

The equine gut model that we have developed and describe has the potential to facilitate the exploration of how the equine gut microbiota is affected by diet, disease, and medication. It is a convenient, cost-effective, and welfare-friendly alternative to in vivo research models.
KEYWORDS: equine, gut model, microbiota, fermentation, metabolites
ABSTRACT
The intestinal microbiota of the horse, an animal of huge economic and social importance worldwide, is essential to the health of the animal. Understanding the intestinal ecosystem and its dynamic interaction with diet and dietary supplements currently requires the use of experimental animals, with consequent welfare and financial constraints. Here, we describe the development and assessment, using multiple analytical platforms, of a three-vessel, continuous-flow, in vitro model of the equine hindgut. After inoculation of the model with fresh horse feces, the bacterial communities established in each vessel had a taxonomic distribution similar to that of the source animal. Short-chain fatty acid (SCFA) and branched-chain fatty acid (BCFA) production within the model at steady state was consistent with the expected bacterial function, although higher concentrations of some SCFA/BCFA relative to those in the ex vivo gut content were apparent. We demonstrate the intermodel repeatability and the ability of the model to capture some aspects of individual variation in bacterial community profiles. The findings of this proof-of-concept study, including recognition of the limitions of the model, support its future development as a tool for investigating the impact of disease, nutrition, dietary supplementation, and medication on the equine intestinal microbiota.
IMPORTANCE The equine gut model that we have developed and describe has the potential to facilitate the exploration of how the equine gut microbiota is affected by diet, disease, and medication. It is a convenient, cost-effective, and welfare-friendly alternative to in vivo research models.
INTRODUCTION
Commensal bacteria that reside within the large intestine of the equine gastrointestinal tract are vital for the horse to be able to utilize a forage-based diet. Some of these bacteria produce short-chain fatty acids (SCFA), which are absorbed through the gut wall and contribute to the energy requirements of the horse. Sequencing of bacterial 16S rRNA genes from the gastrointestinal content or feces has been used to profile the gut microbiota of healthy horses (1–5) and to assess the impact of diseases, such as colitis and grass sickness, on equine fecal bacterial communities (6, 7). In common with the majority of human microbiota studies, most disease-related studies have characterized the equine fecal microbiota due to ease of access to samples. The communities in equine feces have been shown to be broadly representative of the bacterial communities within distal parts of the large colon (1) and to more orad parts of the intestinal tract.
To accurately analyze bacterial communities within the gastrointestinal tract of the horse, direct sampling of each region of interest would be ideal. While this can be done postmortem or with fistulated horses, both techniques have limited application due to financial, ethical, and welfare implications. Therefore, a fermentation model representing the microbiota of the equine large intestine in vitro would facilitate this important area of equine health research, enhancing our ability to explore the impact of diet, pathogens, novel foodstuffs, dietary supplements, and drugs on the equine gut microbiota.
Models of the human gut have been a valuable and widely used research tool (8). Studies have shown how an intervention with galactooligosaccharide (GOS) has effects on the microbial community in vitro similar to those on the microbial community in a human study population (9). In vitro and in vivo approaches were taken to study the effect of the prebiotic GOS on the gut microbiota of children with autism (10, 11). Both approaches demonstrated an increase in butyrate production (in the gut model and human feces, respectively), suggesting that the model is metabolically valid. Gut models have provided preliminary insights into the effects of dietary supplements or drugs on the human gut microbiota before commencing costly human trials.
In vitro fermentation models developed to represent bacterial activity in the equine gastrointestinal tract have been reported previously (12–20). However, the majority of these models have used a simple medium or equine feed to maintain fecal bacterial populations and do not replicate the continuous flow of ingesta through the gastrointestinal tract (12, 13, 16–22). These models often use gas production as a marker of bacterial function and measure the kinetic properties of the bacterial populations cultured (13, 17–21). Sequencing of bacterial DNA from fermentation samples allows for a more comprehensive overview of the bacterial communities present; however, this approach has so far been reported in only one equine fermentation study (14).
In this study, we report on the development and performance of a three-stage fermentation model designed to simulate bacterial communities in the equine large intestine. Two experiments were designed: (i) a concordance study of three individual gut models, each inoculated with feces from a different horse (postmortem), to evaluate the extent to which the gut bacterial profiles of individual horses are replicated, and (ii) a repeatability study of two gut models inoculated with feces from the same horse (premortem). To analyze samples taken from the vessels of the gut model, we used fluorescence in situ hybridization (FISH) analysis to assess total bacterial numbers and 16S rRNA bacterial gene sequencing to characterize the microbial community profile in vitro compared to that in the samples taken ex vivo. Proton nuclear magnetic resonance (1H NMR) spectroscopy and gas chromatography were used to measure a broad range of metabolites to evaluate the functional activity of the model microbiota. Specifically, we report on (i) the model concordance with in vivo gut bacterial populations, (ii) the repeatability of the model, and (iii) the ability of the model to capture individual variation in the bacterial community profile.
RESULTS
The gut model medium was adapted to reflect a normal equine diet.
The acid detergent fiber (ADF) and starch contents of gastrointestinal content samples were analyzed to inform the compositional changes made to the gut model medium. We aimed to replicate the diet of the average UK leisure horse in the medium used to feed the model. A large percentage of the gastrointestinal content of the equine ileum, large colon, and feces was ADF (see Fig. S1 in the supplemental material). Feces had the highest percentage of ADF (42.9% ± 3.9%) and the lowest percentage of starch (0.56% ± 0.49%), whereas the ileal content had the lowest percentage of ADF (25.9% ± 10.8%) and the highest percentage of starch (3.8% ± 3.3%). The analysis of ADF and starch from the ileum content informed the cellulose and starch content of the gut model medium used to feed the model.
The equine in vitro gut model can support a metabolically functioning bacterial population.
The concentrations of five short-chain fatty acids (SCFA) and branched-chain fatty acids (BCFA) produced in each vessel of the in vitro gut models were measured during the concordance study to assess the metabolic output of bacterial fermentation. Figure 1 shows the concentrations of acetate, propionate, butyrate, isobutyrate, and valerate in the three vessels as a mean for the three separate gut models. The concentrations of acetate and propionate rose quickly in all three vessels after initial inoculation (Fig. 1A and B). The concentrations of butyrate, isobutyrate, and valerate increased only marginally between samples taken immediately after inoculation (referred to as turnover −1 [T−1]) and turnover 0 (T0; which was after batch culture for 24 h) but then increased sharply after the flow of medium was started at T0 (Fig. 1C to E). The mean concentration for all SCFA/BCFA measured was observed to stabilize in all three vessels by turnover 5 (T5), as there was no significant change in concentration between T3 and T5 (P > 0.05).
FIG 1.

Demonstration of the biological functionality of the gut models in the concordance study. The SCFA/BCFA production of the three separate gut models inoculated with feces from three different horses during the concordance study is shown as the mean and standard deviation for the three vessels of the model. The SFCA/BCFA measured were acetate (A), propionate (B), butyrate (C), isobutyrate (D), and valerate (E). T, turnover; V, vessel.
The metabolic profiles of the three gut models in the concordance study were compared to those of the gastrointestinal content of the donor horses. Figure 2 shows the principal-component analysis (PCA) model (R2 = 0.64), built with metabolic data from the gastrointestinal contents, feces from the rectum, and all samples taken from the three gut models. The PCA scores plot shows that the largest source of variation within the data set (the first principal component [PC1], representing 50% of the total variation in the data set) was explained by samples taken from the model after T1 having higher concentrations of acetate (Fig. 2A and B). Within the second principal component (PC2) of this PCA model, samples of gastrointestinal content clustered away from samples from the gut models (representing 14% of the total variance; Fig. 2A). This was due to the content of the large colon having a higher concentration of acetate than the gut model (Fig. 2C).
FIG 2.

Biochemical variation (measured by 1H NMR spectroscopy) between the gut contents of donor horses and the in vitro gut model supernatant from the concordance study. (A) PCA scores plot for PC1 and PC2 of the model (R2 = 0.64). (B and C) The loading plot for PC1, describing 50% of the total variance associated with the higher concentration of acetate in model supernatant at time points T1 to T5 (B), and the loading plot for PC2, describing 14% of the total variance associated with the higher acetate concentration in gut contents (C). PC, principal component; T, turnover.
A PCA model was constructed with the metabolic profiles of the gastrointestinal content and samples taken from the three concordance study models at T5 (R2 = 0.64). There was a separation of points representing samples from the two groups in PC1 (explaining 48% of the variance in the data set) in the PCA scores plot (Fig. S2A), indicating metabolic differences between samples of gastrointestinal content and the gut model. When these differences were explored further, samples of gastrointestinal content had higher concentrations of acetate, whereas the samples from the gut model at T5 had higher levels of valerate and propionate (Fig. S2B). An orthogonal projection to latent structures-discriminant analysis (OPLS-DA) model was also built with the metabolic spectra from these two groups of samples (Q2Y = 0.82). This supervised model highlighted differences in the levels of acetate, propionate, and valerate (which were also evident in the PCA model) alongside higher levels of diethylene glycol (DEG), ethanol, trimethylamine (TMA), and isovalerate in samples from the gut models (Fig. S2C).
The equine gut model can replicate part of the equine large intestinal microbiota.
An estimate of the total number of bacteria was created using FISH analysis to assess the bacterial concentration within the gut model after fecal inoculation. The mean total numbers of bacteria per milliliter for the three gut models of the concordance study were lower in the vessels of the gut model at T−1 (2.68 × 108, 2.86 × 108, 2.21 × 108 bacteria per ml) than in the gastrointestinal content (3.93 × 108, 3.40 × 108, 3.32 × 108 bacteria per ml). At T0, bacterial counts had risen in all three vessels (2.81 × 108, 4.02 × 108, 3.69 × 108 bacteria per ml). By T5 (steady state), bacterial proliferation had plateaued, with vessel 1 (V1) having the highest counts (5.51 × 108 bacteria per ml) and vessel 3 having the lowest counts (3.74 × 108 bacteria per ml). Bacterial alpha diversity (measured as the operational taxonomic units [OTUs], which are bacterial groups with different taxonomic classifications, observed) within the gut models at T0 and T5 was significantly lower than the bacterial diversity of the gastrointestinal content and the models at T−1 (P < 0.05; Fig. S3A).
Unsurprisingly, the bacterial community profile at the phylum level of the model at T−1 (immediately after inoculation) was similar to that of the gastrointestinal contents of the horses that provided feces for inoculation (Fig. 3). However, the abundance of Verrucomicrobia was greater in the in vitro gut models immediately after inoculation (T−1) than in the gastrointestinal content (14% versus 1%, P < 0.05). After the gut models were batch cultured for 24 h (T0), an increased abundance of Proteobacteria (from an average of 2% to 27%) and Firmicutes (from an average of 40% to 64%) was observed in all vessels (P < 0.05 from T−1 to T0 for both bacterial phyla). However, by T5 the relative abundance of Firmicutes and Bacteroidetes was comparable to that of the gastrointestinal content and the relative abundance of Proteobacteria was much reduced relative to that at T0 (from an average of 37% to 8%, P < 0.05).
FIG 3.

Relative abundance of the bacterial phyla (assigned by bacterial 16S rRNA gene sequencing) identified in the gastrointestinal (GI) content from donor horses and the three gut models from the concordance study. The key indicates whether bacterial phyla were identified in both gastrointestinal and gut model samples at T5 (black), only in gastrointestinal samples (blue), or only in gut model samples (red). C, cecum; RVC, right ventral colon; LVC, left ventral colon; V, vessel; T, turnover; M, model; WPS-2, “Candidatus Eremiobacterota.”
On average, samples taken directly from the cecum were composed of 40% Bacteroidetes and 46% Firmicutes (as the mean for the three horses sampled), whereas in the gut model, these proportions of bacteria were 21% and 59% (as an average for vessel 1 for the three models of the concordance study), respectively. For the right ventral colon, the proportions were 40% and 47%, respectively, for the gut contents and 15% and 53%, respectively, in the gut model (vessel 2), and for the left ventral colon, the gut content proportions were 37% and 50%, respectively, and the gut model proportions were 25% and 48%, respectively (vessel 3).
Interestingly, the percentage of the bacterial community within the in vitro gut model that was identified as Bacteroidetes decreased further down the model (from vessel 1 to vessel 3), similar to the changes observed in aboral regions of the large colon. There were higher levels of Bacteroidetes in vessel 3 of model 1 (M1V3) than in vessel 3 of the other two models (M2V3 and M3V3). There was a higher relative abundance of Synergistetes in the gut model at T5 (mean for all three vessels = 14%) than in the gastrointestinal content (<1%). There were also slight differences in the bacterial communities in the three models (M1, M2, and M3) inoculated with feces from different horses. M2 had a significantly higher percentage of reads identified as Firmicutes than M3 (P < 0.05), and M3 had a significantly higher percentage of Verrucomicrobia and Synergistetes bacteria than the other two models (P < 0.05).
The mean SCFA/BCFA concentration and bacterial phylum count for each vessel of the concordance study gut models were used to build regression models. A number of the models built showed a positive correlation between SCFA/BCFA and bacterial phyla with a good model fit (R2 > 0.5). However, only a few of these associations were significant (P < 0.05); these included Fusobacteria and acetate (V1), Synergistetes and valerate (all vessels), and Synergistetes and isobutyrate (V1 and V3). For the R2 and P values for the regression analyses, see Table S2.
Bacterial community profiles were also illustrated at the class and order levels (Fig. S4). The bacterial class Clostridia (on average, 47% in gastrointestinal samples and 53% in all vessels of the gut model at T5) and the order Clostridiales (47% and 53%, respectively) were present at abundances similar to those of the Firmicutes phylum (48% and 53%, respectively), and the class Bacteroidia (37% and 21%, respectively) and the order Bacteroidales (37% and 21%, respectively) were present at abundances similar to those of the Bacteroidetes phylum (39% and 21%, respectively) in all gastrointestinal and gut model samples. Differences associated with a different relative abundance of Clostridia/Clostridiales and Bacteroidia/Bacteroidales were observed between the gut models inoculated with feces from different horses.
Venn diagrams were produced to visualize the number of distinct OTUs that were shared between the three vessels of the gut models (at T5) of the concordance study and their corresponding region of the equine large intestine (Fig. 4). Vessel 3 shared the highest number of OTUs, with 57 OTUs (59% of the total OTUs identified in samples from vessel 3) being common between vessel 3 and the content from the left ventral colon. Vessel 2 shared 53 OTUs (56%) with the right ventral colon samples, and vessel 1 shared 47 OTUs (57%) with cecum samples.
FIG 4.
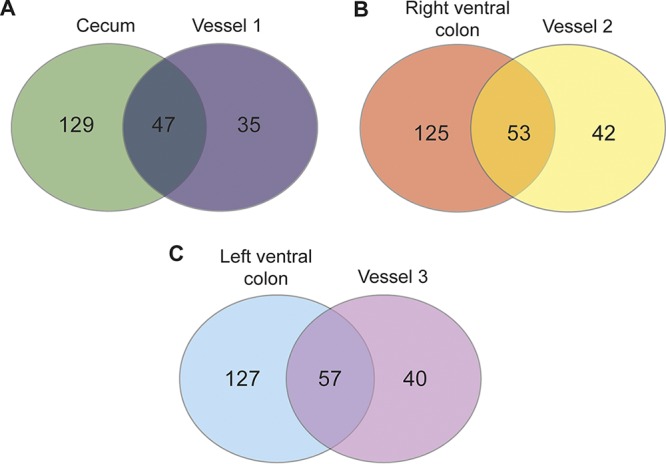
Venn diagram showing the total number of distinct OTUs that are unique and shared between samples taken from the areas of the equine large intestine and their corresponding vessels from the gut models. These models were from the concordance study used to capture individual variation.
The metabolic and microbial profile created by the equine gut model is repeatable.
The concentrations of the five SCFA/BCFA measured using gas chromatography rose significantly between T−1 and T1 in all of the vessels of the two gut models of the repeatability study, before plateauing (P < 0.05; Fig. 5). As with the concordance study, the models from the reproducibility study were deemed to be stable by T5 (P > 0.05 for all SCFA/BCFA between T3 and T5). The concentrations of butyrate and valerate slightly decreased after steady state was reached (between T5 and T7), but this decrease was not significant (P > 0.05). Furthermore, the concentrations of the five SCFA/BCFA measured by gas chromatography were not significantly different in the two models of the repeatability study (P > 0.05 for all SCFA/BCFA at all turnovers).
FIG 5.

Demonstration of the repeatability of biological function between models. SCFA/BCFA production (measured by gas chromatography) in each vessel of the models inoculated with the same feces (repeatability study) was determined. Each line represents the level of an SCFA/BCFA in a single vessel of one of the two gut models of the repeatability study. The SFCA/BCFA measured were acetate (A), propionate (B), butyrate (C), isobutyrate (D), and valerate (E). T, turnover; V, vessel; M, model.
The reproducibility of the metabolic signatures of the two gut models was assessed by building a PCA model with the 1H NMR spectra gained from the two gut models of the repeatability study (R2 = 0.75; Fig. S5). The largest source of variation within this data set (PC1, representing 63% of the total variance in the model) was the increase in the concentrations of propionate and acetate over the first 4 days of the model (T−1 to T1). Points representing samples from the two different models clustered together after T1, indicating that the two models were metabolically similar.
Total bacterial counts calculated by FISH were similar in the two gut models of the repeatability study (M4 and M5). M4 had slightly more bacteria in all three vessels at T5 than M5 (mean number of bacteria per milliliter, 4.02 × 108 for M4 and 3.88 × 108 for M5), but this was not statistically significant (P > 0.05). There was a slight increase in the total number of bacteria per milliliter between T5 and T7 in all of the vessels of the two gut models of the repeatability study (on average, the total number of bacteria per milliliter of all vessels increased by 3.74 × 107), but this increase was not statistically significant (P > 0.05).
The bacterial alpha diversity (measured as observed OTUs) within the models at T0, T5, and T7 was significantly lower than the bacterial diversity of the fecal sample used as the inoculum in the model at T−1 (P < 0.05; Fig. S3B). However, the bacterial diversity in the models did not significantly change between T5 and T7 (P > 0.05). Bacterial community profiles were constructed for the two gut models of the repeatability study to assess the reproducibility of the equine gut model (Fig. 6). There were no significant differences between the two models when the bacterial phyla identified in each of the three vessels of the models at T5 were compared (P > 0.05). There were also no significant differences between the bacterial phylum profile of each vessel at T5 and T7 (P > 0.05). The bacterial community profiles at the class and order levels were similar between the two models of the repeatability study and stable between T5 and T7 (Fig. S6).
FIG 6.

Demonstration of the repeatability of model bacterial communities. Taxonomic assignments (the phyla determined from bacterial 16S rRNA gene sequencing) are from the repeatability study; two models were inoculated with feces from the same horse. M, model; V, vessel; T, turnover.
DISCUSSION
We have presented a description of an in vitro model of the equine hindgut and have examined it against intestinal content samples taken from euthanized horses. This allowed for the assessment of the equine gut model’s ability to maintain bacterial communities representative of those found in vivo, the repeatability of the gut model setup, and the model’s ability to capture the individual variation that exists in the gut microbial communities of horses. Models, by definition, are not exact representations of reality. We acknowledge that a model of the equine microbiota will never be an exact replica of the real bacterial community within the equine large intestine. The model will, however, replicate aspects of the microbial community to allow interrogation and experimental manipulation in a more convenient and less invasive way. The data presented in this paper demonstrate the strengths and limitations of this novel gut model system and its potential to extend our understanding of equine intestinal health.
The three equine gut models of the concordance study showed that the fecal bacteria introduced into the gut model were able to metabolize the gut model medium and produce SCFA/BCFA. Immediately after inoculation with feces, the abundance of acetate and propionate within the model increased sharply. The concentrations of butyrate, isobutyrate, and valerate were not observed to notably increase over the first 24 h. However, the concentrations of these three SCFA/BCFA rose sharply in all three vessels during the first 3 days of being fed with medium. Differences in the timing of SCFA/BCFA appearance may reflect the differential acclimatization of the fecal bacterial communities to the gut model environment, with bacterial communities producing acetate and propionate adapting to the environment more rapidly than those producing butyrate, isobutyrate, and valerate. By T5, the production of all five SCFA/BCFA had stabilized and the models were deemed to have entered steady state. In human gut model studies, this is the point at which a food supplement or drug intervention to be studied is added (10, 23–26).
Multivariate statistical models were used to identify metabolic differences between the gastrointestinal contents and the gut models. Ethanol, valerate, isovalerate, propionate, DEG, and TMA were present at higher concentrations in the gut model at steady state than in the gastrointestinal contents. Choline can be degraded by the gut microbiota to liberate ethanol and TMA. Ethanol can also be produced via the metabolism of keto acids and saturated fatty acids following the microbial breakdown of amino acids. Similarly, isovalerate arises from the microbial degradation of leucine. These findings indicate that the functional capacity of the equine gut microbiota to liberate amino acids from ingesta is preserved in the gut model system. The accumulation of theses metabolites is most likely due to the lack of absorption in the vessels of the gut model system. However, acetate was present in smaller amounts in the equine gut model than in the gastrointestinal contents. Acetate is a product of bacterial carbohydrate fermentation and is usually absorbed by the gut wall so that it can undergo further metabolism to generate ATP. Acetate concentrations in V1 of the concordance study models were found to be significantly positively correlated with the presence of the known acetate-producing bacterial phylum Fusobacteria (27, 28). Lower levels of acetate in the gut model suggest that carbohydrate availability may be less in the gut model or that some of the carbohydrate-fermenting bacteria cannot be supported. Alternatively, the acetate produced could be washed through the gut model system into the waste by the constant flow of medium. The high levels of acetate in gastrointestinal contents emphasizes the importance of acetate-generating bacteria to enable horses to liberate energy from their diet.
FISH analysis of samples of gastrointestinal contents and the vessels of the concordance study gut models provided an estimate of the total number of bacteria in the two sample types. The mean concentration of bacteria in vessel 1 was higher than that seen in the cecum, but this was also the vessel in which the total number of bacteria varied the most between the three gut models from that in the concordance study. The differences in the total number of bacteria between the three vessels were similar to those seen in the three sample sites of the large intestine, although they were more pronounced in the gut model. This may be due to the progressive nutrient depletion as medium passes through the model system.
We chose to use 16S rRNA gene sequencing for taxonomic characterization of the bacterial communities present in the model. The advantages of this method are efficiency and breadth of coverage, but we also recognize the disadvantage of limited resolution and specificity. A more balanced approach for future studies might include the use of quantitative PCR for targeted taxonomic (and functional) markers of interest. Future characterization of this model system could also include evaluation of the virome and mycobiome.
The bacterial community profiles of the gastrointestinal content samples that we used were consistent with those reported previously (5) and dominated by Bacteroidetes and Firmicutes. Immediately after inoculation, all three vessels of the gut models had a composition similar to that of the fecal samples. After the first 24 h of fermentation, between T−1 and T0, there was a shift in the bacterial populations, where Proteobacteria and Firmicutes became dominant. However, by T5 the percentage of Firmicutes and Bacteroidetes bacteria in the three vessels did not significantly differ from that in the equivalent regions of the large intestine and the initial Proteobacteria overgrowth was reduced.
It is worth noting, however, that not all the populations present in gut content samples were restored in the model on reaching steady state. This situation will largely be dictated by the diet of the horse providing samples for inoculation of the model system instead of the nutrients available within the model medium. Varying the carbon and nitrogen sources in the medium could aid the growth of a more diverse range of organisms. Changes in bacterial community dynamics have been observed in human gut models by varying the medium composition and the model retention time (8). In equine and human gut model systems, the medium flowing through the model means that different nutrient availability occurs in different regions, enabling different environmental conditions to be modeled, supporting the growth of different microbial populations at different enteric sites. The bacterial community within the equine model system could be treated with different foods, bacteria, or antibiotics to determine the impact of dietary changes on the microbiota; this could therefore aid in determining whether certain changes may be of benefit or not to a horse. Indeed, the advantage of a model system is that the diet is very well controlled; therefore, if a test substrate is added after steady state has been reached, subsequent changes will be known to be a result of the substrate.
The bacterial profile for vessel 3 of model 1 (M1V3) seen in Fig. 3 had a higher percentage of Bacteroidetes than expected; the percentage of Bacteroidetes usually decreases from vessel 2 to vessel 3 (as can be seen in M2 and M3). This high level of Bacteroidetes likely relates to the starting bacterial community in this horse, as all models were treated identically, except for inoculation with feces from different horses. The ability of this in vitro system to replicate aspects of the bacterial community profile representative of the equine large intestine is consistent at the phylum, class, and order levels.
Many of the low-abundance bacterial taxa identified in the gastrointestinal content were absent from the gut model. Significant differences were found when comparing the OTUs identified in the gut models at T5 to the areas in the large intestine that they represent. These OTUs may correspond to nonviable bacteria that were detected in the gastrointestinal content and feces of horses but that were not able to survive and proliferate in the gut model. Alternatively, it may reflect the failure of the model to provide the exact environmental conditions needed to support the vast number of low-abundance bacterial groups normally found within the horse large intestine. Clearly, a limitation of this model is that it does not support the growth and function of all the taxonomic diversity present in the large intestine of horses. To allow a more diverse bacterial community to establish, other features of the equine large intestine may need to be added to the model, such as epithelial cells and gut wall secretions. By visualizing the unique OTUs that are shared between the gut model vessels and the gastrointestinal content in a Venn diagram, we can see that the gut models were able to maintain the bacteria that contribute to 39 to 43% of the unique OTUs identified in the gastrointestinal samples of horses.
Our studies have indicated that the equine gut model may capture some aspects of interindividual variation seen in the gut microbiota of equine populations. Inoculating models with feces from different horse donors resulted in different bacterial community profiles by the time that steady state was reached. This important aspect of the model is worthy of further exploration, as it may allow the investigation of individual responses to food supplements and susceptibility to diseases that are mediated via the gut microbiota.
In the repeatability study, two identical gut models were inoculated with feces from the same horse. The SCFA/BCFA levels measured by gas chromatography showed that their production was similar in the two gut models of the repeatability study. 1H NMR analysis of supernatants from these two models showed that the metabolic profiles of the vessels were similar at all time points after T1. FISH analysis estimating the total number of bacteria maintained by the two models showed that the differences in the total number of bacteria between the three vessels were similar across the models. Bacterial DNA sequencing of samples taken from the three vessels of these two models at T5 showed comparable taxonomic profiles at both the phylum and the class levels. The bacterial community profiles were stable for the two models between T5 and T7. The repeatability study provides preliminary evidence that models inoculated with the same fecal matter will produce microbial communities that are both structurally and functionally similar.
We have reported on the analyses used to assess the microbial and metabolic output of a three-stage equine gut model. The model is metabolically functional and is able to support a bacterial community which replicates aspects of that found in the equine large intestine. This model is not an exact replication of the in vivo bacterial community of the equine large intestine, but it does provide an in vitro alternative to studies that involve invasive or postmortem sampling of horses. These may be particularly valuable for studying cecal microbiota, for which feces are not an adequate proxy. This model has the potential to aid in providing an understanding of how the equine gut microbiota is affected by diet, disease, and drugs.
MATERIALS AND METHODS
Equine gastrointestinal content collection.
Samples of gastrointestinal contents were obtained from horses, free of gastrointestinal disease, no more than 3 h after euthanasia. The whole gastrointestinal tract was removed from the carcass, and the large intestine was identified. Two 12-ml tubes were filled with the contents from the following areas of each horse: cecum, left ventral colon, right ventral colon, and feces from the end of the small colon. Measurement of the pH of the fresh gut contents at all sample sites was achieved using a calibrated, hand-held pH monitor. Samples were transported on dry ice and then stored at −80°C prior to analysis. Descriptive details of all horses sampled can be found in Table S1 in the supplemental material.
Compositional analysis of gastrointestinal content.
To inform the makeup of the medium, the ADF and starch contents of gastrointestinal samples from three UK leisure horses (horses 1, 2, and 3 in Table S1) were analyzed. All samples were freeze-dried to remove all moisture. The ADF contents of the free-dried samples were analyzed using the filter bag technique (Ankom Technology). The starch contents of the samples were analyzed by converting starch into glucose by treatment of the hot water extract with amyloglucosidase, followed by acid hydrolysis (29). Total reducing sugars were measured colorimetrically as described by Fuller (30).
Three-stage continuous culture system.
The human gut model developed by Macfarlane et al. (8) was adapted with the aim of replicating the microbiota of the cecum, right ventral colon, and left ventral colon of the equine large intestine (Fig. 7). This continuous culture system constituted three vessels (V1, V2, and V3) with respective volumes of 300 ml, 400 ml, and 200 ml to provide a scaled-down version of their respective regions of the equine gastrointestinal tract. The temperature (held at 38°C with a circulating water bath; Optima) and pH (controlled by Broadly James F695 pH probes and a Fermac 260 pH measurement and control module; Electrolab) were automatically controlled as described by Macfarlane and Englyst (31). V1 and V2 were maintained at pH 6.3 to 6.5 and V3 was maintained at pH 6.6 to 6.8 to reflect the pH of the cecum, right ventral colon, and left ventral colon, respectively. All vessels and the medium reservoir were stirred by the use of magnetic fleas and maintained under anaerobic conditions by a continuous flow of anaerobic mix gas (80% N2, 10% CO2, 10% H2). A single-channel, variable-speed peristaltic pump (Electrolab) fed vessel 1 with medium from the medium reservoir at a rate of 900 ml every 3 days, which represents the scaled flow rate of ingesta through the equine gastrointestinal tract. The medium then flowed from vessel 1 to vessel 2 and from vessel 2 to vessel 3 through the waste pipe of each vessel.
FIG 7.

Three-stage, in vitro fermentation model designed to replicate the microbiota of the equine cecum, right ventral colon, and left ventral colon (method modified from Macfarlane et al. [8]).
The human gut model medium from Macfarlane et al. (8) was altered to mirror the ADF and fiber content seen in compositional analysis of the gastrointestinal content (Fig. S1). To represent the normal diet of a horse, the percentages of starch and fiber were increased and the percentage of protein was decreased. The composition of this medium in distilled water was as follows: cellulose (15 g liter−1), yeast extract (5 g liter−1), NaCl (5 g liter−1), KCl (5 g liter−1), mucin (4 g liter−1), raffinose (3.5 g liter−1), starch (2 g liter−1), peptone water (1.5 g liter−1), tryptone (1.5 g liter−1), NaHCO3 (1.5 g liter−1), MgSO4·7H2O (1.25 g liter−1), arabinogalactan (1 g liter−1), xylan (0.835 g liter−1), cysteine·HCl (0.8 g liter−1), KH2PO4 (0.5 g liter−1), K2HPO4 (0.5 g liter−1), bile salts (0.4 g liter−1), CaCl2·6H2O (0.15 g liter−1), hemin (0.05 g liter−1), FeSO4·H2O (0.005 g liter−1), Tween 80 (0.5 ml liter−1), and vitamin K (0.01 ml liter−1).
To inoculate the gut model, a sample of feces was collected from the rectum postmortem or immediately after defecation for noninvasive sample acquisition and placed in a sealed jar with an anaerobic atmosphere-generating sachet (Oxoid AnaeroGen 2.5-liter sachet; Thermo Fisher) for transportation for a maximum of 2 h. A 20% fecal slurry (in 1× phosphate-buffered saline [PBS]) was made in a strainer stomacher bag (Seward) by homogenizing fecal balls manually within an anaerobic cabinet (Whitley A35 anaerobic workstation). One hundred milliliters, 133 ml, and 67 ml of the fecal slurry were decanted and added, respectively, to each vessel (samples taken immediately after inoculation were designated T−1). After the fecal slurry was added, the model was allowed to equilibrate as a batch culture for 24 h. The medium pump was started after 24 h, designated T0. The short period of batch culture allowed the fecal bacteria to acclimatize to their new environment and reduce the washing out of bacteria when the flow of medium was started.
A sample of 5 ml was taken through the sample port of each vessel at T−1, T0, and every subsequent full turnover of medium through the model (every 3 days). Two aliquots of 1 ml each were centrifuged at 11,337 × g, the supernatant was stored at −20°C, and the pellets were stored at −80°C. For preparation of FISH slides, an aliquot of 375 μl from each sample was added to 1,125 μl 4% paraformaldehyde (PFA), and the mixture was incubated at 5°C for 4 h. PFA was washed off with 1× PBS, and the pellet was resuspended in 150 μl 1× PBS and 150 μl ethanol and stored at −20°C until required. Processing of all samples was carried out in an anaerobic cabinet (Whitley A35 anaerobic workstation).
Concordance and repeatability studies.
The concordance study used three gut models (M1, M2, and M3) inoculated with feces from three different horses to assess the similarity between the gastrointestinal samples and the stabilized fermentation model of the same horse. Modeling of three different individuals allowed assessment of whether the model could capture the interindividual variation seen in vivo. Samples of gastrointestinal contents were taken postmortem from the cecum, right ventral colon, left ventral colon, and feces from the rectum of three horses (horses 4, 5, and 6 in Table S1). These samples were transported on dry ice and then stored at −80°C until they were defrosted for analyses. A further 100 g of feces was taken from each horse and stored in an anaerobic jar (Oxoid) with an anaerobic gas-generating sachet until it was used to make a 20% fecal slurry, used to inoculate all vessels. Each gut model was sampled every turnover until steady state was reached (T5, when SCFA/BCFA production stabilized).
Subsequent to this, two gut models (M4 and M5) were run at the same time with feces from the same horse for the repeatability study. This aimed to assess the reproducibility of the metabolic and bacterial signatures between different gut models. Freshly voided feces were collected from a healthy horse (horse 7 in Table S1) and transported in a sealed container with an anaerobic gas-generating sachet for use as the fecal slurry inoculum. These models were run until turnover 7 (T7) to assess the stability of the model after steady state (T5) was reached, and samples were taken and processed as described above. An overview of the inoculation and sampling of the concordance and repeatability studies can be seen in Fig. 8.
FIG 8.
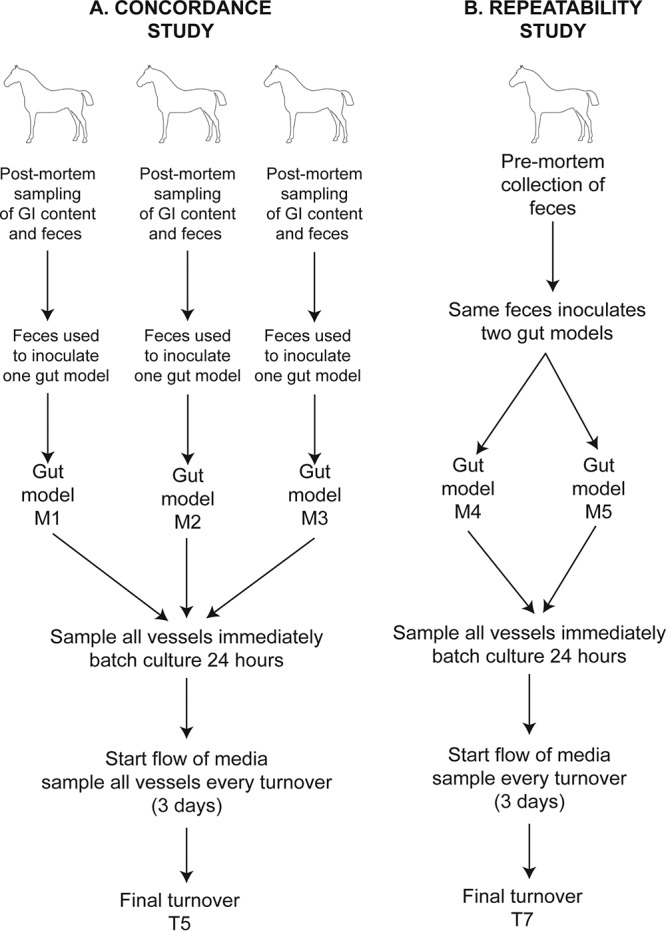
Schematic showing the sampling scheme and the experimental design for the concordance study to demonstrate the capture of interindividual variation (A) and the repeatability study to demonstrate the consistency of models inoculated with the same feces (B).
SCFA/BCFA analysis by gas chromatography.
Supernatants from all gut models at all time points underwent gas chromatography to analyze the levels of acetate, propionate, butyrate, isobutyrate, and valerate. These samples were analyzed using an acidification method adapted from the method of Zhao et al. (32). Samples were filtered through a 0.2-mm-pore-size polyvinylidene difluoride filter and acidified to pH 2 using sulfuric acid. An internal standard of 2-ethyl butyrate at 5 mM was added to each sample. A fused-silica capillary column with a free fatty acid phase (DB-FFAP column) was used with helium as the carrier gas at a flow rate of 14.4 ml/min. The initial oven temperature was 100°C, which was held for 2 min, before being raised to 180°C at 8°C per minute and held at 180°C for a further 2 min, and then the temperature was increased to 200°C at 20°C per minute and held at 200°C for 5 min. A range of standards was used at concentrations ranging from 0.5 mM to 100 mM. These contained acetate, propionate, butyrate, isobutyrate, valerate, and isovalerate. Analysis of peak areas was done using ChemStation (version B.03.01) software (Agilent Technologies).
Human gut models are often run for eight turnovers (24 days) before they are deemed to reach steady state (assessed by the stabilization of SCFA/BCFA levels) and a drug or feed intervention is added (e.g., see the work of Grimaldi et al. [10]). We chose to use SCFA/BCFA production as a marker for model stability, with the plateauing of production indicating the achievement of steady state (Fig. 1). Accordingly, community profiling models were run until T5 (15 days after medium flow was started), as SCFA/BCFA production was deemed to be stable at this time point; the repeatability study was run for a further two turnovers after this point to further assess the stability of the model. Gut models maintained for longer time periods (data not reported here) have confirmed the stability of SCFA/BCFA production until T7 (21 days after medium flow was started).
Kruskal-Wallis tests were performed to assess the stability of the SCFA/BCFA produced by the vessels of the concordance study gut models from T3 to T5 to assess if steady state had been reached. Kruskal-Wallis tests were also used to assess the stability of SCFA/BCFA production in the repeatability study gut models from T3 to T5 and from T5 to T7 and whether there were any differences in SFCA/BCFA production between the two models created with the same fecal inoculum (repeatability study).
1H NMR spectroscopy.
Sample supernatants taken from the model were prepared for 1H NMR analysis by adding 200 μl of phosphate buffer (pH 7.4; 100% D2O) containing 1 mM the internal standard 3-trimethylsilyl-1-[2,2,3,3-2H4] propionate (TSP) to 400 μl of each sample and transferred into 5-mm NMR tubes. Spectroscopic analysis of all samples was carried out on a 700-MHz Bruker NMR spectrometer equipped with a cryoprobe. Standard one-dimensional 1H NMR spectra were acquired for all samples with water peak suppression using a standard pulse sequence. For each sample, 8 dummy scans were followed by 256 scans, for which 64,000 data points were collected. Chemical shifts in the fecal spectra were referenced to the TSP singlet at δ 0.0. A spectral width of 20 ppm and an acquisition time per scan of 3.12 s were used.
1H NMR spectra were analyzed in the Matlab environment (release R2014a; MathWorks) with in-house scripts. Spectra were initially aligned and normalized (probabilistic quotient method), before multivariate models were built to compare the metabolic profiles of the gastrointestinal content to those of the sample supernatants from the gut model. Initial unsupervised PCA models were constructed to identify metabolites that explain the largest sources of variation within the data set. OPLS-DA models were constructed for pairwise comparisons of gastrointestinal content and gut model supernatants. The predictive ability (Q2Y) of the OPLS-DA models was calculated using 7-fold cross validation. Metabolites were assigned to peaks using the database of equine metabolites found in the work of Escalona et al. (33).
FISH analysis for total numbers of bacteria.
FISH was used alongside 16S rRNA gene sequencing in order to enumerate the bacteria. It provides a validated approach for counting total microbial numbers using probes targeting specific bacterial taxa. FISH analyses were carried out by a trained operator; quality assurance was provided by duplicate counting by a second operator, which gave rise to comparable results (greater than 95% confidence of similarity). The total bacterial count per milliliter was calculated by utilizing a EUBmix FISH probe. This is a mixture of three EUB338 probes (probes I, II, and III; Sigma-Aldrich) with the dye Cy3 tagged at the 5′ end. Hybridization was performed as described by Daims et al. (34). All samples prepared for FISH were diluted to 1 in 100 (in PBS-sodium dodecyl sulfate [SDS]), and 20 μl was added to the well of a Teflon- and poly-l-lysine-coated 6-well slide (Tekdon Inc.). The slides were dried on a plate incubator for 15 min at 46 to 50°C, dehydrated in 30%, 80%, and 96% ethanol for 3 min each, and then dried for 2 min. The hybridization mixture (0.9 M NaCl, 0.02 M Tris HCl [pH 8], formamide, 10% SDS, 4.55-ng ml−1 probe) was added to each well, and the slides were placed on a sealed tray and put in a hybridization oven for 4 h at 46°C. Once hybridization was complete, the slides were placed into wash buffer (0.9 M NaCl, 0.02 M Tris HCl [pH 8], 0.005 M EDTA solution [pH 8], H2O) for 10 to 15 min at 48°C. After washing, the slides were dipped into cold water for 2 to 3 s and dried using compressed air. Once they were dry, antifade solution (Dabco) was added to each well and a coverslip was applied. The fixed bacteria were visualized using fluorescence microscopy (Nikon Eclipse Ni-U microscope), the number of bacteria in 15 random fields of view per well was counted, and a mean was calculated. To calculate the number of bacteria per milliliter the following equation was used: number of bacteria per milliliter = 0.8 × mean number of bacteria × 6,788.42 × 50 × 100.
Preparation of DNA and analysis by bacterial DNA sequencing.
DNA was extracted from frozen gastrointestinal samples and pellets from the gut model using a PSP spin stool DNA plus kit (Invisorb) using the manufacturer’s protocol. Extracted DNA was resuspended in 100 μl of elution buffer. Further preparation and DNA sequencing were carried out by the Centre of Genomic Research, Liverpool, United Kingdom. The extracted DNA was amplified using PCR of the V4 region of the 16S rRNA gene using the primers F515 (GTGCCAGCMGCCGCGGTAA) and 806R (GGACTACHVGGGTWTCTAAT) (35), with reverse primers containing individual Golay barcodes. For a final volume of 20 μl to be subjected to V4 region PCR, 1 μl of the extracted DNA from each sample was added to 7 μl of molecular water, 10 μl of a NEBNext high-fidelity master mix (New England Biolabs), 1 μl of forward primer (3 μM), and 1 μl of an individually barcoded reverse primer (3 μM). This mixture was made for each individual sample, which then underwent PCR under the following conditions: 30 s at 98°C and 25 cycles of 10 s at 98°C, 30 s at 55°C, and 30 s at 72°C. This was followed by a final period of 5 min at 72°C, and then the mixture was kept at 4°C until it was processed. PCR products were evaluated by electrophoresis in a 2% agarose gel stained with Midori green. Successfully amplified DNA was cleaned using AMPure XP beads (Agencourt) and quantified using a Quant-iT PicoGreen double-stranded DNA assay (Life Technologies). The PCR products were pooled at an equimolar ratio and size selected using a 2% agarose gel cassette run in a Pippin Prep machine (Sage Science), where fragments were eluted at 254 bp and kept for sequencing. Sequencing was carried out on a MiSeq Illumina platform.
Analysis of the sequencing data was carried out on a remote Linux server provided by the University of Surrey with QIIME2 (36) installed. Zipped read files were uploaded to the Linux server and converted into a QIIME artifact (qiime tools import), and a summary was generated (qiime demux summarize). Quality control was carried out using the DADA2 program (37), and the ends of the reads were trimmed at positions 15 and 220 of the reads (qiime dada2 denoise-single). A rooted phylogenetic tree was generated for diversity analyses (qiime alignment mafft, qiime alignment mask, qiime phylogeny fasttree, and qiime phylogeny midpoint-root). Diversity core metrics were generated at a sampling depth of 47,000 reads (qiime diversity core-metrics-phylogenetic), and box plots displaying alpha diversity (measure as observed OTUs) were generated (qiime diversity alpha-group-significance). The reference database Greengenes (38) was downloaded, and the taxonomic classifiers were trained on this specific sample preparation and these sequencing parameters (qiime feature-classifier classify-sklearn). Taxa summary bar plots were generated (qiime taxa barplot).
Mann-Whitney U tests were used to assess whether the read numbers for the identified bacterial phyla were statistically significantly different when those in the gastrointestinal content were compared to those in the gut model and at the time points of the model. Venn diagrams were generated to visualize how many identifiable and uniquely named OTUs were shared between the vessels of the three biological replicate gut models and their corresponding gastrointestinal compartments. Regression models were built with the mean SCFA/BCFA concentration and bacterial phyla count for each vessel of the concordance study at T−1, T0, and T5. Correlations identified by the models were deemed to be significant when R2 was 0.5 and P was <0.05. Venn diagrams were generated to visualize how many identifiable and uniquely named OTUs were shared between the vessels of the three biological replicate gut models and their corresponding gastrointestinal compartments.
Data availability.
The raw sequencing reads analyzed in this study have been submitted to the European Nucleotide Archive and can be found in project ERP107630.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff at the Veterinary Pathology Centre and the University of Surrey for help with equine gastrointestinal content sample acquisition during postmortem examinations.
This project was funded by the Petplan Charitable Trust and the University of Surrey.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Dougal K, Harris PA, Edwards A, Pachebat JA, Blackmore TM, Worgan HJ, Newbold CJ. 2012. A comparison of the microbiome and the metabolome of different regions of the equine hindgut. FEMS Microbiol Ecol 82:642–652. doi: 10.1111/j.1574-6941.2012.01441.x. [DOI] [PubMed] [Google Scholar]
- 2.Costa MC, Silva G, Ramos RV, Staempfli HR, Arroyo LG, Kim P, Weese JS. 2015. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Vet J 205:74–80. doi: 10.1016/j.tvjl.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Proudman CJ, Hunter JO, Darby AC, Escalona EE, Batty C, Turner C. 2015. Characterisation of the faecal metabolome and microbiome of Thoroughbred racehorses. Equine Vet J 47:580–586. doi: 10.1111/evj.12324. [DOI] [PubMed] [Google Scholar]
- 4.Costa MC, Stämpfli HR, Allen-Vercoe E, Weese JS. 2016. Development of the faecal microbiota in foals. Equine Vet J 48:681–688. doi: 10.1111/evj.12532. [DOI] [PubMed] [Google Scholar]
- 5.Ericsson AC, Johnson PJ, Lopes MA, Perry SC, Lanter R. 2016. A microbiological map of the healthy equine gastrointestinal tract. PLoS One 11:e0166523-17. doi: 10.1371/journal.pone.0166523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa MC, Arroyo LG, Allen-Vercoe E, Stämpfli HR, Kim PT, Sturgeon A, Weese JS. 2012. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS One 7:e41484. doi: 10.1371/journal.pone.0041484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leng J, Proudman C, Darby A, Blow F, Townsend N, Miller A, Swann J. 2018. Exploration of the fecal microbiota and biomarker discovery in equine grass sickness. J Proteome Res 17:1120–1128. doi: 10.1021/acs.jproteome.7b00784. [DOI] [PubMed] [Google Scholar]
- 8.Macfarlane GT, Macfarlane S, Gibson GR. 1998. Validation of a three-stage compound continuous culture system for investigating the effect of retention time on the ecology and metabolism of bacteria in the human colon. Microb Ecol 35:180–187. doi: 10.1007/s002489900072. [DOI] [PubMed] [Google Scholar]
- 9.Walton GE, Van Den Heuvel E, Kosters MHW, Rastall RA, Tuohy KM, Gibson GA. 2012. Randomised crossover study investigating the effects of galacto-oligosaccharides on the faecal microbiota in men and women over 50 years of age. Br J Nutr 107:1466–1475. doi: 10.1017/S0007114511004697. [DOI] [PubMed] [Google Scholar]
- 10.Grimaldi R, Cela D, Swann JR, Vulevic J, Gibson GR, Tzortzis G. 2017. In vitro fermentation of B-GOS: impact on faecal bacterial populations and metabolic activity in autistic and non-autistic children. FEMS Microbiol Ecol 93:fiw233. doi: 10.1093/femsec/fiw233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimaldi R, Gibson GR, Vulevic J, Giallourou N, Castro-Mejía JL, Hansen LH, Leigh Gibson E, Nielsen DS, Costabile A. 2018. A prebiotic intervention study in children with autism spectrum disorders (ASDs). Microbiome 6:133–113. doi: 10.1186/s40168-018-0523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowman RS, Theodorou MK, Hyslop JJ, Dhanoa MS, Cuddeford D. 1999. Evaluation of an in vitro batch culture technique for estimating the in vivo digestibility and digestible energy content of equine feeds using equine faeces as the source of microbial inoculum. Anim Feed Sci Technol 80:11–27. doi: 10.1016/S0377-8401(99)00039-5. [DOI] [Google Scholar]
- 13.Desrousseaux G, Santos AS, Pellikaan WF, Van der Poel AFB, Cone JW, Guedes CMV, Ferreira LMM, Rodrigues MAM. 2012. Effect of collection time on the fermentative activity of microbes in equine faeces. Anim Feed Sci Technol 178:183–189. doi: 10.1016/j.anifeedsci.2012.09.016. [DOI] [Google Scholar]
- 14.Biddle AS, Black SJ, Blanchard JL. 2013. An in vitro model of the horse gut microbiome enables identification of lactate-utilizing bacteria that differentially respond to starch induction. PLoS One 8:e77599-13. doi: 10.1371/journal.pone.0077599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andoh A, Tsujikawa T, Sasaki M, Mitsuyama K, Suzuki Y, Matsui T, Matsumoto T, Benno Y, Fujiyama Y. 2009. Faecal microbiota profile of Crohn’s disease determined by terminal restriction fragment length polymorphism analysis. Aliment Pharmacol Ther 29:75–82. doi: 10.1111/j.1365-2036.2008.03860.x. [DOI] [PubMed] [Google Scholar]
- 16.Moore-Colyer M, O'Gorman DM, Wakefield K. 2014. An in vitro investigation into the effects of a marine-derived, multimineral supplement in simulated equine stomach and hindgut environments. J Equine Vet Sci 34:391–397. doi: 10.1016/j.jevs.2013.07.016. [DOI] [Google Scholar]
- 17.Murray J, Bice RKT, Moore-Colyer M. 2010. The effect of particle size on the in vitro fermentation of different ratios of high-temperature dried lucerne and sugar beet pulp incubated with equine faecal inocula. Anim Feed Sci Technol 162:47–57. doi: 10.1016/j.anifeedsci.2010.09.001. [DOI] [Google Scholar]
- 18.Murray J, McMullin P, Handel I, Hastie PM. 2012. The effect of freezing on the fermentative activity of equine faecal inocula for use in an in vitro gas production technique. Anim Feed Sci Technol 178:175–182. doi: 10.1016/j.anifeedsci.2012.09.013. [DOI] [Google Scholar]
- 19.Murray JMD, McMullin P, Handel I, Hastie PM. 2014. Comparison of intestinal contents from different regions of the equine gastrointestinal tract as inocula for use in an in vitro gas production technique. Anim Feed Sci Technol 187:98–103. doi: 10.1016/j.anifeedsci.2013.10.005. [DOI] [Google Scholar]
- 20.Murray J, Scott B, Hastie PM. 2009. Fermentative capacity of equine faecal inocula obtained from clinically normal horses and those predisposed to laminitis. Anim Feed Sci Technol 151:306–311. doi: 10.1016/j.anifeedsci.2009.01.011. [DOI] [Google Scholar]
- 21.Abdouli H, Ben Attia S. 2007. Evaluation of a two-stage in vitro technique for estimating digestibility of equine feeds using horse faeces as the source of microbial inoculum. Anim Feed Sci Technol 132:155–162. doi: 10.1016/j.anifeedsci.2006.03.005. [DOI] [Google Scholar]
- 22.Elghandour MMY, Vázquez Chagoyán JC, Salem AZM, Kholif AE, Martínez Castañeda JS, Camacho LM, Buendía G. 2014. In vitro fermentative capacity of equine fecal inocula of 9 fibrous forages in the presence of different doses of Saccharomyces cerevisiae. J Equine Vet Sci 34:619–625. doi: 10.1016/j.jevs.2013.11.013. [DOI] [Google Scholar]
- 23.Hobden MR, Martin-Morales A, Guérin-Deremaux L, Wils D, Costabile A, Walton GE, Rowland I, Kennedy OB, Gibson GR. 2013. In vitro fermentation of NUTRIOSE FB06, a wheat dextrin soluble fibre, in a continuous culture human colonic model system. PLoS One 8:e77128-7. doi: 10.1371/journal.pone.0077128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costabile A, Walton GE, Tzortzis G, Vulevic J, Charalampopoulos D, Gibson GR. 2015. Effects of orange juice formulation on prebiotic functionality using an in vitro colonic model system. PLoS One 10:e0121955. doi: 10.1371/journal.pone.0121955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pérez-López E, Cela D, Costabile A, Mateos-Aparicio I, Rupérez P. 2016. In vitro fermentability and prebiotic potential of soyabean Okara by human faecal microbiota. Br J Nutr 116:1116–1124. doi: 10.1017/S0007114516002816. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Gibson GR, Walton GE. 2017. A three-stage continuous culture approach to study the impact of probiotics, prebiotics and fat intake on faecal microbiota relevant to an over 60 s population. J Funct Foods 32:238–247. doi: 10.1016/j.jff.2017.02.035. [DOI] [Google Scholar]
- 27.Gharbia SE, Shah HN. 1991. Pathways of glutamate catabolism among Fusobacterium species. J Gen Microbiol 137:1201–1206. doi: 10.1099/00221287-137-5-1201. [DOI] [PubMed] [Google Scholar]
- 28.Rogers AH, Chen J, Zilm PS, Gully NJ. 1998. The behaviour of Fusobacterium nucleatum chemostat-grown in glucose- and amino acid-based chemically defined media. Anaerobe 4:111–116. doi: 10.1006/anae.1997.0140. [DOI] [PubMed] [Google Scholar]
- 29.Macrae JC, Armstrong DG. 1968. Enzyme method for determination of alpha-linked glucose polymers in biological materials. J Sci Food Agric 19:578. doi: 10.1002/jsfa.2740191006. [DOI] [Google Scholar]
- 30.Fuller KW. 1966. Automated determination of sugars. Autom Anal Chem 2:57–. [Google Scholar]
- 31.Macfarlane GT, Englyst HN. 1986. Starch utilization by the human large intestinal microflora. J Appl Bacteriol 60:195–201. doi: 10.1111/j.1365-2672.1986.tb01073.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhao G, Nyman M, Jönsson JÅ. 2006. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed Chromatogr 20:674–682. doi: 10.1002/bmc.580. [DOI] [PubMed] [Google Scholar]
- 33.Escalona EE, Leng J, Dona AC, Merrifield CA, Holmes E, Proudman CJ, Swann JR. 2015. Dominant components of the Thoroughbred metabolome characterised by 1H-nuclear magnetic resonance spectroscopy: a metabolite atlas of common biofluids. Equine Vet J 47:721–730. doi: 10.1111/evj.12333. [DOI] [PubMed] [Google Scholar]
- 34.Daims H, Bruhl A, Amann R, Schleifer K, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 444:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 35.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolyen E, Rideout J, Dillon M, Bokulich N, Abnet C, Al-Ghalith G, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope E, Da Silva R, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley G, Janssen S, Jarmusch AK, Jiang L, Kaehler B, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, et al. . 2018. QIIME 2: reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 6:e27295v2. doi: 10.7287/peerj.preprints.27295v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Callahan BJ, Mcmurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: High resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing reads analyzed in this study have been submitted to the European Nucleotide Archive and can be found in project ERP107630.