

Abstract
Background
Childhood hearing impairment affects language and cognitive development. Profound congenital sensorineural hearing impairment can be due to an abnormal cochleovestibular nerve (CVN) and cochleovestibular malformations, however, the etiology of these conditions remains unclear.
Methods
We used a trio‐based exome sequencing approach to unravel the underlying molecular etiology of a child with a rare nonsyndromic CVN abnormality and cochlear hypoplasia. Clinical and imaging data were also reviewed.
Results
We identified a de novo missense variant [p(Asn174Tyr)] in the DNA‐binding Homeodomain of SIX1, a gene which previously has been associated with autosomal dominant hearing loss (ADHL) and branchio‐oto‐renal or Branchio‐otic syndrome, a condition not seen in this patient.
Conclusions
SIX1 has an important function in otic vesicle patterning during embryogenesis, and mice show several abnormalities to their inner ear including loss of inner ear innervation. Previous reports on patients with SIX1 variants lack imaging data and nonsyndromic AD cases were reported to have no inner ear malformations. In conclusion, we show that a de novo variant in SIX1 in a patient with sensorineural hearing loss leads to cochleovestibular malformations and abnormalities of the CVN, without any other abnormalities. Without proper interventions, severe to profound hearing loss is devastating to both education and social integration. Choosing the correct intervention can be challenging and a molecular diagnosis may adjust intervention and improve outcomes, especially for rare cases.
Keywords: absent cochlear nerve, cochleovestibular nerve abnormalities, genetics of absent cochlear nerve, pediatric hearing loss
Our work describes a novel variant of SIX1 identified to likely be associated with congenital abnormality of the cochleovestibular nerve and congenital pediatric hearing loss.

1. INTRODUCTION
Congenital sensorineural hearing loss (SNHL) affects 2–3 per 1,000 live births (CDC, n.d.). Of those children born with profound SNHL, it is estimated that approximately 3% of these children will have an abnormal cochleovestibular nerve (CVN; i.e., cochlear nerve aplasia/hypoplasia/deficiency). While many of these children have concomitant cochleovestibular malformations, the etiology of both of these findings is not well understood. A classification system for malformations of the labyrinth has been widely adopted and is based on the various stages of embryogenesis (Jackler, Luxford, & House, 1987; Sennaroglu & Saatci, 2002). Several classification systems for abnormal CVNs have been proposed (Birman, Powell, Gibson, & Elliott, 2016; Buchman et al., 2011; Casselman et al., 1997; Govaerts et al., 2003; Kari, Go, Loggins, Emmanuel, & Fisher, 2018), but no one single system has been widely adopted. More recent literature (Birman et al., 2016; Kari et al., 2018) has proposed classifying abnormal CVNs based on the number of nerve bundles seen in the lateral internal auditory canal (IAC) on high‐resolution T2 structural magnetic resonance imaging (sMRI). Normal individuals have 4 and the cochlear nerve is considered normal caliber if its diameter is roughly equal to that of the facial nerve. Abnormal is considered any individual who has less than 4 or when the cochlear nerve is considered small (i.e., hypoplastic).
Cochlear implants (CIs) have been developed to restore hearing to a deaf ear by directly stimulating the auditory nerve. The hearing and language outcomes in children implanted before the age of 2 can vary but overall are extremely promising in allowing deaf children to hear near normally or normally and communicate like their normal hearing peers (Eisenberg, Fink, & Niparko, 2006; Fink et al., 2007; Niparko, 2004; Niparko et al., 2010). Children with cochleovestibular malformations had once been considered poor CI candidates but research has shown that their hearing and language outcomes are similar to their peers with normal labyrinthine architecture (Buchman et al., 2004, 2011). Children with abnormal CVNs, however, demonstrate much less robust outcomes with CIs. Despite the advent of auditory brainstem implants (ABIs) that bypass the CVN entirely and stimulate the cochlear nucleus directly at the brainstem, hearing, and language outcomes in children with abnormal CVNs who receive ABIs are similarly variable and at times no different than that of those with CIs (Birman et al., 2016; Buchman et al., 2004, 2011; Dettman et al., 2011; Farhood et al., 2017; Kari et al., 2018; Merkus et al., 2014; Sennaroglu et al., 2011; Young, Kim, Ryan, Tournis, & Yaras, 2012).
Children with abnormal CVNs who receive CIs exhibit outcomes that range from absolutely no benefit with no sound awareness at all to children who are able to achieve spoken language and open set speech recognition (Buchman et al., 2011; Freeman & Sennaroglu, 2018; Kari et al., 2018; Young et al., 2012). Unfortunately, there are no preoperative audiological, neuroimaging or neurophysiological data that can predict a child's response and outcomes with a CI or ABI. Consequently, children must undergo several interventions during a limited and precious window of time for acquisition of spoken language. Any delays in their ability to access sound has devastating consequences for their spoken language development.
Our prior work has identified de novo genetic variants in GREB1L in children with abnormal CVNs (Schrauwen et al., 2018). Of note, this work demonstrated that two individuals shared different loss‐of‐function variants in the same affected gene and similar phenotypes. Whereas the labyrinthine architecture and hearing status of children with abnormal CVNs are extremely variable, in these two individuals, their lack of any benefit from a CI and their absence of a CVN intraoperatively were similar (likely only facial nerve was present on both sides of both individuals). In other words, these children presented with more severe variants of CVN abnormalities, both had profound bilateral SNHL, both had no benefit from a CI, and neither had significant comorbid conditions or syndromes. The finding that these two individuals had mutations in the same gene was significant. Understanding the molecular biology of these and other cases could potentially lead to a genetic test preoperatively that may portend poor CI outcome and potentially save several individuals the need for sequential interventions that may not benefit the child.
In this paper, we evaluated the underlying molecular etiology of a child with a nonsyndromic malformation of the inner ear and CVN via trio‐based exome sequencing.
2. MATERIAL & METHODS
2.1. Ethical compliance
Informed consent was obtained from all participants. This study was done following the guidelines of the Institutional review board (IRB), and approval for human research was obtained (University of Southern California #HS‐14‐00513‐CR002 and Western IRB #20120512).
2.2. Clinical evaluation
This child was identified with hearing loss with the newborn hearing screen and was later evaluated for his hearing loss by a neurotologist at a tertiary academic referral CI center. The evaluation included a comprehensive history and neurotologic physical exam. His evaluation for a CI then also included a comprehensive audiological evaluation in which the testing performed was age appropriate and included otoacoustic emission testing, auditory brainstem response testing as well as behavioral audiometry. Pure tone audiometric testing and age appropriate speech understanding were evaluated. As part of his CI evaluation, he also underwent imaging with a magnetic resonance imaging (MRI) of the IACs. Clinical characteristics and imaging findings were reviewed by a neurotologist and an experienced neuroradiologist.
2.3. Exome sequencing
A DNA sample from the affected individual (male) and parents (family CN8_0013) was collected using the iSWAB DNA buccal collection kit (Mawi DNA Technologies) followed by extraction with the DNeasy Blood & Tissue kit (Qiagen). Exome sequencing was performed on both the affected individual and the parents. Exomic libraries were prepared with the TruSeq Exome Library Prep Kit, following the manufacturer's protocol (Illumina Inc). Sequencing was performed by 100 bp paired‐end sequencing on a HiSeq2500 instrument (Illumina Inc), with an average target coverage of 40x. Alignment to the Human genome (Hg19/GRCh37) was performed using the Burrows–Wheeler algorithm (BWA‐MEM) (Li & Durbin, 2010). Reads were sorted, polymerase chain reaction (PCR) duplicates were removed, base quality recalibration and indel realignment were performed using Picard and the Genome Analysis Toolkit (GATK) (McKenna et al., 2010). Variants were called jointly with HaplotypeCaller and recalibrated with GATK, and annotation was performed with ANNOVAR (Wang, Li, & Hakonarson, 2010). Variants with a high allele frequency in the Genome Aggregation Database (gnomAD) were removed (MAF >0.005). Bioinformatic prediction scores from dbNSFPv3.5 and dbscSNV1.1 were used for variant evaluation (Jian, Boerwinkle, & Liu, 2014; Liu, Wu, Li, & Boerwinkle, 2016). We considered various inheritance models, including de novo, autosomal recessive and X‐linked. The gender of each sample was confirmed by evaluating the zygosity of the X‐chromosome variants. Maternal and paternal relationships were verified by assessing the percentage of shared variants between parent/child.
Sanger sequencing was performed to validate variants of interest. In short, a PCR was performed and direct sequencing of the PCR product was performed on an ABI3130XL sequencer (Applied Biosystems Inc.).
3. RESULTS
3.1. Clinical findings
Here we describe an otherwise healthy male child who was born with bilateral profound SNHL, his bilateral unaided pure tone thresholds ranged 100–115 dB HL from 500 to 8000 Hz. The patient demonstrated no abnormalities of his genitourinary system, cardiac system or other organ system. Renal ultrasound and electrocardiogram testing confirmed no abnormalities. Imaging (see Figure 1) demonstrated a hypoplastic cochlear bud with an incomplete basal turn, absent modiolus and absent or bony cochlear aperture. The vestibular system was also noted to be abnormal with a small lateral semicircular canal. IAC diameters were normal bilaterally. The child had three nerves in both lateral IACs, normal being four nerves. He underwent a left‐sided CI and continued to use a hearing aid on his right ear. Aided testing with his hearing aid and CI used together demonstrated mild hearing loss thresholds (25–40 dB) and language assessment demonstrated ability to acquire closed set speech recognition at only 3 months postop from his CI surgery (52% Word Intelligibility on Picture Identification, WIPI).
Figure 1.
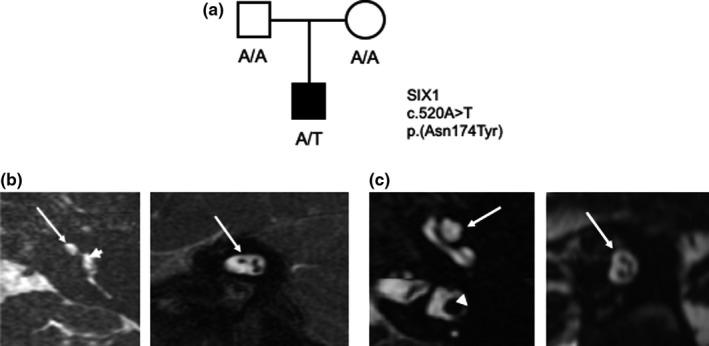
(a) Pedigree showing the de novo variant identified in SIX1 in the affected child. (b) MRI high‐resolution axial T2 sequences showing the (right) hypoplastic cochlear bud (white arrow) emanating from the vestibule (white arrowhead) and (left) oblique cuts perpendicular through the IAC showing the three nerve bundles in the lateral IAC (normal is 4). (c) MRI high‐resolution axial T2 sequences showing (right) normal cochlear architecture (white arrow) and the normal vestibule (white arrowhead) and (left) oblique cuts perpendicular through the IAC showing four nerve bundles in the lateral IAC. IAC, internal auditory canal; MRI, magnetic resonance imaging
3.2. Genetic screening
We identified a novel de novo variant (NM_005982:c.520A>T:p.[Asn174Tyr]) in SIX1, which previously has been associated with autosomal dominant hearing loss (ADHL) and branchio‐oto‐renal (BOR) or Branchio‐otic (BO) syndrome (Mosrati et al., 2011; Ruf et al., 2004). Both maternity and paternity were confirmed, and the de novo variant was verified via Sanger sequencing. This variant is located in the DNA‐binding homeodomain (HD) and is absent from genomic databases (gnomAD). It is predicted damaging by various bioinformatic tools (Table S1), and is located at a residue that is highly conserved amongst species (GREP++RS: 5.96; phastCons20waymam: 1.0) (Liu et al., 2016). The Combined Annotation‐Dependent Depletion score of this variant is 29.1, which indicates it is in the top ~0.1% of the most deleteriousness variants across genome‐wide assessed variants.
4. DISCUSSION
In the current study, we performed exome sequencing to identify a de novo variant in SIX1 in a patient with a nonsyndromic rare malformation of the inner ear and CVN. SIX1 is a homeobox protein that controls organ development and is a key regulator of otic vesicle patterning during embryogenesis. Six1‐deficient embryos lack inner ear structures, including a cochlea and vestibule, whereas their endolymphatic sac was enlarged (Yajima et al., 2014). Disruption of SIX1 leads to aberrant numbers of vestibulocochlear ganglion neurons and hair cells (Kirby & Collazo, 2006). In addition, SIX1 is widely expressed in neural crest cells that colonize the pre‐otic mesenchyme and a variety of cephalic neural crest and mesoderm‐derived cell types and tissues (Fonseca, Couly, & Dupin, 2017).
Variants in SIX1 have previously been involved in both ADHL and BOR/BO spectrum syndrome. SIX1 has two evolutionary conserved domains, the N‐terminal SIX1 domain and the HD, which are mainly involved in protein–protein interactions (including EYA1) and protein‐DNA binding, respectively. Variants in the HD, however, have been reported to diminish both SIX1‐EYA1 protein binding and SIX1‐DNA binding (Kochhar et al., 2008). Almost all variants that have been reported so far in ADHL and BOR/BO are located in either of these two evolutionary conserved domains (Table 1), and are mainly missense variants. The SIX1 variant we identified, p.(Asn174Tyr), is located in the DNA‐binding HD domain, is predicted damaging by various bioinformatic tools (Table S1), and lies adjacent to an amino acid that was found altered in a BOR patient (Table 1).
Table 1.
Overview of variants reported in SIX1 and the associated phenotypes
| Exon | Domain | Variant (NM_005982.3) | Inheritance | References | Phenotype | Treatment(s) |
|---|---|---|---|---|---|---|
| 1 | SIX domain | c.50T>A; p.(Val17Glu) | AD | Kochhar et al., (2008); Patrick, Schiemann, Yang, Zhao, and Ford, (2009) | BOR/BO | Unknown |
| 1 | SIX domain | c.218A>C; p.(His73Pro) | AD | Kochhar et al., (2008); Patrick et al., (2009) | BOR/BO | Hearing aids |
| 1 | SIX domain | c.317T>G; p.(Val106Gly) | AD | Kochhar et al., (2008); Patrick et al., (2009) | BOR/BO | Unknown |
| 1 | SIX domain | c.328C>T; p.(Arg110Trp) | AD | Kochhar et al., (2008); Patrick et al., (2009); R. G. Ruf et al., (2004) | BOR/BO | Hearing aids |
| 1 | SIX domain | c.329G>A; p.(Arg110Gln) | AD | Kochhar et al., (2008); Patrick et al., (2009) | BOR/BO | Unknown |
| 1 | SIX domain | c.334C>T; p.(Arg112Cys) | Unknown | Kochhar et al., (2008); Patrick et al., (2009) | BOR/BO | Unknown |
| 1 | SIX domain | c.364T>A; p.(Trp122Arg) | AD | Sanggaard et al., (2007) | BOR/BO | Cochlear implant |
| 1 | Homeodomain | c.373G>A; p.(Glu125Lys) | AD | Mosrati et al., (2011); Yan et al., (2016) | ADHL | Unknown |
| 1 | Homeodomain | c.386A>G; p.(Tyr129Cys) | De novo, AD | Ito et al., (2006); Krug et al., (2011); Patrick et al., (2009); Ruf et al., (2004); Yang et al., (2014) | BOR/BO | Unknown |
| 1 | Homeodomain | c.397_399delGAG; p.(Glu133del) | AD | Häfner et al., (2000); Patrick et al., (2009); Ruf et al., (2004) | BOR/BO; ADHLa | Unknown |
| 1 | Homeodomain | c.519G>C; p.(Lys173Asn) | Unknown | Unzaki et al., (2018) | BOR/BO | Unknown |
| 1 | Homeodomain | c.520A>T; p.(Asn174Tyr) | De novo | This study | ADHL due to cochlear and CVN abnormality | Hearing aid and cochlear implant |
| 1 | c.560+3A>T; splicing | Unknown | Krug et al., (2011) | BOR/BO | Unknown |
Variant c.746C>T; p.(Pro249Leu) in exon 2 was reported in a patient with BOR (Krug et al., 2011), however was not included here due to its higher than expected heterozygote frequency found later in gnomAD.
Abbreviations: AD, autosomal dominantly inherited or presumed autosomal dominantly inherited variant based on family history; Unknown: unknown inheritance but heterozygous germline variant; BOR/BO, Branchio‐otic (BO) or branchio‐oto‐renal (BOR) syndrome spectrum disorders.
Previously reported as nonsyndromic hearing loss, but a patient was found to have renal involvement later (Ruf et al., 2004).
Our patient carries a de novo variant in SIX1, and presents with bilateral profound SNHL, a hypoplastic cochlear bud, a small lateral semicircular canal and only three nerves in both lateral IACs. All previously identified nonsyndromic ADHL variants are also located in the same HD domain (Table 1), however, some of these ADHL patients were previously investigated via temporal bone computed tomography and showed no inner ear malformations (Mosrati et al., 2011). Additional prior studies often do not describe any imaging data of the inner ear, although one patient with BOR was reported to have enlarged vestibular aqueducts and some BOR patients were reported to have inner ear defects without any description of these malformations (Ito, Noguchi, Yashima, & Kitamura, 2006; Sanggaard et al., 2007). Hypoplastic cochleae and labyrinths have been described in some cases of BOR with unknown genetic background (Kemperman et al., 2001, 2002; Ritter & Martin, 2018). Six1−/− mice display malformations of the outer, middle, and inner ears (Zheng et al., 2003). Six1−/− embryos also show malformations of cranial sensory ganglia, including a loss of the VIIIth and distal VIIth sensory ganglia. No VIIIth nerve projection could be identified, and a misrouting of the VII branchial motoneurons was found as well in these embryos (Zou, 2004).
This child underwent a CI on the left side at the age of 4 and continued to use a hearing aid on his right. He was able to achieve closed set speech recognition (52% word intelligibility by picture identification, or WIPI) only 3 months after his CI surgery and anticipate he will continue to progress in his language acquisition.
Very little is known clinically of the human clinical findings in children with SIX1 variants and CVN abnormalities as the genetics of abnormal CVNs is poorly understood and many children with congenital hearing loss are not identified with known genetic aberrations.
In this study, we show that a de novo variant in SIX1 leads to nonsyndromic SNHL, cochleovestibular malformations, and abnormalities of the CVN (i.e., cochlear nerve aplasia/deficiency).
CONFLICTS OF INTEREST
The authors have no conflicts of interest related to the work in this manuscript.
Supporting information
ACKNOWLEDGMENTS
The authors thank the families for participating in this study. This study was supported by private donations to TGen's Center for Rare Childhood Disorders (https://www.tgen.org/giving/tgen-foundation/), the American Hearing Research Foundation to I.S. (http://american-hearing.org/), National Institutes of Health R01 010856, RO1 DC003594 and R01 DC011651 (https://www.nih.gov/) to S.M.L. and the Mills Auditory Foundation (http://millsauditoryfoundation.org/) to R.A.F.
Kari E, Llaci L, Go JL, et al. A de novo SIX1 variant in a patient with a rare nonsyndromic cochleovestibular nerve abnormality, cochlear hypoplasia, and bilateral sensorineural hearing loss. Mol Genet Genomic Med. 2019;7:e995 10.1002/mgg3.995
Funding information
This study was supported by private donations to TGen's Center for Rare Childhood Disorders (https://www.tgen.org/giving/tgen-foundation/), the American Hearing Research Foundation to I.S. (http://american-hearing.org/), National Institutes of Health R01 010856, RO1 DC003594 and R01 DC011651 (https://www.nih.gov/) to S.M.L. and the Mills Auditory Foundation (http://millsauditoryfoundation.org/) to R.A.F.
Contributor Information
Elina Kari, Email: ekari@ucsd.edu.
Isabelle Schrauwen, Email: isabelle.schrauwen@gmail.com.
REFERENCES
- Birman, C. S. , Powell, H. R. F. , Gibson, W. P. R. , & Elliott, E. J. (2016). Cochlear implant outcomes in cochlea nerve aplasia and hypoplasia. Otology & Neurotology, 37(5), 438–445. 10.1097/MAO.0000000000000997 [DOI] [PubMed] [Google Scholar]
- Buchman, C. A. , Copeland, B. J. , Yu, K. K. , Brown, C. J. , Carrasco, V. N. , & Pillsbury, H. C. (2004). Cochlear implantation in children with congenital inner ear malformations. The Laryngoscope, 114(February), 309–316. 10.1097/00005537-200402000-00025 [DOI] [PubMed] [Google Scholar]
- Buchman, C. A. , Teagle, H. F. B. , Roush, P. A. , Park, L. R. , Hatch, D. , Woodard, J. , … Adunka, O. F. (2011). Cochlear implantation in children with labyrinthine anomalies and cochlear nerve deficiency: Implications for auditory brainstem implantation. The Laryngoscope, 121(9), 1979–1988. 10.1002/lary.22032 [DOI] [PubMed] [Google Scholar]
- Casselman, J. W. , Offeciers, F. E. , Govaerts, P. J. , Kuhweide, R. , Geldof, H. , Somers, T. , & D'Hont, G. (1997). Aplasia and hypoplasia of the vestibulocochlear nerve: Diagnosis with MR imaging. Radiology, 202, 773–781. 10.1148/radiology.202.3.9051033 [DOI] [PubMed] [Google Scholar]
- CDC . (n.d.). Hearing loss in children. Retrieved from http://www.cdc.gov/ncbddd/hearingloss/data.html
- Dettman, S. , Sadeghi‐Barzalighi, A. , Ambett, R. , Dowell, R. , Trotter, M. , & Briggs, R. (2011). Cochlear Implants in forty‐eight children with cochlear and/or vestibular abnormality. Audiology and Neurotology, 16(4), 222–232. 10.1159/000320608 [DOI] [PubMed] [Google Scholar]
- Eisenberg, L. S. , Fink, N. E. , & Niparko, J. K. (2006). Childhood development after cochlear implantation. ASHA Leader, 11(16‐November), 28–29. [Google Scholar]
- Farhood, Z. , Nguyen, S. A. , Miller, S. C. , Holcomb, M. A. , Meyer, T. A. , & Rizk, H. G. (2017). Cochlear implantation in inner ear malformations: Systematic review of speech perception outcomes and intraoperative findings. Otolaryngology–head and Neck Surgery, 156(5), 783–793. 10.1177/0194599817696502 [DOI] [PubMed] [Google Scholar]
- Fink, N. E. , Wang, N.‐Y. , Visaya, J. , Niparko, J. K. , Quittner, A. , Eisenberg, L. S. , & Tobey, E. A. (2007). Childhood Development after Cochlear Implantation (CDaCI) study: Design and baseline characteristics. Cochlear Implants International, 8(2), 92–116. 10.1179/cim.2007.8.2.92 [DOI] [PubMed] [Google Scholar]
- Fonseca, B. F. , Couly, G. , & Dupin, E. (2017). Respective contribution of the cephalic neural crest and mesoderm to SIX1‐expressing head territories in the avian embryo. BMC Developmental Biology, 17(1), 13 10.1186/s12861-017-0155-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, S. R. , & Sennaroglu, L. (2018). Management of cochlear nerve hypoplasia and aplasia. Advances in Oto‐Rhino‐Laryngology, 81, 81–92. 10.1159/000485542 [DOI] [PubMed] [Google Scholar]
- Govaerts, P. J. , Casselman, J. , Daemers, K. , De Beukelaer, C. , Yperman, M. , & De Ceulaer, G. (2003). Cochlear implants in aplasia and hypoplasia of the cochleovestibular nerve. Otology and Neurotology, 24(6), 887–891. 10.1097/00129492-200311000-00011 [DOI] [PubMed] [Google Scholar]
- Häfner, F. M. , Salam, A. A. , Linder, T. E. , Balmer, D. , Baumer, A. , Schinzel, A. A. , … Leal, S. M. (2000). A novel locus (DFNA24) for prelingual nonprogressive autosomal dominant nonsyndromic hearing loss maps to 4q35‐qter in a large Swiss German kindred. American Journal of Human Genetics, 66(4), 1437–1442. 10.1086/302865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, T. , Noguchi, Y. , Yashima, T. , & Kitamura, K. (2006). SIX1 mutation associated with enlargement of the vestibular aqueduct in a patient with branchio‐oto syndrome. The Laryngoscope, 116(5), 796–799. 10.1097/01.mlg.0000209096.40400.96 [DOI] [PubMed] [Google Scholar]
- Jackler, R. K. , Luxford, W. M. , & House, W. F. (1987). Congenital malformations of the inner ear: A classification based on embryogenesis. The Laryngoscope, 97(Suppl 40), 2–14. 10.1288/00005537-198703001-00001 [DOI] [PubMed] [Google Scholar]
- Jian, X. , Boerwinkle, E. , & Liu, X. (2014). In silico prediction of splice‐altering single nucleotide variants in the human genome. Nucleic Acids Research, 42(22), 13534–13544. 10.1093/nar/gku1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari, E. , Go, J. L. , Loggins, J. , Emmanuel, N. , & Fisher, L. M. (2018). Abnormal cochleovestibular anatomy and hearing outcomes: Pediatric patients with a questionable cochleovestibular nerve status may benefit from cochlear implantation and/or hearing AIDS. Audiology and Neurotology, 23(1), 48–57. 10.1159/000488793 [DOI] [PubMed] [Google Scholar]
- Kemperman, M. H. , Koch, S. M. P. , Joosten, F. B. M. , Kumar, S. , Huygen, P. L. M. , & Cremers, C. W. R. J. (2002). Inner ear anomalies are frequent but nonobligatory features of the branchio‐oto‐renal syndrome. Archives of Otolaryngology–head & Neck Surgery, 128(9), 1033 10.1001/archotol.128.9.1033 [DOI] [PubMed] [Google Scholar]
- Kemperman, M. H. , Stinckens, C. , Kumar, S. , Huygen, P. L. , Joosten, F. B. , & Cremers, C. W. (2001). Progressive fluctuant hearing loss, enlarged vestibular aqueduct, and cochlear hypoplasia in branchio‐oto‐renal syndrome. Otology and Neurotology, 22(5), 637–643. 10.1097/00129492-200109000-00014 [DOI] [PubMed] [Google Scholar]
- Kirby, M. , & Collazo, A. (2006). Nodose placode contributes autonomic neurons to the heart in the absence of cardiac neural crest. Journal of Neuroscience, 8(4), 1089–1095. 10.1523/jneurosci.1025-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochhar, A. , Orten, D. J. , Sorensen, J. L. , Fischer, S. M. , Cremers, C. W. R. J. , Kimberling, W. J. , & Smith, R. J. H. (2008). SIX1 mutation screening in 247 branchio‐oto‐renal syndrome families: A recurrent missense mutation associated with BOR. Human Mutation, 29(4), 565–565. 10.1002/humu.20714 [DOI] [PubMed] [Google Scholar]
- Krug, P. , Morinière, V. , Marlin, S. , Koubi, V. , Gabriel, H. D. , Colin, E. , … Heidet, L. (2011). Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio‐oto‐renal syndrome calls into question the pathogenic role of SIX5 mutations. Human Mutation, 32(2), 183–190. 10.1002/humu.21402 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England), 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Wu, C. , Li, C. , & Boerwinkle, E. (2016). dbNSFP v3. 0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkus, P. , Lella, F. D. , Trapani, G. D. , Pasanisi, E. , Beltrame, M. A. , Zanetti, D. , … Sanna, M. (2014). Indications and contraindications of auditory brainstem implants: Systematic review and illustrative cases. European Archives of Oto‐Rhino‐Laryngology, 271(1), 3–13. 10.1007/s00405-013-2378-3 [DOI] [PubMed] [Google Scholar]
- Mosrati, M. A. , Hammami, B. , Rebeh, I. B. , Ayadi, L. , Dhouib, L. , Ben mahfoudh, K. , … Masmoudi, S. (2011). A novel dominant mutation in SIX1, affecting a highly conserved residue, result in only auditory defects in humans. European Journal of Medical Genetics, 54(5), e484–e488. 10.1016/J.EJMG.2011.06.001 [DOI] [PubMed] [Google Scholar]
- Niparko, J. K. (2004). Speech, language, and reading skills after early cochlear implantation. JAMA, 291(19), 2378–2380. 10.1001/jama.291.19.2378 [DOI] [PubMed] [Google Scholar]
- Niparko, J. K. , Tobey, E. A. , Thal, D. J. , Eisenberg, L. S. , Wang, N.‐Y. , Quittner, A. L. , & Fink, N. E. (2010). Spoken language development in children following cochlear implantation. JAMA, 303(15), 1498–1506. 10.1001/jama.2010.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick, A. N. , Schiemann, B. J. , Yang, K. , Zhao, R. , & Ford, H. L. (2009). Biochemical and functional characterization of six SIX1 Branchio‐oto‐renal syndrome mutations. Journal of Biological Chemistry, 284(31):20781‐20790. 10.1074/jbc.M109.016832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter, K. E. , & Martin, D. M. (2018). Neural crest contributions to the ear: Implications for congenital hearing disorders. Hearing Research, 376, 22–32. 10.1016/J.HEARES.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf, R. G. , Xu, P.‐X. , Silvius, D. , Otto, E. A. , Beekmann, F. , Muerb, U. T. , … Hildebrandt, F. (2004). SIX1 mutations cause branchio‐oto‐renal syndrome by disruption of EYA1‐SIX1‐DNA complexes. Proceedings of the National Academy of Sciences of the United States of America, 101(21), 8090–8095. 10.1073/pnas.0308475101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanggaard, K. M. , Rendtorff, N. D. , Kjaer, K. W. , Eiberg, H. , Johnsen, T. , Gimsing, S. , … Tranebjærg, L. (2007). Branchio–oto–renal syndrome: Detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. European Journal of Human Genetics, 15(11), 1121–1131. 10.1038/sj.ejhg.5201900 [DOI] [PubMed] [Google Scholar]
- Schrauwen, I. , Kari, E. , Mattox, J. , Llaci, L. , Smeeton, J. , Naymik, M. , … Friedman, R. A. (2018). De novo variants in GREB1L are associated with non‐syndromic inner ear malformations and deafness. Human Genetics, 137(6–7), 459–470. 10.1007/s00439-018-1898-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennaroglu, L. , Colletti, V. , Manrique, M. , Laszig, R. , Offeciers, E. , Saeed, S. , … Konradsson, K. (2011). Auditory brainstem implantation in children and non‐neurofibromatosis type 2 patients: A consensus statement. Otology and Neurotology, 32(2), 187–191. 10.1097/MAO.0b013e318206fc1e [DOI] [PubMed] [Google Scholar]
- Sennaroglu, L. , & Saatci, I. (2002). A new classification for cochleovestibular malformations. The Laryngoscope, 112(12), 2230–2241. 10.1097/00005537-200212000-00019 [DOI] [PubMed] [Google Scholar]
- Unzaki, A. I. , Morisada, N. , Nozu, K. , Ye, M. J. , Ito, S. , Matsunaga, T. , … Iijima, K. (2018). Clinically diverse phenotypes and genotypes of patients with branchio‐oto‐renal syndrome. Journal of Human Genetics, 63(5), 647–656. 10.1038/s10038-018-0429-8 [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima, H. , Suzuki, M. , Ochi, H. , Ikeda, K. , Sato, S. , Yamamura, K.‐I. , … Kawakami, K. (2014). Six1 is a key regulator of the developmental and evolutionary architecture of sensory neurons in craniates. BMC Biology, 12(1), 40 10.1186/1741-7007-12-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, D. , Tekin, D. , Bademci, G. , Foster, J. , Cengiz, F. B. , Kannan‐Sundhari, A. , … Tekin, M. (2016). Spectrum of DNA variants for non‐syndromic deafness in a large cohort from multiple continents. Human Genetics, 135(8), 953–961. 10.1007/s00439-016-1697-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Muzny, D. M. , Xia, F. , Niu, Z. , Person, R. , Ding, Y. , … Eng, C. M. (2014). Molecular findings among patients referred for clinical whole‐exome sequencing. JAMA, 312(18), 1870–1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, N. M. , Kim, F. M. , Ryan, M. E. , Tournis, E. , & Yaras, S. (2012). Pediatric cochlear implantation of children with eighth nerve deficiency. International Journal of Pediatric Otorhinolaryngology, 76(10), 1442–1448. 10.1016/j.ijporl.2012.06.019 [DOI] [PubMed] [Google Scholar]
- Zheng, W. , Huang, L. , Wei, Z.‐B. , Silvius, D. , Tang, B. , & Xu, P.‐X. (2003). The role of Six1 in mammalian auditory system development. Development (Cambridge, England), 130(17), 3989–4000. 10.1242/dev.00628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, D. (2004). Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development, 131(22), 5561–5572. 10.1242/dev.01437 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials