nutrients (Table S1). A positive correlation was also observed between glucose and glutamine consumption, indicating an unexpected coordination between glutaminolysis and glucose consumption (Figure 1B).
Normalizing lactate secretion to glucose consumption (Lac/Glc ratio) provides an estimate of each cell line’s preference for aerobic glycolysis. The cell lines displayed an 8-fold range of Lac/Glc ratios, indicating that NSCLC cell lines are diverse in the extent to which they discard glucose carbon as lactate (Table S1). Lac/Glc correlated significantly but weakly with the growth rate in complete medium (Day3/Day1 ratio; p=0.04) (Table S2). To identify associations between Lac/Glc and transcriptional programs, we derived single sample gene set enrichment analysis (ssGSEA) scores for each cell line based on the C2-CGP gene sets representing expression signatures of genetic and chemical perturbations from MSigDB (Subramanian et al., 2005). Enrichment scores from ssGSEA represent the degree to which genes in a particular gene set are coordinately up- or down-regulated (Barbie et al., 2009). Almost all (25/29) enrichment scores generated by hypoxia-related signatures are positively associated with Lac/Glc (Figure 1C); an example gene set is shown in Figure 1D (Winter et al., 2007). This gene set includes LDHA and several other glycolytic genes as defined by REACTOME (Fabregat et al., 2018; van Wijk and van Solinge, 2005). Individual genes in this set exhibit moderate but overall positive correlations with Lac/Glc (Figure 1E). In contrast, we found a negative correlation between Lac/Glc and ssGSEA scores derived from gene sets related to neuronal processes, which tend to be expressed in cells with neuroendocrine differentiation (Ionescu et al., 2007). We derived neuroendocrine scores for our cell lines based on a 50-gene signature (Zhang et al., 2018) and found that cells with high scores are invariably low in Lac/Glc (Figure 1F). Altogether these data indicate that hypoxia gene sets correlate with glycolytic metabolism even when cells are cultured in normoxia, and that cell lines with neuroendocrine-like signatures release relatively little lactate per glucose consumed.
We also normalized glutamate secretion to glutamine uptake (Glu/Gln ratio) as a surrogate for the release of carbon derived from glutamine. Glu/Gln ranged from essentially no glutamate released to a 1:1 ratio of glutamate release per glutamine consumed. Glu/Gln correlates positively with GLS mRNA, which encodes the glutaminolytic enzyme glutaminase (GLS) (Figure 1G). GLS2, which encodes another glutaminase isoform, did not correlate with Glu/Gln at the transcript level, and GLS did not correlate with GLS2.
To understand relationships between nutrient utilization, nutrient dependence and cell growth, we examined pairwise correlations amongst these features in a correlation heatmap (Figure 1H). The two cell growth features (Day3/Day1 and Day5/Day1) are strongly correlated, as expected. The six features related to glucose and glutamine dependence (Day1-G, Day3-G, Day5-G, Day1-Q, Day3-Q, Day5-Q) also correlate with each other, indicating that cells sensitive to glucose deprivation also tend to be sensitive to glutamine deprivation and vice versa. Growth rates correlate negatively with nutrient dependence, indicating that cell lines which grow rapidly under nutrient replete conditions perish more rapidly upon either glucose or glutamine withdrawal. However, neither glucose nor glutamine consumption correlate with growth rates, suggesting that these nutrients are used for processes other than or in addition to biomass assimilation.
In some cases, partial correlations produced stronger associations than direct pairwise correlations. Figure 1I provides an example where the negative association between Lac and Day5-G is more significant in a partial correlation controlling for Glc than in a pairwise correlation, indicating that for a given rate of glucose uptake, the more lactate produced, the more sensitive the cell line is to glucose deprivation. In Figure 1J, the negative association between Lac and Gln is more significant in a partial correlation controlling for Glc than in a pairwise correlation. In other words, with the same amount of glucose uptake, the more lactate a cell produces, the less glutamine it consumes.
Diversity in metabolic pathway utilization inferred by mass isotopologue distributions.
Mass isotopologue distributions (MIDs) report the fates of 13C-labeled fuels, providing a view of metabolism that cannot be achieved from steady-state metabolite levels (Buescher et al., 2015; Jang et al., 2018). MID analysis has been used to assess nutrient contributions to metabolic intermediates in the TCA cycle and other pathways. For example, oxidative and reductive pathways of glutamine metabolism are readily differentiated using MID of citrate following culture with [U-13C]glutamine. Glutamine oxidation, a major source of anaplerosis, generates m+5 labeling in α-ketoglutarate and m+4 labeling in other TCA cycle metabolites (Figure 2A). Glutamine-dependent reductive carboxylation (GDRC) via isocitrate dehydrogenase-1 or -2 (IDH1, IDH2) generates α-ketoglutarate m+5, citrate m+5, acetyl-CoA m+2, and m+3 labeling in other TCA cycle intermediates (Figure 2B).
Figure 2. Differential pathway utilization inferred by mass isotopologue distributions.
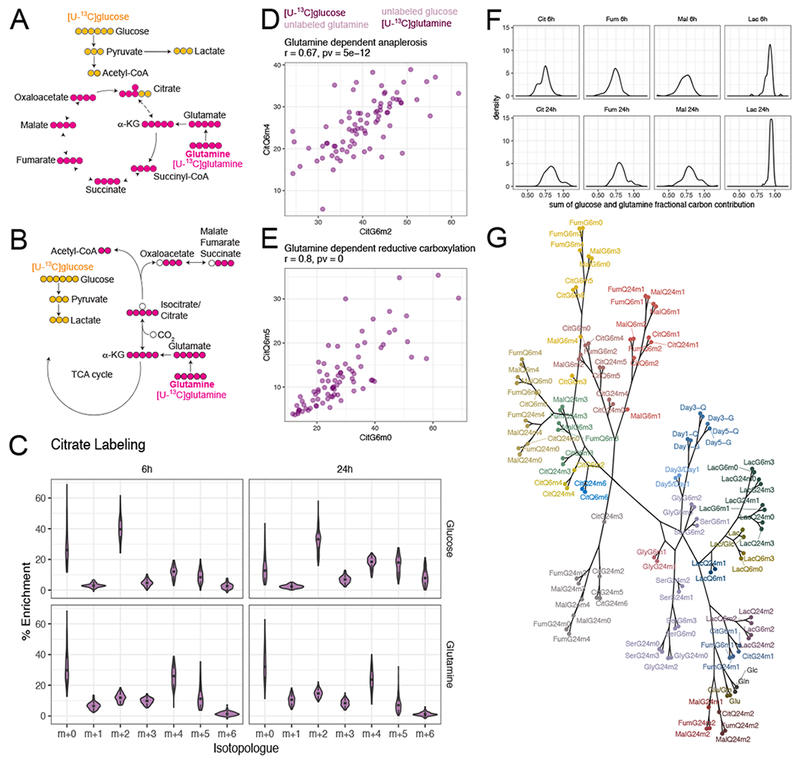
(A and B). Schematics of representative isotopologues generated from [U-13C]glucose or [U-13C]glutamine labeling through glutamine-dependent anaplerosis (A) or glutamine-dependent reductive carboxylation (B).
(C). Violin plots showing distribution of citrate isotopologues from different tracers and different labeling durations. See also Figure S2.
(D and E). Scatterplots showing that enrichment of signature isotopologues produced from two different tracers for the same metabolic pathway (glutamine-dependent anaplerosis in D and glutamine-dependent reductive carboxylation in E) are highly correlated with each other. Figure title provides Pearson correlation coefficient and p-value.
(F). Distribution of the sum of glutamine and glucose fractional carbon contribution into different metabolites.
(G). Dendrogram of all metabolic features from hierarchical clustering with absolute Pearson correlation-based distance using Ward’s minimum variance method. The branches were arbitrarily colored.
Abbreviations: Cit, citrate; Fum, fumarate; Mal, malate; Lac, lactate; Ser, serine; Gly, glycine.
To our knowledge, there have been no previous attempts to use parallel-tracer MID analysis to capture metabolic diversity in a panel of cell lines as large as ours. We named 13C labeling features using an abbreviation for the metabolite, followed by the tracer, labeling duration and number of 13C nuclei. For example, m+2 citrate after 6 hours of [U-13C]glucose labeling is named CitG6m2. The data revealed remarkable cell-autonomous diversity in isotope labeling among the cell lines, but good consistency among replicates (Figure S1B–E).
Examination of citrate MIDs after labeling with either [U-13C]glucose or [U-13C]glutamine reveals several interesting features (Figure 2C). First, MID distributions are generally conserved between the two time points, indicating that most labeling occurs within the first 6 hours, as expected for glycolysis and the TCA cycle (Jang et al., 2018). The modest changes at 24 hours included reduced prominence of unlabeled citrate and increased abundance of isotopologues derived from multiple rounds of the TCA cycle (e.g. m+4 and m+6 from [U-13C]glucose). Second, labeling patterns are heterogeneous across the panel, with some citrate MIDs (particularly those involving m+0 and m+2 from [U-13C]glucose and m+0, m+4 and m+5 from [U-13C]glutamine) covering about one third of the possible MID range. This indicates heterogeneity of intrinsic pathway preference even when cell lines are cultured under identical conditions.
Third, the abundant m+2 isotopologues from [U-13C]glucose, coupled with the m+4 isotopologues from [U-13C]glutamine, demonstrate the expected prominence of glucose-dependent acetyl-CoA formation and glutamine-dependent anaplerosis (DeBerardinis et al., 2007). In this common pathway, glucose and glutamine metabolism converge on citrate synthesis (Figure 2A). Although CitG6m2 and CitQ6m4 fractions varied widely across the panel, they correlated well with each other (Figure 2D). MIDs from other TCA cycle intermediates agreed with the prominence of glutamine-dependent anaplerosis (Figure S2A–E; standard deviations are in Figure S2F). Other pairs of glucose- and glutamine-derived isotopologues also demonstrated strong correlations. One of the most striking was the positive correlation between CitQ6m5 and CitGlc6m0, two isotopologues characteristic of GDRC (Figure 2E). Some cells had as much as 35% m+5 labeling in citrate from [U-13C]glutamine; this was unexpected considering that GDRC is observed in hypoxia and other conditions of impaired glucose and glutamine oxidation (Gameiro et al., 2013; Metallo et al., 2011; Mullen et al., 2011; Rajagopalan et al., 2015). Given the prominent contribution of glucose and glutamine to central metabolism, we also determined the overall fraction of carbons in citrate, fumarate, malate and lactate derived from these nutrients. The mean fractional contribution of glucose and glutamine to carbons in TCA cycle intermediates exceeded 75% (Figure 2F).
Relationships among MIDs and other features are summarized in Figure S3 and Table S2 and illustrated as a dendrogram using absolute correlation-based distances (Figure 2G). The same isotopologues are strongly correlated at 6h and 24h, indicating that the cell lines approached isotopic steady state within these labeling periods (Figure S3, Table S2). Strong correlations are enriched between isotopologues from metabolites in the same pathway, as observed among TCA cycle intermediates or between serine and glycine. Surprisingly, there was a paucity of associations between 13C labeling and nutrient utilization, cell growth or nutrient dependence. Thus, even though pathways reported by 13C labeling contribute to growth, the prominence of a given set of labeling features predicts neither the growth rate nor nutrient dependence. Despite the overall paucity of associations between 13C and non-13C features, we captured some expected associations, including correlations between Lac, LacG6m3 and LacG24m3 (Figure S3).
Glucose-dependent anaplerosis inferred from [U-13C]glucose labeling predicts dependence on pyruvate carboxylase.
Alternative positional labeling of precursors can simplify interpretation of metabolite labeling. For example, [3,4-13C]glucose is a preferred tracer to detect glucose-dependent anaplerosis via pyruvate carboxylase (PC). This tracer differentiates PC-dependent vs. PDH-dependent entry of glucose carbon into the TCA cycle, because [3,4-13C]glucose is converted to [1-13C] pyruvate. PDH removes the labeled carbon as 13CO2, whereas PC transfers the label to OAA so that it is retained in TCA cycle intermediates as m+1 isotopologues (Figure 3A). When [U-13C]glucose is the precursor, glycolysis produces [U-13C]pyruvate, and both PDH and PC transfer label from [U-13C]pyruvate to the TCA cycle, producing m+2 or m+3 isotopologues on the first turn. However, combined contribution of PDH and PC, coupled with multiple turns of the cycle, complicate interpretation of MIDs from [U-13C]glucose (Figure 3B and S4A). It would be advantageous to infer PC activity from [U-13C]glucose, because a) anaplerosis contributes to biomass assimilation and cell growth; b) PC is the primary anaplerotic route in some tumors in vivo and accounts for glutamine-independent growth in some cells (Cheng et al., 2011; Sellers et al., 2015); and c) the high cost of [3,4-13C]glucose relative to [U-13C]glucose limits use of the former tracer in human in vivo experiments.
Figure 3. Inferring pyruvate carboxylase contribution from enrichment of malate isotopologues.
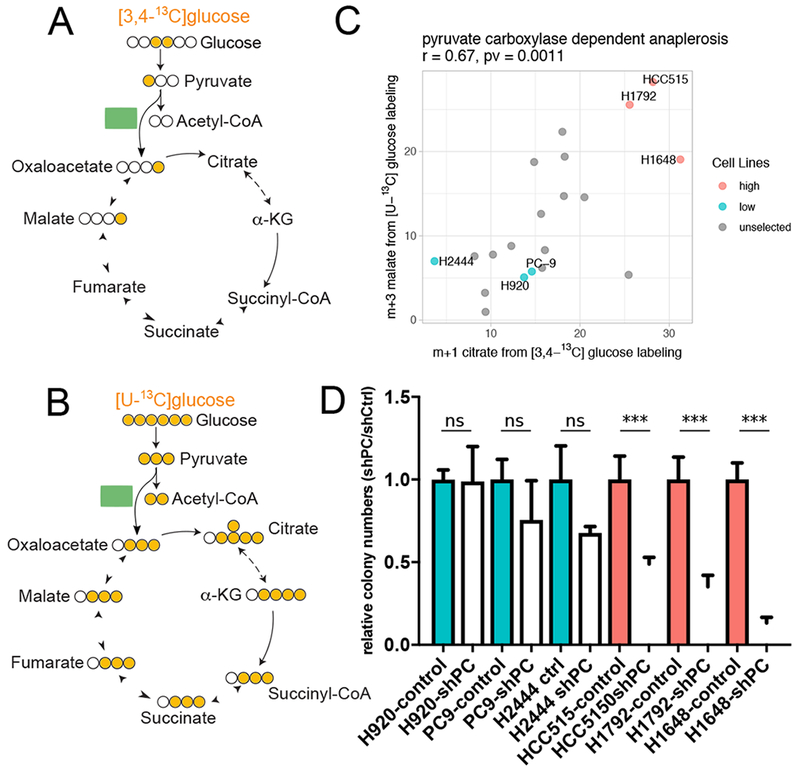
(A and B). Signature isotopologues produced from pyruvate carboxylation with [3,4-13C]glucose (A) or [U-13C]glucose (B) labeling.
(C). Scatter plot showing correlation between m+1 citrate from [3,4-13C]glucose and m+3 malate from [U-13C]glucose. Cell lines selected for PC dependence testing in (D) are indicated with labels. Correlation coefficient and p-value from Pearson correlation are provided in title.
(D). Effect of PC silencing on colony formation in cells lines predicted to have high (red) or low (blue) PC-dependent anaplerosis. Data are mean ± SEM. Statistical significance based on t-test, ***p < 0.005.
See also Figure S4.
To test whether [U-13C]glucose tracing can reliably report PC’s contribution to the TCA cycle, we cultured 20 cell lines in [3,4-13C]glucose and compared the resulting m+1 isotopogues to m+3 isotopologues derived from [U-13C]glucose. Specifically, we compared citrate m+1 with malate m+3; the latter species arises through malate dehydrogenase-dependent exchanges with OAA m+3, which was not abundant enough to detect in our samples. The strong positive correlation between these isotopologues indicates that [U-13C]glucose tracing reports contributions from PC (Figure 3C). MalG6m3 also correlates with PC mRNA abundance (Figure S4B). To remove the confounding effect of MalG6m3 arising from multiple turns of the TCA cycle, we fitted a linear regression model using CitG6m4, which also arises from multiple TCA cycle turns, to predict the portion of MalG6m3 arising independently of PC (Figure S4C). The residuals from this fit, which we take to arise from PC, correlate much better with PC mRNA expression (Figure S4D).
To test whether cells with high PC-dependent labeling of TCA cycle intermediates also had enhanced PC dependence, we selected three cell lines each with high (HCC515, H1792, H1648) or low (H920, PC9, H2444) PC-dependent labeling (Figure 3C). Each cell line was modified to express a control shRNA or an shRNA directed against PC. PC silencing reduced soft agar colony formation in cells with high but not low PC-dependent labeling (Figure 3D). Thus, mass isotopologues from [U-13C]glucose can be used to predict PC’s contribution to the TCA cycle, PC gene expression and dependence on PC for growth in soft agar.
Associations between MIDs and oncogenotypes.
Next we explored relationships between metabolic phenotypes and oncogenotypes. We clustered cells based on citrate MIDs after [U-13C]glucose labeling for 6h and examined mutations in recurrently-mutated NSCLC genes, including EGFR, KRAS and STK11 (Figure 4A–F). Generally, stronger associations were observed when site-specific rather than site-agnostic mutations were considered (Figure 4A). Examination of CitG6m0 demonstrates this point. Cells with EGFR mutations tended to have higher fractional contents of CitG6m0, but this was particularly striking for exon 19 deletions (Figure 4A,B). We then used our database of intra-operative [U-13C]glucose infusions in NSCLC patients to examine citrate labeling in human NSCLC. In these studies, patients with NSCLC receive [U-13C]glucose infusions during resection of the tumor (Faubert et al., 2017; Hensley et al., 2016). Fragments from EGFR-mutant tumors had higher citrate m+0 fractions than tumors with wild-type EGFR, but no differences were noted for adjacent lung samples from the same patients (Figure 4F). These findings indicate that some subtype-selective labeling phenotypes translate from cell lines to primary human tumors.
Figure 4. Associations between oncogenotypes and citrate mass isotopologues.
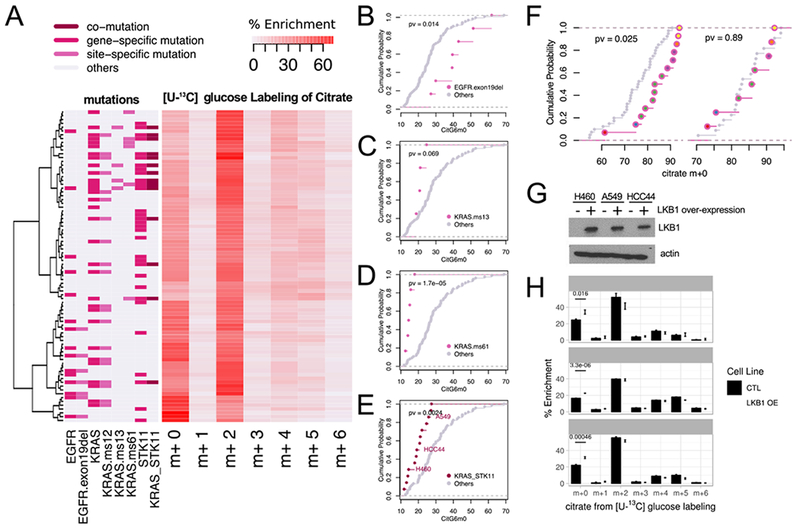
(A). Heatmap with hierarchical clustering of cell lines and citrate mass isotopologues from [U-13C]glucose labeling for 6h. Clustering was based on Ward’s minimum variance method. Relevant oncogenotypes are indicated.
(B-E). Cumulative distribution function plots showing different levels of CitG6m0 in cell lines with EGFR exon 19 deletion (B); KRAS missense mutation of the 13th codon (C); KRAS missense mutation of the 61st codon (D); and concurrent KRAS/STK11 mutation (E) compared to the rest of the cell lines (p-values based on K-S test). See also Table S3.
(F). Left, cumulative distribution function plots comparing citrate m+0 fractions between tumor fragments with or without EGFR mutations. Right, cumulative distribution function plots of the adjacent lung samples from the same patients. We compared 14 tumor fragments from 6 patients with EGFR mutations to 40 tumor fragments from 22 EGFR WT patients, whereas for the adjacent lung, we compared 8 fragments from 6 patients with EGFR mutations to 22 fragments from 22 EGFR WT patients. Patient origins of the EGFR mutant fragments are indicated by different colors inside the circles. (p-values based on K-S test)
(G). Western blot for LKB1 (encoded by STK11) in three cell lines with co-mutant KRAS and STK11. OE, over-expression.
(H). Re-expression of LKB1 in cell lines with co-mutant KRAS and STK11 increases CitG6m0 after labeling with [U-13C]glucose. Data are mean ± SEM.
Cells with KRAS mutations at Q61 have low CitG6m0, and KRAS G13 mutants display a similar trend (Figure 4A,C,D). KRAS-mutant cells with concurrent loss of function STK11 mutations have distinct vulnerabilities from cell lines with mutations in KRAS alone (Kim et al., 2017; Liu et al., 2013), and tumors with this combination of mutations have enhanced aggressiveness (Calles et al., 2015; Ji et al., 2007). We find that cells with concurrent KRAS/STK11 mutations have low CitG6m0 content, indicating a propensity to supply the TCA cycle with glucose carbon (Figure 4E). Re-constituting three co-mutant cell lines with wild-type STK11 resulted in a small but significant increase in CitG6m0, indicating that STK11 regulates glucose’s contribution to the TCA cycle (Figure 4G,H), consistent with previous studies (Faubert et al., 2014; Kim et al., 2013).
GDRC is associated with an epithelial state and sensitivity to EGFR inhibitors.
The unexpected prominence of GDRC in some NSCLC cells (Figure 2E) prompted us to examine orthogonal data for novel associations with this pathway. We used GSEA to identify transcriptional programs associated with GDRC, focusing on CitG6m0 and CitQ6m5. This yielded positive associations with genes targeted by the epithelial-mesenchymal transition (EMT)-related transcription repressor ZEB1 (Aigner et al., 2007) and negative associations with genes related to resistance to the EGFR inhibitor Gefitinib (Coldren et al., 2006) (Figure 5A,B and Figure S5A,B). These associations implied a relationship between GDRC and an epithelial phenotype characterized by EGFR signaling. Consistent with this idea, reversed-phase proteomics arrays (RPPA) revealed positive correlations between GDRC and both beta-catenin and E-cadherin, two components of the catenin-cadherin complex that maintains epithelial integrity (Figure 5C). GDRC also correlated with EGFR phosphorylation at Y1173 (pY1173), a marker for EGFR signaling (Figure 5C). Finally, comparing metabolism to drug sensitivity data (McMillan et al., 2018) revealed that GDRC cell lines tend to be sensitive to the EGFR inhibitor Erlotinib; these cell lines have low areas under the Erlotinib dose-response curve (AUC) (Figure 5C).
Figure 5. Reductive carboxylation is associated with an epithelial state and is enriched in cell lines sensitive to EGFR inhibitors.
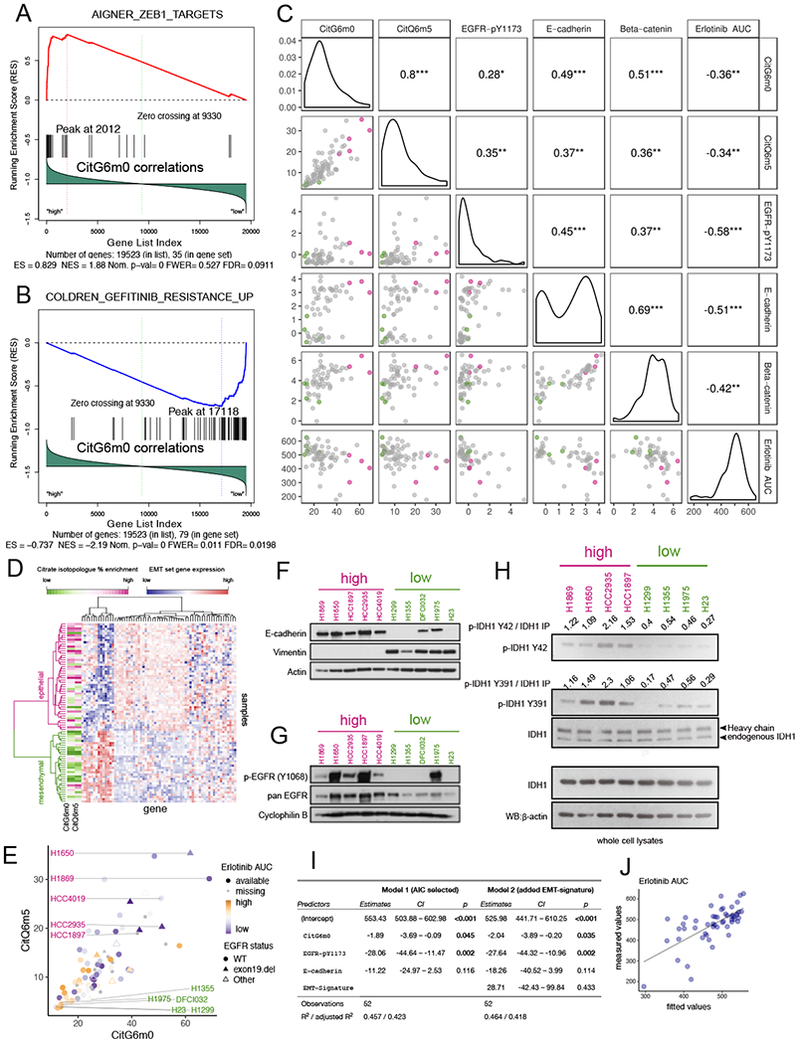
(A-B). Gene Set Enrichment Analysis (GSEA) identified CitG6m0 as positively correlated with ZEB1 target genes (A) and negatively correlated with Gefitinib resistance genes (B).
(C). Scatterplot and pairwise Pearson correlation among GDRC metabolic features CitG6m0, CitQ6m5; RPPA features beta-catenin, E-cadherin and EGFR-pY1173; and compound sensitivity feature Erlotinib AUC (higher area under the dosing curve represents higher resistance). The color scheme for points in the scatterplot is explained in the legend for (E). ***, p<=0.001; **, p<=0.01; *, p<=0.05.
(D). Heatmap with hierarchical clustering of samples and EMT signature genes. Clustering was based on Ward’s minimum variance method. The CitG6m0 and CitQ6m5 fractions are indicated by the color scale. Higher levels of GDRC metabolic features were observed for the epithelial cluster.
(E). Scatter plot of CitG6m0 and CitQ6m5 with EGFR mutation status marked by different symbols and EGFR inhibitor sensitivity indicated by different color and shapes. Five GDRC-high cell lines (magenta lettering) and five GDRC-low cell lines (green lettering) were selected for further characterization. These cell lines are also indicated by coloring in (C).
(F-G). Validation of EMT status and EGFR activation by western blot in 10 selected cell lines. In (F), the epithelial marker E-cadherin is expressed in GDRC-high cell lines, whereas the mesenchymal marker vimentin is expressed GDRC-low cell lines. In (G), p-EGFR (Y1068) indicative of EGFR activation is more prominent in GDRC-high cell lines.
(H). Higher phosphorylation of IDH1 on Y42 and Y391 in cell lines with high GDRC.
(I). Coefficients and p-values from multiple regression models predicting inhibitor sensitivity from different feature sets. Model 1 was obtained from stepwise feature selection based on Akaike information criterion (AIC) with input features including CitG6m0, CitQ6m5, EGFR-pY1173, E-cadherin, beta-catenin and EMT class. Model 2 adds the EMT-signature (EMT class) into Model 1. Note that p-values for CitG6m0 are significant in both models while controlling for the RPPA features or gene expression-derived EMT feature.
(J). Scatterplot of fitted values from model 1 in (I) and the measured value (Erlotinib AUC).
Sensitivity to EGFR inhibitors is predicted by an EMT signature defined by expression of 76 genes, with mesenchymal cells demonstrating resistance to EGFR inhibitors regardless of EGFR mutation status (Byers et al., 2013). Using this signature, we found that cells with high GDRC isotopologues were over-represented in the epithelial-like group (Figure 5D and Figure S5C). Lac/Glc and Glu/Gln features were similar between the two groups (Figure S5C). We selected five GDRC-high and five GDRC-low cell lines for further analysis (Figure 5E and Table S3). On western blotting, GDRC-high cells had high E-cadherin and low vimentin levels, consistent with the predicted epithelial state (Figure 5F). They also had abundant EGFR Y1068 phosphorylation, despite the fact that only two (H1650 and HCC1935) had detectable EGFR mutations (Figure 5G). In GDRC-low lines, Y1068 phosphorylation was only observed in H1975, one of the two EGFR-mutant lines selected in this class (Figure 5G). We also assessed the correlation between CitG6m0 and E-cadherin at the level of gene methylation, mRNA and protein expression and found the best correlation with protein expression (Figure S5D).
Phosphorylation of two IDH1 residues, Y42 and Y391, fosters GDRC (Chen et al., 2019). Y42 phosphorylation promotes substrate binding and formation of catalytically active IDH1 dimers, while Y391 phosphorylation promotes cofactor binding. These modifications result from a cooperative signaling cascade stimulated by recombinant EGF. The association of GDRC with EGFR signaling in our panel prompted us to compare Y42 and Y391 phosphorylation between cells with high and low GDRC-dependent citrate labeling. Among 8 cell lines, all four with high GDRC have prominent phosphorylation of IDH1 Y42 and Y391, and all four low-GDRC lines have little phosphorylation of these residues (Figure. 5H). Because mutating either Y42 or Y391 to phenylalanine reduces IDH1’s ability to engage in GDRC (Chen et al., 2019), the data suggest that tonic IDH1 phosphorylation in cells with high EGFR signaling may contribute to GDRC.
To exploit the potential of metabolic features as predictors of EGFR inhibitor sensitivity, we fitted a multiple regression model starting from a feature set that includes GDRC-related isotopologues (CitG6m0 and CitQ6m5); epithelial/mesenchymal classification based on EMT signature clustering; and the RPPA features EGFR-pY1173, E-cadherin and beta-catenin. Then we performed stepwise feature selection, with the model selected using Akaike information criterion (AIC). Features remaining in the final multiple regression model included CitG6m0, EGFR-pY1173 and E-cadherin, with significant p-values for CitG6m0 and EGFR-pY1173 (Figure 5I, Model 1 and Figure 5J). Importantly, when the EMT-signature binary classification feature was added to the model, the p-value for CitG6m0 remained significant, indicating that this metabolic feature predicts EGFR inhibitor sensitivity independently of features based on gene and protein expression (Figure 5I, Model2).
De novo serine synthesis is associated with pemetrexed sensitivity.
Serine has garnered increasing attention because of its roles in methionine, lipid, folate and nucleotide metabolism, all of which are important in cancer cells (Mattaini et al., 2016). In de novo serine synthesis, [U-13C]glucose is converted to serine m+3 via the glycolytic intermediate 3-phosphoglycerate (3PG). Serine m+3 is converted to glycine m+2 by serine hydroxymethyltransferases (SHMT), resulting in a high correlation between serine SerG6m3 and GlyG6m2 isotopologues (Figure 6A). A subset of these data were used to discover regulation of serine biosynthesis by NRF2 and ATF4 (DeNicola et al., 2015). Indeed, many transcripts involved in serine biosynthesis correlate with SerG6m3 (Figure S6A).
Figure 6. De novo serine synthesis from glucose is associated with sensitivity to pemetrexed.
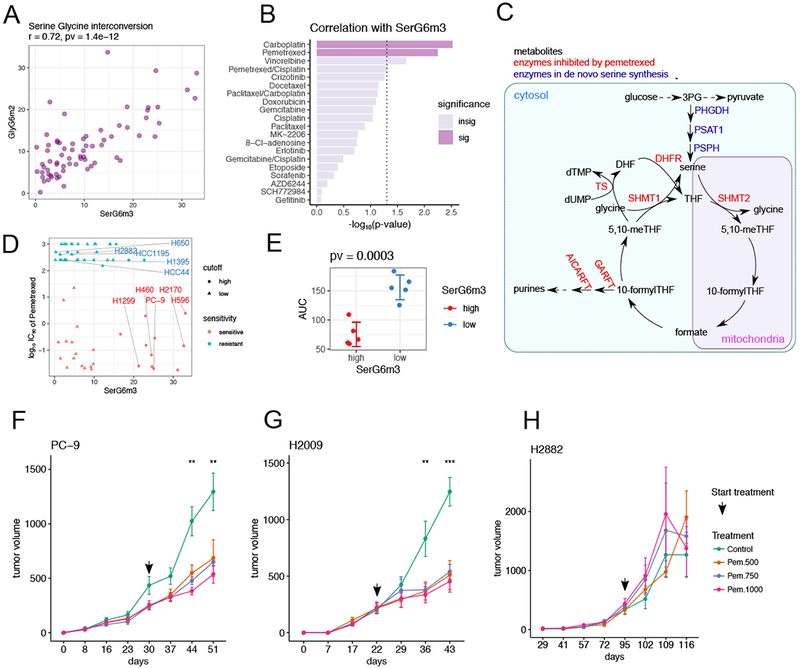
(A). Scatterplot showing positive correlation between SerG6m3 and GlyG6m2. Correlation coefficient and p-value from Pearson correlation are provided in the title.
(B). Drug sensitivity correlations with SerG6m3. −log10 (p-values) from Pearson correlations are plotted and the hits ranked by decreasing statistical significance. The dashed line demarcates the nominal p-value cut-off of 0.05 after −log10 transformation, and the darkly-colored bars denote statistical significance after multiple comparison controlled by beta uniform mixture modeling of p-values. See also Table S4.
(C). Schematic of serine biosynthesis feeding into one-carbon metabolism. Metabolites are in black, serine de novo synthesis enzymes are in blue and enzymes reportedly targeted by pemetrexed are in red.
(D). Relationship between SerG6m3 and pemetrexed IC50, and selection of cell lines for further characterization. Note that almost all the cell lines with high SerG6m3 are sensitive to pemetrexed. Cell lines are colored based on pemetrexed sensitivity. 5 pemetrexed sensitive cell lines with high SerG6m3 and 5 pemetrexed resistant cell lines with low SerG6m3 were selected for further characterization.
(E). Validation of pemetrexed sensitivity between selected cell lines with high and low SerG6m3 fractions. P-value from t-test is displayed in the title. Each dot represents average from triplicates for a single cell line; error bars denote mean ± SD for each group. See also Figure S6.
(F-H). In vivo testing of pemetrexed sensitivity in xenograft mouse models. PC-9 (F) and H2009 (G), cell lines sensitive to pemetrexed in culture, are also sensitive to pemetrexed in vivo, whereas the pemetrexed-resistant cell line H2882 (H) retains this resistance in vivo. The arrow indicates the time when pemetrexed therapy was initiated. Data are mean ± SEM. Statistical significance at each time point was determined by one-way ANOVA. ***, p<=0.001; **, p<=0.01. 4-5 mice were used for each treatment condition.
A search for correlations between SerG6m3 and therapeutic sensitivities (Table S4) identified the antifolate pemetrexed (Figure 6B). This was interesting because pemetrexed inhibits several folate-dependent reactions from one-carbon metabolism and hence impacts nucleotide biosynthesis (Curtin and Hughes, 2001; Daidone et al., 2011; Ducker and Rabinowitz, 2017) (Figure 6C). We selected 5 pemetrexed-sensitive/SerG6m3-high cell lines and 5 pemetrexed-resistant/SerG6m3-low cell lines for further characterization (Figure 6D) and validated their pemetrexed sensitivities (Figure 6E). Despite the link between serine metabolism and nucleotide synthesis, SerG6m3 was not associated with cell proliferation or BrdU staining (Figure S6B,C). Nutrient consumption and secretion were no different between the groups (Figure S6D–G), and no differences were observed in intracellular serine content or extracellular serine uptake (Figure S6H–J). These findings link serine biosynthesis to pemetrexed sensitivity through mechanisms that cannot fully be explained by cell proliferation rates or simple markers of nutrient exchange.
To assess whether SerG6m3 also predicts pemetrexed sensitivity in vivo, three cell lines (two with high and one with low SerG6m3) were implanted into nude mice. Pemetrexed treatment was initiated when the mice developed palpable tumors. Pemetrexed reduced tumor growth in both SerG6m3-high cell lines, but not in the SerG6m3-low cell line (Figure 6F–H).
Discussion
A growing appreciation of metabolic heterogeneity in cancer has increased the need to identify mechanisms that regulate metabolic preferences and dependencies in tumors. Many relationships between cancer genotypes and metabolism have been established with isogenic systems in which gain or loss of a single mutation is used to identify metabolic differences. These approaches are productive but cannot fully account for the impact of germline and somatic genetic heterogeneity on metabolism. Combining even two mutations can dramatically alter metabolism compared to either mutation alone. We performed an unbiased assessment of metabolism in a large panel of genetically diverse cell lines cultured under identical conditions. Therefore, metabolic features associated with a characteristic of interest (e.g. a single mutation) are by definition robust enough to withstand the mitigating effects of private differences.
A few limitations warrant mention. We used 21% oxygen and conventional media with a non-physiological nutrient composition. Recent studies have demonstrated the benefits of media formulated to mimic physiological conditions (Cantor et al., 2017; Vande Voorde et al., 2019). We used conventional culture so that we could integrate metabolic data with orthogonal data obtained under these same conditions. We are encouraged that several predictions arising from our phenotyping were validated in vivo. But studies in physiological media would have added value. Also, complementing our dataset with metabolomics would likely produce additional associations, as observed in other profiling studies (Li et al., 2019; Ortmayr et al., 2019).
NSCLC cells are remarkably diverse in the rate of glucose utilization and pathways supplied by glucose. Although the average Lac/Glc ratio was 1.35, the ratio was as low as 0.3 and varied 8-fold across the panel. Because this ratio reports the fraction of glucose carbon secreted as lactate, it is a surrogate for the Warburg effect. The Warburg effect is therefore not a universal feature of these cells, and Lac/Glc correlated only weakly with growth rates. Glucose consumption did not correlate with the growth rate, consistent with a previous study (Jain et al., 2012), but it did correlate with glutamine consumption, emphasizing the cooperative utilization of these two major nutrients in cancer cells.
The TCA cycle supports growth by producing intermediates for bioenergetics and biosynthesis. Despite the consistency of culture conditions, we observed diverse forms of the TCA cycle. All cell lines had extensive anaplerotic input into the TCA cycle, but no single anaplerotic pathway correlated with growth rates. As expected, each cell line demonstrated blending of multiple forms of the TCA cycle, and it will be interesting to study whether this reflects multiple activities operating concurrently in the same cell or heterogeneity across the population in the dish (e.g. different metabolic activities regulated by cell cycle stage). The most dominant pattern was the use of glucose as a precursor for acetyl-CoA and glutamine as a precursor for α-ketoglutarate, as indicated by the high CitG6m2 and CitQ6m4 fractions. Pyruvate carboxylation was also detected and was studied this pathway in detail because it is activated in human NSCLC (Sellers et al., 2015). Isotopologue signatures related to PC predicted both PC expression and dependence on PC for colony formation. Reductive metabolism of glutamine was surprisingly prominent given the evidence that this pathway is induced by metabolic stressors that suppress acetyl-CoA levels or electron transport chain function, none of which were anticipated in NSCLC cells growing in nutrient-replete conditions. A lack of strong correlations between GDRC labeling and either lactate secretion or the Lac/Glc ratio suggested that GDRC was not stimulated by major defects in oxidative metabolism in these cells.
Phenotyping these well-annotated cell lines provides the opportunity to observe metabolic features in aggregate and discover factors contributing to metabolic heterogeneity. Interestingly, while we identified some correlations between metabolic features and single oncogenic drivers, we found more with orthogonal data including DNA methylation, transcriptomics and protein expression. This implies that cell-autonomous control of cancer metabolism arises from the cumulative effects of cell lineage, epigenetics, the constitutional genome and multiple somatically-acquired mutations rather than from single drivers. Among the detected associations, cells with co-occurring mutations in KRAS and STK11 displayed higher overall labeling in citrate from glucose (i.e. lower CitG6m0) than the rest of the panel. This association was stronger than associations with either mutant KRAS or mutant STK11 alone, indicating metabolic cooperativity between these mutations as reported elsewhere (Kim et al., 2013; Kim et al., 2017; Liu et al., 2013). In contrast, EGFR mutations were associated with GDRC, indicating a different strategy to produce TCA cycle intermediates than in KRAS/STK11 co-mutants. We also noted a high content of unlabeled citrate in EGFR-mutant tumors from NSCLC patients subjected to intraoperative [U-13C]glucose infusions, increasing our confidence in the translational potential of the data set. To our knowledge, this is the first example of a correlation between an oncogenic driver and 13C labeling features that translates from cell culture to human tumors. Note that none of these patients were infused with 13C-glutamine, so it remains to be seen if GDRC is active in EGFR-mutant NSCLCs in patients.
In some cases, metabolism correlated with therapeutic sensitivity. GDRC was accompanied by enhanced sensitivity to EGFR inhibitors, as predicted by the epithelial signatures in these cells. Importantly, GDRC correlated with sensitivity to EGFR inhibition even in cells lacking canonical EGFR mutations. This is potentially important because intra-operative isotope infusions in cancer patients may support in situ detection of metabolic phenotypes relevant to therapy. Predicting sensitivity to conventional chemotherapy is challenging, as we lack markers to guide deployment of these agents. Metabolic phenotyping may provide such markers. A recent study identified a relationship between sensitivity to the antifolate methotrexate and histidine catabolism, which consumes tetrahydrofolate and renders cells susceptible to the impact of methotrexate on folate pools (Kanarek et al., 2018). We find that sensitivity to pemetrexed, another antifolate, is predicted by de novo serine/glycine synthesis from glucose. This association may arise from the fact that many of the reactions supplied by serine’s contribution to the folate pool are thought to be inhibited by pemetrexed.
Metabolic phenotypes arise from the complex interplay of factors intrinsic and extrinsic to cancer cells. It is remarkable that the cell lines studied here had such heterogeneous phenotypes despite our use of uniform culture conditions to isolate cell-autonomous regulation of metabolism. Our findings emphasize that diverse metabolic activities can support rapid proliferation of malignant cells. We anticipate that defining cell-autonomous metabolic diversity will contribute to ongoing efforts to understand metabolic phenotypes of human tumors in vivo.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ralph J. DeBerardinis (Ralph.DeBerardinis@UTSouthwestern.edu).
Experimental Model and Subject Details
Cell culture
Most NSCLC lines used in this study were part of the NCI and HCC (Hamon Cancer Center at UT Southwestern) series, with the exception of A549, Calu-1, Calu-6 (American Type Culture Collection; ATCC), DFCI024, DFCI032 (Dana Farber Cancer Institute, courtesy of Pasi Janne) and PC-9 (Johns Hopkins University School of Medicine, courtesy of Bert Vogelstein). Cell lines were DNA fingerprinted with PowerPlex Fusion24 (Promega). All cell lines were cultured in Roswell Park Memorial Institute medium (RPMI) with 10 mM glucose and 2 mM glutamine supplemented with 5% FBS (Sigma). Nutrient deprivation and metabolic labeling experiments were conducted in RPMI supplemented with 5% dialyzed FBS (Hyclone), sodium bicarbonate (42.5 mM), HEPES (25 mM), Penicillin/Streptomycin (10 U and 10 μg/mL, respectively) and glucose/glutamine as indicated.
Xenograft studies
Female NOD/SCID mice were obtained from Wakeland laboratories, UT Southwestern Mouse Breeding Core (Dallas, TX) at approximately 6-8 weeks of age. 1x106 cancer cells were injected subcutaneously into the shaved right flank and monitored by calipers (v = π/6*|*w2; v, volume; l, length, w, width). All mouse procedures were performed in compliance with UT Southwestern IACUC policies.
Method Details
Stable Isotope labeling
Dishes of 80–90% confluent cells were rinsed twice in PBS, then overlaid with medium containing the isotopically enriched nutrient and cultured for 6 hours or 24 hours. Note that 4mM glutamine was used because some cell lines depleted the medium of glutamine within 24h if 2mM was used. For analysis of intracellular metabolites by GC-MS, cells labeled in 6-cm dishes were rinsed in ice-cold normal saline and lysed with three freeze-thaw cycles in cold 50% methanol. The lysates were centrifuged to remove precipitated protein, a standard (50 nmols of sodium 2-oxobutyrate) was added, and the samples were evaporated and derivatized by trimethylsilylation (Tri-Sil HTP reagent, Thermo Scientific). Three microliters of the derivatized material were injected onto an Agilent 6970 gas chromatograph equipped with a fused silica capillary GC column and networked to an Agilent 5973 mass selective detector. Retention times of all metabolites of interest were validated using pure standards. The abundance of the following ions was monitored: m/z 245-249 for fumarate, m/z 335-339 for malate, m/z 219-222 for lactate, m/z 306-309 for serine, m/z 276-278 for glycine, and m/z 465–471 for citrate.
Nutrient utilization rates
To measure metabolic rates, one million cells were plated into 6-cm dishes and cultured until 90% confluent. At time 0, the cells were rinsed in PBS, fed with 1.5 mL of RPMI with 10 mM glucose, 2 mM glutamine and dialyzed FBS, and cultured. End-point experiments proceeded for 7 hours, then the medium was collected and analyzed for metabolite abundance. Concentrations of glucose, lactate, glutamine, and glutamate were determined from 0.6-mL aliquots of medium using an automated electrochemical analyzer (BioProfile Basic-4 analyzer; NOVA). Metabolic rates were determined by normalizing absolute changes in metabolite abundances to final protein content. For estimated metabolic rates normalized to the average protein content over the 7 hour culture period, we assumed exponential growth throughout the 7 hour period and derived an average protein content from the Day3/Day1 and Day5/Day1 cell proliferation data.
Cell growth and nutrient dependence assay
To monitor proliferation, cells were seeded at 5,000/well in 48-well plates. The next day, cells were replenished with 0.5 ml of test medium (complete RPMI, RPMI without glucose or RPMI without glutamine). After 1, 3 and 5 days, DNA content was determined by adding 0.25 ml water to each well and freezing at −80°C for 2 hr. The cells were then warmed to room temperature, and 0.5 ml of 0.1 μg/ml Hoechst 33258 in TNE buffer (2 M NaCl, 10 mM Tris-HCl [pH 7.4], and 1 mM EDTA) was added. The plate was incubated in the dark at room temperature, and O.D. at 350 nm was measured using a plate reader. Cell proliferation and survival measurements were made by normalizing DNA content of cells from Day 3 and Day 5 to that of Day 1.
Protein expression
Whole-cell lysates were prepared in RIPA buffer and quantified using the BCA protein assay (Thermo Scientific). Protein was separated by SDS/PAGE and transferred to a PVDF membrane. The membrane was blocked overnight at 4°C in PBS with Tween 20 (PBST) containing 5% milk, then probed with primary antibodies overnight at 4°C.
LKB1 over-expression
The MigCD8t control and LKB1 overexpression plasmids were kindly provided by Dr. Russell Jones (Van Andel Research Institute). Selected cell lines were infected with supernatants containing MigCD8t or MigCD8t-LKB1 retrovirus. Three days later, flow cytometry was used to enrich for infected cells expressing the CD8t surface marker (Nicolas Loof, the Moody Foundation Flow Cytometry Facility of UTSW).
RNA Interference
shPC was cloned into pLKO.1 backbone purchased from Open Biosystems. Lentiviral particles were produced by co-transfecting 293FT cells with the lentiviral construct, pCMVΔR8.91, and pMD2.G using polyjet (SignaGen Laboratories). Virus-containing supernatant was collected 2 days after the transfection and used to infect cells. Puromycin (1 μg/mL) was added 2 days after infection, and selection was continued for 7 days before any experiments.
Soft agar colony formation assay
1000 cells were plated into 12-well plates with 0.33% Noble agar (Difco). After 2 weeks, the cultures were stained with 0.05% crystal violet in 20% methanol. Colonies >200 μm in diameter were counted.
Immunoprecipitation
Cell lysates (1–2 mg) were incubated with anti-IDHl antibody overnight at 4°C. After incubation, protein G-Sepharose was used for precipitation for 2 hours. The beads were then washed 3 times with 1× TBS and eluted by boiling in SDS sample buffer for Western blotting.
BrdU staining
Cells were cultured with 10μΜ BrdU for one hour and fixed in 70% ethanol at −20°C. After washing with phosphate/citric buffer (40ml Na2HPO4 with 4ml 0.1M citric acid), the cells were incubated with anti-BrdU antibody for one hour, then stained with propidium iodide (PI). For PI/RNase staining, cells were incubated with 0.5 mL of PI/RNAse staining buffer (BD Pharmingen) at 25°C for 15 minutes, then analyzed by flow cytometry.
Free amino acid concentration quantitation
Consumption and secretion of amino acids were measured by HPLC (Hitachi; L8900). One million cells were cultured in 6cm dishes and switched to RPMI medium with dialyzed FBS for 7 hours. Medium samples from before and after culture were combined 1:1 with 1M sulfosalicylic acid, followed by centrifugation at 10000 rpm for 15 min at 4°C. The supernatant was then combined with an AEC standard (1:10 by volume) and analyzed by HPLC.
Pemetrexed sensitivity assay
Selected cell lines were treated with different doses (0.01, 0.05, 0.1 and 1 μM) of pemetrexed for 3 days. A standard 4-parameter log-logistic fit between the survival rate and the dosage was generated by the “drm” function from the R package “drc” (Ritz et al., 2015). Areas under the fitted dose response curve were calculated and compared by t-test.
Quantification and Statistical Analysis
All of the statistical details of experiments can be found in the figure legends, results and code deposited to GitHub. Majority of the statistical analyses were conducted in R (Team, 2010).
Normalization of nutrient utilization to average protein content
If the initial cell number is n0 and the doubling time is a, assuming exponential growth, the cell number at time t can be calculated by the following equation:
Our dataset includes two measurements that estimate proliferation rates, Day3/Day1 and Day5/Day1. Doubling times inferred from both measurements were averaged. Nutrient utilization rates were reestimated to account for drift in cell number during the 7-hr growth using this doubling time estimate (by multiplying a factor of to normalize each rate to the average rather than final protein content). This average protein content normalized nutrient utilization rates were compared to the final protein content normalized nutrient utilization rates in Figure S1A.
Stable isotope labeling data analysis
The measured distribution of mass isotopologues was corrected for natural abundance of 13C using Metran (Yoo et al., 2008). Results from multiple experiments were averaged. Fractional carbon contribution was calculated by , where n is the number of carbon atoms in the metabolite of interest, i is the number of 13C nuclei in an isotopologue for that metabolite, and s is the relative fraction of each isotopologue.
Correlation and partial correlation
R package “Hmisc”(Frank E Harrell Jr, 2018) was used for calculating correlations and “ppcor”(Kim, 2015) was used for calculating partial correlations, and R package “corrplot” was used for correlation matrix visualization (Simko, 2017).
Gene set enrichment analysis (GSEA) and single sample GSEA (ssGSEA)
The C2 (curated gene sets) library was downloaded from Molecular Signatures Database (MSigDB). GSEA was performed in continuous mode with the Spearman rank correlation as the ranking metric. The analysis used an R script adapted from R-GSEA. Scores from ssGSEA were calculated by implementing the “gsva” function from the R package “GSVA” with method specification as “ssgsea” (Hanzelmann et al., 2013).
Model based classification of pemetrexed sensitive and resistant cell lines
R package “mclust” (Scrucca et al., 2016) was used for classification of pemetrexed sensitive and resistant lines based on Gaussian mixture model clustering.
Stepwise regression
Feature selection for the Erlotinib sensitivity prediction model was performed by stepwise regression using “stepAIC” function with “both,” as directed by the R package “MASS”. Coefficient tables from multiple regression models were visualized with the “tab_model” function from the R package “sjPlot”.
Web Application
Web application was written in R Markdown (Grolemund, 2018; Iannone, 2019). Code has been deposited to https://github.com/cailing20/NSCLC_Metabolism_web/.
Other Statistical Analyses
Hierarchical clustering, two-sided two-sample t-tests, two-sided two-sample Kolmogorov-Smirnov (K-S) test and density calculations were performed using the “stats”(Team, 2010) package in R. Heatmaps were generated by R packages “heatmap3”(Shyr, 2015) and “ComplexHeatmap” (Gu et al., 2016). R packages “ggplot2”(Wickham, 2016), “GGally”(Barret Schloerke, 2018), “ggpubr”(Kassambara, 2018), “ggrepel”(Slowikowski), “cowplot”(Wilke, 2019) and “gridExtra”(Auguie, 2017) were also used for visualization. R package “RColorBrewer”(Neuwirth, 2014) was used for color specification. For multiple comparison adjustment, beta-uniform mixture model was fitted by the “Bum” function in R package “ClassComparison”(Coombes, 2018) and the false discovery rate (FDR) was set at 0.05 to determine the number of significant p-values. Enhanced visualization of dendrogram was generated by “fviz_dend” function from R package “factoextra”(Mundt, 2017). R package “openxlsx”(Walker, 2018) was used for reading and writing data.
Data and Code Availability
The datasets and code generated during this study are available at GitHub: https://github.com/cailing20/NSCLC_Metabolism
Supplementary Material
Table S1. Metabolic features, related to Figure 1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin | Cell signaling | 4970S |
| Cyclophilin B | Proteintech | 11607-1-AP |
| LKB1 | Cell signaling | 3047S |
| E-cadherin | Cell signaling | 3195P |
| Vimentin | Cell Signaling | 5741P |
| p-EGFR (Y1068) | Cell signaling | 3777P |
| Brdu | ThermoFisher | BDB347583 |
| IDH1 | Shanghai Genomics, Inc. | N/A |
| p-IDH1 (Y42) | Shanghai Genomics, Inc. | N/A |
| p-IDH1 (Y391) | R & D Systems | Cat# MAB7049 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| pemetrexed (cell line validation) | Medchem Express | Cat# HY-10820A |
| pemetrexed (xenograft validation) | Eli Lilly | N/A |
| [U-13C] glucose | Cambridge Isotopes | Cat# CLM-1396 |
| [3,4-13C] glucose | Cambridge Isotopes | CLM-6750 |
| [U-13C] glutamine | Cambridge Isotopes | CLM-1822 |
| Puromycin | Sigma | Cat# P8833 |
| Critical Commercial Assays | ||
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat# 23225 |
| Deposited Data | ||
| NSCLC cell line microarray data | (Kim et al., 2016) | GEO: GSE32036 |
| NSCLC cell line mutation data | (McMillan et al., 2018) | described in table S4 |
| NSCLC cell line RPPA data | This paper | described in table S4 |
| NSCLC cell line methylation data | (Walter et al., 2012) | described in table S4 |
| NSCLC cell line Erlotinib sensitivity data | (McMillan et al., 2018) | N/A |
| NSCLC cell line Pemetrexed sensitivity data | This paper | described in table S4 |
| Experimental Models: Cell Lines | ||
| NSCLC cell line collection | This paper | described in table S4 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJArc | UT Southwestern | RRID:IMSR_ARC:NSG |
| Recombinant DNA | ||
| pLKO.1 | Open Biosystems | N/A |
| pCMVΔR8.91 | (Cheng et al., 2011) | N/A |
| pMD2.G | (Cheng et al., 2011) | N/A |
| MigCD8t | (Faubert et al., 2014) | N/A |
| MigCD8t-LKB1 | (Faubert et al., 2014) | N/A |
| Oligonucleotides | ||
| shPC (CCGGGCCAAGGAGAACAACGTAGATCTCGAGATCTACGTTGTTCTCCTTGGCTTTTTG) | (Cheng et al., 2011) | N/A |
| Software and Algorithms | ||
| GC MSD Chemstation | Agilent | https://www.agilent.com/en/products/software-informatics/massspec-workstations/gc-msd-chemstation-software |
| R Version 3.3.2 | R Core Team, 2016 | https://www.r-project.org/ |
| Gene Set Enrichment Analysis | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/downloads.jsp |
| GSVA | (Hanzelmann et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| drc | (Ritz et al., 2015) | https://cran.r-project.org/web/packages/drc/index.html |
| Other | ||
| Fetal bovine serum | Gemini Bio-Products | Cat# 100-106 |
| Dialyzed fetal bovine serum | Gemini Bio-Products | Cat# 100-108 |
| Tri-Sil HTP reagent | ThermoFisher | N/A |
| Difco™ Noble Agar | VWR | N/A |
| ECL Western Blotting Substrate | ThermoFisher | Cat# 32106 |
| PolyJet™ In Vitro DNA Transfection Reagent | SignaGen Laboratories | N/A |
Highlights.
Cell-autonomous metabolic diversity is reported in over 80 lung cancer cell lines
Heterogeneous metabolic phenotypes support lung cancer cell growth
Relating metabolic and molecular data uncovers new aspects of metabolic regulation
Some metabolic features predict sensitivity to chemotherapy and targeted agents
Acknowledgments
We thank Maithili Dalvi, Robin Frink, Rachel Greer and Sunny Zachariah for generating compound sensitivity data. We thank Julia Kozlitina for advice about data analysis. Genentech provided DNA methylation data. L.C. was supported by an American Association for Cancer Research Basic Cancer Research Fellowship (15 40 01 CAIL). R.J.D. is supported by the Howard Hughes Medical Institute, National Cancer Institute (NCI, R35CA22044901), Cancer Prevention and Research Institute of Texas (CPRIT, RP160089), Robert A Welch Foundation (Grant I-1733), Robert L. Moody, Sr. Faculty Scholar Endowment and Joel B. Steinberg, M.D. Chair in Pediatrics. J.D.M. is supported by CPRIT (RP160652) and NCI (P50CA70907), which also provided cell line resources for this project.
Declaration of Interests
R.J.D. is an advisor for Agios Pharmaceuticals. J.D.M receives licensing fees from the NCI and UT Southwestern to distribute cell lines. J.V.H is an advisor for AstraZeneca, Boehringer Ingelheim, Exelixis, Genentech, GSK, Guardant Health, Hengrui, Lilly, Novartis, Spectrum, EMD Serono, and Syntal; receives research support from AstraZeneca, Bayer, GlaxoSmithKline, Spectrum, Takeda; and receives royalty and licensing fees from Spectrum, Biotree. L.A.B receives research support from AbbVie, AstraZeneca, GenMab, Tolero Pharmaceuticals and Sierra Oncology. M.A.W. and E.A.M. are currently employed by Pfizer Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P et al. (2007). The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26, 6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs LK, and DeBerardinis RJ (2015). Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, DeBerardinis RJ, Feron O, Frezza C, Ghesquiere B et al. (2015). A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol 34, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. (2013). An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calles A, Sholl LM, Rodig SJ, Pelton AK, Hornick JL, Butaney M, Lydon C, Dahlberg SE, Oxnard GR, Jackman DM, et al. (2015). Immunohistochemical Loss of LKB1 Is a Biomarker for More Aggressive Biology in KRAS-Mutant Lung Adenocarcinoma. Clin Cancer Res 21, 2851–2860. [DOI] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr., Lewis CA, and Sabatini DM (2017). Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272 e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xia S, Wang M, Lin R, Li Y, Mao H, Aguiar M, Famulare CA, Shih AH, Brennan CW, et al. (2019). Mutant and Wild-Type Isocitrate Dehydrogenase 1 Share Enhancing Mechanisms Involving Distinct Tyrosine Kinase Cascades in Cancer. Cancer Discov 9, 756–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, and DeBerardinis RJ (2011). Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A 108, 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coldren CD, Helfrich BA, Witta SE, Sugita M, Lapadat R, Zeng C, Baron A, Franklin WA, Hirsch FR, Geraci MW, et al. (2006). Baseline gene expression predicts sensitivity to gefitinib in non-small cell lung cancer cell lines. Mol Cancer Res 4, 521–528. [DOI] [PubMed] [Google Scholar]
- Curtin NJ, and Hughes AN (2001). Pemetrexed disodium, a novel antifolate with multiple targets. Lancet Oncol 2, 298–306. [DOI] [PubMed] [Google Scholar]
- Daidone F, Florio R, Rinaldo S, Contestabile R, di Salvo ML, Cutruzzola F, Bossa F, and Paiardini A (2011). In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. Eur J Med Chem 46, 1616–1621. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, and Thompson CB (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104, 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. (2004). Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64, 3892–3899. [DOI] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. (2014). Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A 111, 2554–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS, Mueckler MM, Usher P, and Lodish HF (1987). Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 235, 1492–1495. [DOI] [PubMed] [Google Scholar]
- Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P et al. (2013). In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab 17, 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat, Lv et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu DN, Treaba D, Gilks CB, Leung S, Renouf D, Laskin J, Wood-Baker R, and Gown AM (2007). Nonsmall cell lung carcinoma with neuroendocrine differentiation--an entity of no clinical or prognostic significance. Am J Surg Pathol 31, 26–32. [DOI] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, and Mootha VK (2012). Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Chen L, and Rabinowitz JD (2018). Metabolomics and Isotope Tracing. Cell 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al. (2007). LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810. [DOI] [PubMed] [Google Scholar]
- Kanarek N, Keys HR, Cantor JR, Lewis CA, Chan SH, Kunchok T, Abu-Remaileh M, Freinkman E, Schweitzer LD, and Sabatini DM (2018). Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, Zubovych I, Seo BY, Kim J, Eskiocak B, Chung H et al. (2013). Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 155, 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, Bezwada D, Rodriguez-Canales J, Villalobos P, Lin YF, et al. (2017). CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, and Dang CV (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11, 325–337. [DOI] [PubMed] [Google Scholar]
- Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez D et al. (2019). The landscape of cancer cell line metabolism. Nat Med 25, 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Marks K, Cowley GS, Carretero J, Liu Q, Nieland TJ, Xu C, Cohoon TJ, Gao P, Zhang Y et al. (2013). Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov 3, 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaini KR, Sullivan MR, and Vander Heiden MG (2016). The importance of serine metabolism in cancer. J Cell Biol 214, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan EA, Ryu MJ, Diep CH, Mendiratta S, Clemenceau JR, Vaden RM, Kim JH, Motoyaji T, Covington KR, Peyton M et al. (2018). Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell 173, 864–878 e829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, and DeBerardinis RJ (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortmayr K, Dubuis S, and Zampieri M (2019). Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat Commun 10, 1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan KN, Egnatchik RA, Calvaruso MA, Wasti AT, Padanad MS, Boroughs LK, Ko B, Hensley CT, Acar M, Hu Z et al. (2015). Metabolic plasticity maintains proliferation in pyruvate dehydrogenase deficient cells. Cancer Metab 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D (2015). Dose-Response Analysis Using R. PLoS One 10, e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R et al. (2015). Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 125, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, and Dang CV (1997). c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A 94, 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA, Duran RV, and Gottlieb E (2010). Targeting metabolic transformation for cancer therapy. Nat Rev Cancer 10, 267–277. [DOI] [PubMed] [Google Scholar]
- van Wijk R, and van Solinge WW (2005). The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood 106, 4034–4042. [DOI] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, and Tardito S (2019). Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, and Chandel NS. (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107, 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, Shah KA, Cox GJ, Corbridge RJ, Homer JJ, et al. (2007). Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res 67, 3441–3449. [DOI] [PubMed] [Google Scholar]
- Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna JD, et al. (2018). Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res 7, 32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Metabolic features, related to Figure 1.
Data Availability Statement
The datasets and code generated during this study are available at GitHub: https://github.com/cailing20/NSCLC_Metabolism