

Abstract
Cantú syndrome (CS), characterized by hypertrichosis, distinctive facial features, and complex cardiovascular abnormalities, is caused by pathogenic variants in ABCC9 and KCNJ8 genes. These genes encode gain‐of‐function mutations in the regulatory (SUR2) and pore‐forming (Kir6.1) subunits of KATP channels, respectively, suggesting that channel‐blocking sulfonylureas could be a viable therapy. Here we report a neonate with CS, carrying a heterozygous ABCC9 variant (c.3347G>A, p.Arg1116His), born prematurely at 32 weeks gestation. Initial echocardiogram revealed a large patent ductus arteriosus (PDA), and high pulmonary pressures with enlarged right ventricle. He initially received surfactant and continuous positive airway pressure ventilation and was invasively ventilated for 4 weeks, until PDA ligation. After surgery, he still had ongoing bilevel positive airway pressure (BiPAP) requirement, but was subsequently weaned to nocturnal BiPAP. He was treated for pulmonary hypertension with Sildenafil, but failed to make further clinical improvement. A therapeutic glibenclamide trial was commenced in week 11 (initial dose of 0.05 mg–1 kg–1 day–1 in two divided doses). After 1 week of treatment, he began to tolerate time off BiPAP when awake, and edema improved. Glibenclamide was well tolerated, and the dose was slowly increased to 0.15 mg−1 kg−1day−1 over the next 12 weeks. Mild transient hypoglycemia was observed, but there was no cardiovascular dysfunction. Confirmation of therapeutic benefit will require studies of more CS patients but, based on this limited experience, consideration should be given to glibenclamide as CS therapy, although problems associated with prematurity, and complications of hypoglycemia, might limit outcome in critically ill neonates with CS.
Keywords: BiPAP, cardiomegaly, continuous positive airway pressure, hypertrichosis, osteodysplasia, patent ductus arteriosus, sulfonylurea
1. INTRODUCTION
First recognized as a distinct condition 37 years ago (Cantu, Garcia‐Cruz, Sanchez‐Corona, Hernandez, & Nazar, 1982), Cantú syndrome (CS) is a complex syndrome involving hypertrichosis and distinctive facial features, as well as a low frontal hairline, epicanthal folds, puffy eyelids, flat nasal bridge with broad nasal tip, long philtrum, macroglossia, and prominent mouth with full lips (Grange, Nichols, & Singh, 2014). Patients also exhibit a variety of cardiovascular, lymphatic, and fluid balance complications (Grange et al., 2014; Nichols, Singh, & Grange, 2013). Although the underlying cellular and tissue mechanisms of CS are complex and incompletely understood (Huang et al., 2018), the molecular basis is now clear: CS results from gain‐of‐function variants in the ABCC9 and KCNJ8 genes, which encode the regulatory ABCC9 (SUR2) sulfonylurea receptor and pore‐forming KCNJ8 (Kir6.1) subunits, respectively, of ATP‐sensitive potassium (KATP) channels (Brownstein et al., 2013; Cooper et al., 2014; Harakalova et al., 2012; van Bon et al., 2012). The realization that CS results from gain‐of‐function of SUR2‐dependent KATP channels raises the possibility that sulfonylurea drugs such as glibenclamide, which are potent and specific inhibitors of KATP channel activity (Nichols, 2006), may be appropriate therapy, as indeed they have proven to be for neonatal diabetes resulting from gain‐of‐function mutations in SUR1‐dependent KATP channels (Bowman et al., 2018; Pearson et al., 2006). So far, sulfonylureas have been limited to the treatment of diabetes, where they act to trigger secretion of insulin (Ashcroft, 2005), and have not reached clinical acceptance in cardiovascular diseases. Although the debate is still not resolved (Schramm et al., 2011), there is a possibility that blockade of cardiac KATP channels may be detrimental in conditions of myocardial ischemia, during which KATP channel activity is presumed to be cardioprotective.
Here, we describe our experience with glibenclamide therapy in a baby with CS. Treatment was initiated following incomplete success with surgical and medical intervention for pulmonary hypertension, and ongoing requirements for positive airway pressures to treat respiratory failure.
2. CLINICAL REPORT
2.1. Clinical diagnosis and initial therapy
The male patient was born prematurely at 32 weeks gestation with macrosomia (birthweight 3.6 kg [>>97th centile], length 48 cm [>>90th centile], head circumference 34.2 cm [>90th centile]), after a pregnancy complicated by polyhydramnios that required amnioreduction. There was no significant family history. In addition to polyhydramnios, features consistent with the diagnosis of CS, including hypertrichosis, edema, hypotonia and course facial features were evident at birth (Figure 1a).
Figure 1.
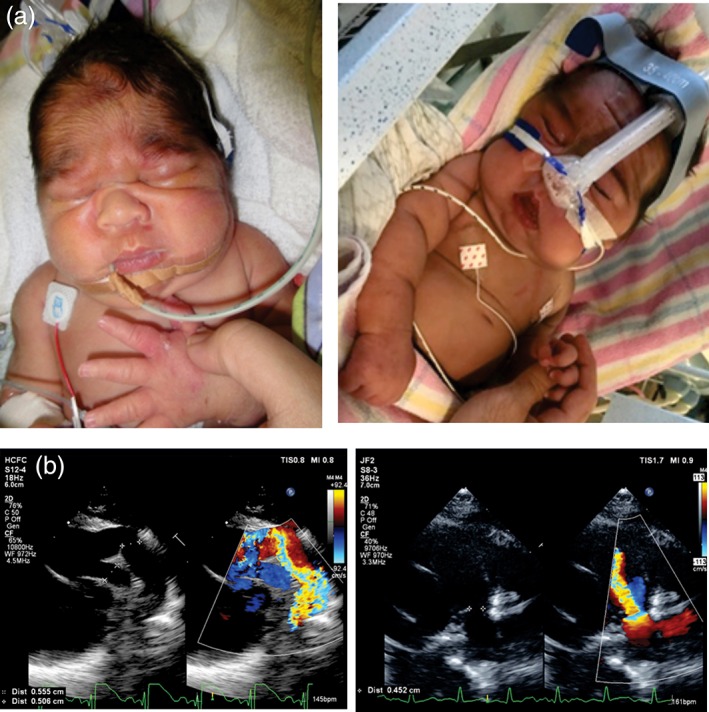
(a) Patient appearance in the newborn period (left), and immediately prior to starting on glibenclamide (right). Note coarse facial features, low anterior hairline, generalized hirsuitism, and prominent generalized edema. (b) Atypical PDA presentation. Patient PDA is tortuous, large and inserts proximally on the main pulmonary artery (left). Typical PDA for comparison is short, with conical insertion near the origin of the left pulmonary artery (right). PDA, patent ductus arteriosus
Initial echocardiogram identified a patent foramen ovale (PFO) with left to right flow and normal intracardiac anatomy. The left atrium was mildly dilated and he had mild mitral regurgitation. Subjectively, he also had mild hypertrophy of the interventricular septum with normal left‐ (LV) and right‐ventricular (RV) systolic function. He had a large tortuous patent ductus arteriosus (PDA) that inserted more proximally into the Main Pulmonary Artery than a typical PDA (Figure 1b). There was low velocity flow across the PDA suggesting elevated pulmonary artery pressures. He was initially managed for persistent pulmonary hypertension and received surfactant and continuous positive airway pressure ventilation; however, due to ongoing respiratory distress, he was invasively ventilated for 4 weeks. He underwent surgical ductal ligation and the echocardiogram immediately thereafter indicated elevated RV pressures, for which he was treated with sildenafil (1.5 mg–1 kg–1day–1). Bilateral inguinal hernias were also noted and surgically repaired, but he reherniated 1 day after his operation. By the third postoperative week, the RV pressure had reduced to less than half systemic pressure, the PFO had closed spontaneously, and he had normal biventricular size and systolic function. However, he had persistent ventilation requirements. Further testing with chromosome microarray analysis, white cell enzymology for lysosomal storage disorders and urine metabolic screen were normal.
2.2. Genetic diagnosis
Clinical genetic analysis was carried out in the New South Wales Health Pathology East Genomics Laboratory. Next‐generation whole exome sequencing was performed with an Ampliseq RDY exome kit, with libraries analyzed on a Life Technologies Proton instrument using a P1 v3 chip (ThermoFisher Scientific, USA). Alignment was performed with TorrentSuite v5.0.5 and data analysis based on Gemini v18 (https://gemini.readthedocs.io/en/latest/), and variants annotated and classified according to American College of Medical Genetics guidelines (Richards et al., 2015). Bioinformatic analysis, restricted to ABCC9 and KCNJ8, identified a heterozygous ABCC9 variant (c.3347G>A; p.(Arg1116His)), previously associated with CS and shown to generate gain‐of‐function in recombinant‐expressed KATP channels (Harakalova et al., 2012), consistent with the clinical diagnosis. The variant was confirmed by direct Sanger sequencing, and parental sequencing further confirmed that it arose de novo in the patient.
We analyzed recombinant KATP channels to assess the mutant channel properties and sulfonylurea sensitivity. Mutations were introduced into a rat SUR2A (pCMV_rSUR2A; GenBank accession No. D83598.1) cDNA construct using site‐directed mutagenesis (McClenaghan et al., 2018). Cultured Cosm6 cells, transfected with wild‐type pcDNA3.1_mKir6.2 (0.6 μg; GenBank accession No. D50581.1) and wild‐type or mutant pCMV_rSUR2A constructs (1 μg) were analyzed by patch clamp (McClenaghan et al., 2018). Dose–response curves (Figure S1A,B) confirmed that, while the intrinsic sensitivity to inhibition by ATP (in the absence of Mg2+) was no different from wild‐type channels, R1116H mutant channels were more strongly activated by MgATP and MgADP, explaining the overall gain‐of‐function. Importantly, some CS mutations are insensitive to glibenclamide. As shown in Figure S1D,F, R1116H channels were still very sensitive to glibenclamide, although ~10‐fold less sensitive than wild‐type channels (see Section 3).
2.3. Sulfonylurea therapy
Despite continuous bilevel positive airway pressure (BiPAP) and sildenafil (1.5 mg–1 kg–1 day–1), the patient failed to make further clinical improvement over the next 4 weeks. Therefore, a therapeutic trial of glibenclamide was commenced at Week 11, starting at a low dose of 0.05 mg–1 kg–1 day in two divided doses. This followed a previously developed protocol for initiation of glibenclamide therapy in SUR1‐related neonatal diabetes (Pearson et al. 2006). After a week of treatment, he was beginning to tolerate time off BiPAP when awake, and his edema had improved (Figure 1b). The dosage was slowly increased to 0.15 mg–1 kg–1 day–1 over the next 12 weeks. This period was complicated by a number of respiratory infections causing further increases in his level of respiratory support. However, he continued to make steady clinical progress and began to tolerate time off BiPAP, with maintained loss of edema and was weaned to nocturnal BiPAP over a 12‐week period. A small number of hypoglycemic episodes were noted, related to increasing his dosage of glibenclamide, but these were sporadic and not considered clinically significant (lowest blood glucose was 40 mg/dL [2.2 mmoL/L], but the majority were over 60 mg/dL [3.3 mmoL/L]). These hypoglycemic events were self‐limiting and spontaneously improved into the normal range. His pulmonary hypertension also improved, and his latest echocardiogram revealed no signs of cardiomegaly.
At the time of this report, the patient is 13 months old, and maintained on glibenclamide at 0.15 mg–1 kg–1 day–1, with fasting random blood glucose levels >76 mg/dL (>4.2 mmoL/L). Due to his clinical improvement, sildenafil was weaned after discharge from hospital at 5 months of age. He remains on BiPAP at night only during sleep, and requirements are 14/6 cm H2O×40 per minute, and there is no facial edema, although hypertrichosis is still evident (Figure 2c,d). Polysomnography was carried out, one‐third duration as diagnostic and two‐thirds duration as pressure titration. The diagnostic component demonstrated evidence of hypoventilation with CO2 rising from 44 mmHg at baseline to a maximum of 56 mmHg in REM sleep. His minimum saturation was 85%. Gas exchange normalized on pressures of 14/6×40/min. Most recent cardiac follow‐up a year post‐PDA ligation identified normal biventricular size and systolic function.
Figure 2.
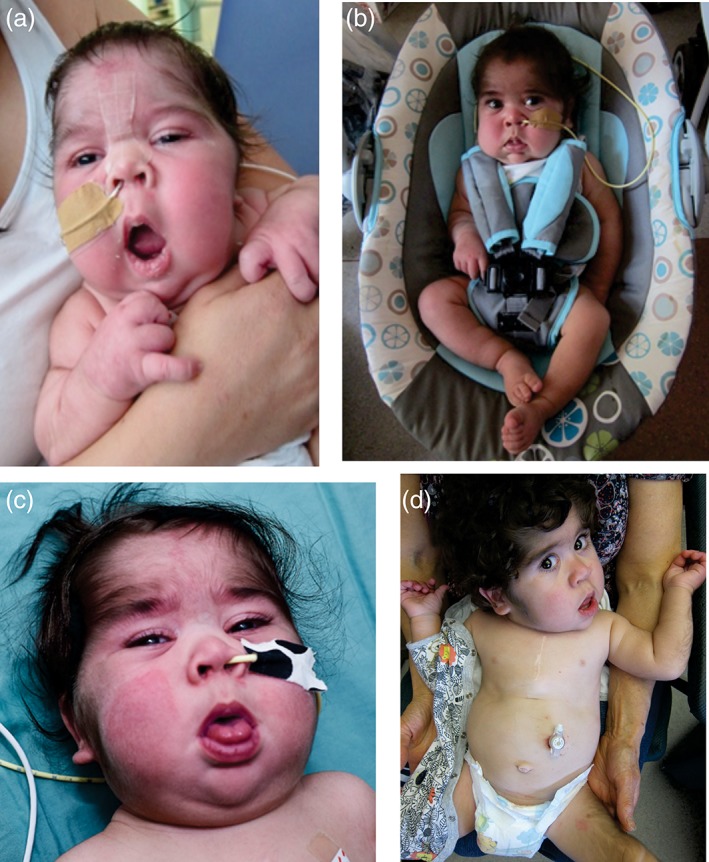
Patient appearance (a) 1 week after glibenclamide, (b) at 10 weeks after glibenclamide initiation, (c) at 5 months of age, 11 weeks after glibenclamide initiation, and (d) at 1 year of age. Note the relative normalization of facial features and markedly reduced edema, but persistent hirsutism throughout
3. DISCUSSION
3.1. The molecular basis of CS
In addition to polyhydramnios in utero, our patient exhibited typical CS features in the neonatal period, including PDA, marked edema, pulmonary hypertension, and evidence of an enlarged right ventricle. Since Kir6.1 and SUR2 subunits are the primary components of KATP channels in smooth muscles, and overactivity of these channels causes smooth muscle relaxation (Huang et al., 2018; Nelson & Quayle, 1995), persistence of the PDA may be explained as a consequence of maintained vessel dilation following birth. Reduced lymphatic smooth muscle tone could also underlie edema, and reduced airway smooth muscle contractility may affect breathing. Marked cardiac enlargement is found in most cases of CS (Levin et al., 2016), and may also be part of secondary compensation for reduced vascular tone (Huang et al., 2018). The reason for excess hair growth remains unclear (Rossi et al., 2012), but may be related to dilation of blood vessels increasing the supply of oxygen, blood, and nutrients to the hair follicle. Thus, while some CS features, that is, those resulting from smooth muscle relaxation, are likely to be a direct consequence of KATP overactivity, others are likely to be secondary pathologies of complex etiology (Huang et al., 2018). As such, they may respond with differing time courses or extent to reversal of KATP overactivity, an important point to consider when considering responsivity to sulfonylurea inhibition.
3.2. Sulfonylurea therapy for CS
Drugs that enhance KATP channel activity (e.g., diazoxide and minoxidil) exhibit side effects which closely parallel the features of CS, including hypertrichosis, pericardial effusion, and edema (Pennisi et al., 1977). Conversely, sulfonylurea drugs inhibit KATP channel activity and may therefore reverse CS features. However, clinical use of sulfonylureas has been historically restricted to the treatment of diabetes, where the therapeutic action is on the pancreatic‐expressed Kir6.2 and SUR1 KATP channel isoforms (Ashcroft & Gribble, 1999). These drugs have proven very effective in treatment of Kir6.2‐ or SUR1‐dependent neonatal diabetes (Pearson et al. 2006). There is a long‐standing debate as to potential negative cardiovascular side effects of these drugs (Gore & McGuire, 2011; Schramm et al., 2011) but the realization that CS results from Kir6.1 and SUR2 KATP channel gain‐of‐function now brings the so far untested opportunity for the use of KATP channel inhibitors as a potential targeted therapy in this syndrome.
In the absence of any controlled studies of sulfonylurea in CS, the decision to initiate glibenclamide as therapy in our patient was made on compassionate grounds, after exhausting conventional approaches to manage respiratory problems. Four weeks of mechanical ventilation and surgery to reverse the PDA, as well as surfactant and BiPAP ventilation provided only partial relief of respiratory distress, and he failed to make further improvement. Bronchopulmonary dysplasia (BPD) has been described previously in CS (Park, Koo, Jung, Lim, & Chung, 2014). In that case, BPD was progressive even after ventilator support with steroid therapy and a tracheostomy, and the baby developed recurrent and refractory pneumothoraces and sepsis, eventually dying of cor pulmonale, sepsis, and pneumothorax at 248 days of age (Park et al., 2014). In the case of our patient, however, 1 week following commencement of glibenclamide treatment, he could tolerate time off BiPAP, and there was clear improvement in edema (Figure 2). With increasing dosage, although complicated by respiratory infections that caused further increase in the level of respiratory support, he clearly made steady progress off BiPAP.
3.3. Efficacy and side effect concerns
It is tempting to conclude that glibenclamide was of benefit to our patient. While this single case is not definitive, clinical improvement of pulmonary hypertension, and lack of signs of cardiomegaly while on glibenclamide is at least suggestive of a normalizing action of the drug. Refractory pulmonary hypertension is a not uncommon finding in CS (Kobayashi, Cook, & Williams, 2010; Park et al., 2014; Scurr et al., 2011), and there is no evidence for otherwise spontaneous reversal of the cardiac enlargement that is typical in CS (Levin et al., 2016).
At the cellular level, there is good evidence that sulfonylureas act more potently on SUR1 channels than on SUR2, and that many CS mutations result in further decrease in SUR2 sensitivity to these drugs (Gribble & Ashcroft, 2000; McClenaghan et al., 2018). Thus glibenclamide or other available sulfonylureas are probably not ideal drugs for CS, and actions on SUR1 in the pancreas could yield significant side effects. There was the expected hypoglycemia upon initiation of glibenclamide in our patient, although this was not considered clinically concerning, and it spontaneously improved, as is also seen with chronic glibenclamide dosing in nondiabetic mice (Remedi & Nichols, 2008). Nevertheless, an agent with greater specificity for SUR2, or Kir6.1 would ultimately be preferable, and efforts to develop SUR2B (or Kir6.1) specific blockers are to be encouraged.
3.4. Prospects
We conclude that glibenclamide is likely to have contributed to clinical improvement of some CS features in our patient. There was no evidence that hypertrichosis was reversed, even after a year of treatment (Figure 2d), suggesting perhaps that this may be a permanent, neonatally induced consequence of CS. However, the drug was well tolerated: hypoglycemic episodes were minimal and spontaneously remitted. Sulfonylurea intervention was carried out after exhausting conventional therapies for the emergent symptoms. Our experience suggests both that sulfonylureas could be considered in the treatment of CS patients, and that it would be worthwhile to carry out controlled drug dosing and escalation trials in a full cohort of CS patients of different ages.
FUNDING INFORMATION
This study was partially supported by funds from US NIH grant R35 HL140024 (to CGN).
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Figure S1 (A) Representative inside‐out excised patch clamp recordings from Cosm6 cells transfected with Kir6.2 with wild type (top, black) or [R1112H] (equivalent to human [R1116H]) SUR2A (bottom, red). (B) Dose response for ATP inhibition of Kir6.2/SUR2A‐WT and Kir6.2/SUR2A[R1112H] channels, as determined from excised patch clamp recordings (n = 5 for WT; n = 6 for R1112H). (C) IC50 for ATP inhibition in the absence of magnesium, mean ± SEM shown as bars. (D) Representative records showing regulation by MgATP, MgADP, and glibenclamide. (E) Summary data show that SUR2A[R1112H] channels exhibit increased activity in the presence of 100 μM MgATP or 100 μM MgATP with 500 μM MgADP; MgATP and MgATP/MgADP. Mean ± SEM shown as bars, * denotes p < 0.05 according to Mann Whitney U Test (n = 5 for both). (F) Summary data show that R1112H channels are ~10‐fold less sensitive to glibenclamide than WT channels. Mean ± SEM shown as bars, * denotes p < 0.05 according to Mann Whitney U Test (n = 5 for both).
Ma A, Gurnasinghani S, Kirk EP, et al. Glibenclamide treatment in a Cantú syndrome patient with a pathogenic ABCC9 gain‐of‐function variant: Initial experience. Am J Med Genet Part A. 2019;179A:1585–1590. 10.1002/ajmg.a.61200
Funding information National Heart, Lung, and Blood Institute, Grant/Award Number: HL140024
This article was published online on 07 June 2019. An error was subsequently identified. This notice is included in the online and print versions to indicate that both have been corrected 20 June 2019.
REFERENCES
- Ashcroft, F. M. (2005). ATP‐sensitive potassium channelopathies: Focus on insulin secretion. The Journal of Clinical Investigation, 115(8), 2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, F. M. , & Gribble, F. M. (1999). ATP‐sensitive K+ channels and insulin secretion: Their role in health and disease. Diabetologia, 42(8), 903–919. [DOI] [PubMed] [Google Scholar]
- Bowman, P. , Sulen, A. , Barbetti, F. , Beltrand, J. , Svalastoga, P. , Codner, E. , … Nøddegård, R. (2018). Effectiveness and safety of long‐term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: An international cohort study. The Lancet Diabetes & Endocrinology, 6(8), 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein, C. A. , Towne, M. C. , Luquette, L. J. , Harris, D. J. , Marinakis, N. S. , Meinecke, P. , … Beggs, A. H. (2013). Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities—Support for the role of K(ATP) channels in this condition. European Journal of Medical Genetics, 56(12), 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu, J. M. , Garcia‐Cruz, D. , Sanchez‐Corona, J. , Hernandez, A. , & Nazar, Z. (1982). A distinct osteochondrodysplasia with hypertrichosis? Individualization of a probable autosomal recessive entity. Human Genetics, 60(1), 36–41. [DOI] [PubMed] [Google Scholar]
- Cooper, P. E. , Reutter, H. , Woelfle, J. , Engels, H. , Grange, D. K. , van Haaften, G. , … Nichols, C. G. (2014). Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Human Mutation, 35(7), 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore, M. O. , & McGuire, D. K. (2011). Resolving drug effects from class effects among drugs for type 2 diabetes mellitus: More support for cardiovascular outcome assessments. European Heart Journal, 32(15), 1832–1834. [DOI] [PubMed] [Google Scholar]
- Grange, D. K. , Nichols, C. G. , & Singh, G. K. (2014). Cantu syndrome and related disorders In Pagon R. A., Adam M. P., Ardinger H. H., Wallace S. E., Amemiya A., Bean L. J. H., et al. (Eds.), GeneReviews®. University of Washington: Seattle, WA. [PubMed] [Google Scholar]
- Gribble, F. M. , & Ashcroft, F. M. (2000). Sulfonylurea sensitivity of adenosine triphosphate‐sensitive potassium channels from beta cells and extrapancreatic tissues. Metabolism, 49(10: Suppl 2), 3–6. [PubMed] [Google Scholar]
- Harakalova, M. , van Harssel, J. J. T. , Terhal, P. A. , van Lieshout, S. , Duran, K. , Renkens, I. , … Cuppen, E. (2012). Dominant missense mutations in ABCC9 cause Cantú syndrome. Nature Genetics, 44(7), 793–796. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , McClenaghan, C. , Harter, T. M. , Hinman, K. , Halabi, C. M. , Matkovich, S. J. , … Nichols, C. G. (2018). Cardiovascular consequences of KATP overactivity in Cantu syndrome. JCI Insight, 3(15), pii: 121153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, D. , Cook, A. L. , & Williams, D. A. (2010). Pulmonary hypertension secondary to partial pulmonary venous obstruction in a child with Cantu syndrome. Pediatric Pulmonology, 45(7), 727–729. [DOI] [PubMed] [Google Scholar]
- Levin, M. D. , Singh, G. K. , Zhang, H. X. , Uchida, K. , Kozel, B. A. , Stein, P. K. , … Nichols, C. G. (2016). KATP channel gain‐of‐function leads to increased myocardial L‐type Ca2+ current and contractility in Cantu syndrome. Proceedings of the National Academy of Sciences of the United States of America, 113(24), 6773–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClenaghan, C. , Hanson, A. , Sala‐Rabanal, M. , Roessler, H. I. , Josifova, D. , Grange, D. K. , … Nichols, C. G. (2018). Cantu syndrome‐associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain‐of‐function by differential mechanisms. The Journal of Biological Chemistry, 293(6), 2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M. T. , & Quayle, J. M. (1995). Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology, 268(4 Pt 1), C799–C822. [DOI] [PubMed] [Google Scholar]
- Nichols, C. G. (2006). KATP channels as molecular sensors of cellular metabolism. Nature, 440, 471–476. [DOI] [PubMed] [Google Scholar]
- Nichols, C. G. , Singh, G. K. , & Grange, D. K. (2013). KATP channels and cardiovascular disease: Suddenly a syndrome. Circulation Research, 112(7), 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. Y. , Koo, S. H. , Jung, Y. J. , Lim, Y. J. , & Chung, M. L. (2014). A patient with Cantu syndrome associated with fatal bronchopulmonary dysplasia and pulmonary hypertension. American Journal of Medical Genetics Part A, 164A(8), 2118–2120. [DOI] [PubMed] [Google Scholar]
- Pearson, E. R. , Flechtner, I. , Njolstad, P. R. , Malecki, M. T. , Flanagan, S. E. , Larkin, B. , … Neonatal Diabetes International Collaborative Group . (2006). Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. New England Journal of Medicine, 355(5), 467–477. [DOI] [PubMed] [Google Scholar]
- Pennisi, A. J. , Takahashi, M. , Bernstein, B. H. , Singsen, B. H. , Uittenbogaart, C. , Ettenger, R. B. , … Fine, R. N. (1977). Minoxidil therapy in children with severe hypertension. The Journal of Pediatrics, 90(5), 813–819. [DOI] [PubMed] [Google Scholar]
- Remedi, M. S. , & Nichols, C. G. (2008). Chronic antidiabetic sulfonylureas in vivo: Reversible effects on mouse pancreatic beta‐cells. PLoS Medicine, 5(10), e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, A. , Cantisani, C. , Melis, L. , Iorio, A. , Scali, E. , & Calvieri, S. (2012). Minoxidil use in dermatology, side effects and recent patents. Recent Patents on Inflammation & Allergy Drug Discovery, 6(2), 130–136. [DOI] [PubMed] [Google Scholar]
- Schramm, T. K. , Gislason, G. H. , Vaag, A. , Rasmussen, J. N. , Folke, F. , Hansen, M. L. , … Torp‐Pedersen, C. (2011). Mortality and cardiovascular risk associated with different insulin secretagogues compared with metformin in type 2 diabetes, with or without a previous myocardial infarction: A nationwide study. European Heart Journal, 32(15), 1900–1908. [DOI] [PubMed] [Google Scholar]
- Scurr, I. , Wilson, L. , Lees, M. , Robertson, S. , Kirk, E. , Turner, A. , … Smithson, S. (2011). Cantu syndrome: Report of nine new cases and expansion of the clinical phenotype. American Journal of Medical Genetics Part A, 155A(3), 508–518. [DOI] [PubMed] [Google Scholar]
- van Bon, B. W. , Gilissen, C. , Grange, D. K. , Hennekam, R. C. , Kayserili, H. , Engels, H. , … Hoischen, A. (2012). Cantu syndrome is caused by mutations in ABCC9. American Journal of Human Genetics, 90(6), 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Representative inside‐out excised patch clamp recordings from Cosm6 cells transfected with Kir6.2 with wild type (top, black) or [R1112H] (equivalent to human [R1116H]) SUR2A (bottom, red). (B) Dose response for ATP inhibition of Kir6.2/SUR2A‐WT and Kir6.2/SUR2A[R1112H] channels, as determined from excised patch clamp recordings (n = 5 for WT; n = 6 for R1112H). (C) IC50 for ATP inhibition in the absence of magnesium, mean ± SEM shown as bars. (D) Representative records showing regulation by MgATP, MgADP, and glibenclamide. (E) Summary data show that SUR2A[R1112H] channels exhibit increased activity in the presence of 100 μM MgATP or 100 μM MgATP with 500 μM MgADP; MgATP and MgATP/MgADP. Mean ± SEM shown as bars, * denotes p < 0.05 according to Mann Whitney U Test (n = 5 for both). (F) Summary data show that R1112H channels are ~10‐fold less sensitive to glibenclamide than WT channels. Mean ± SEM shown as bars, * denotes p < 0.05 according to Mann Whitney U Test (n = 5 for both).