
Figure 1.
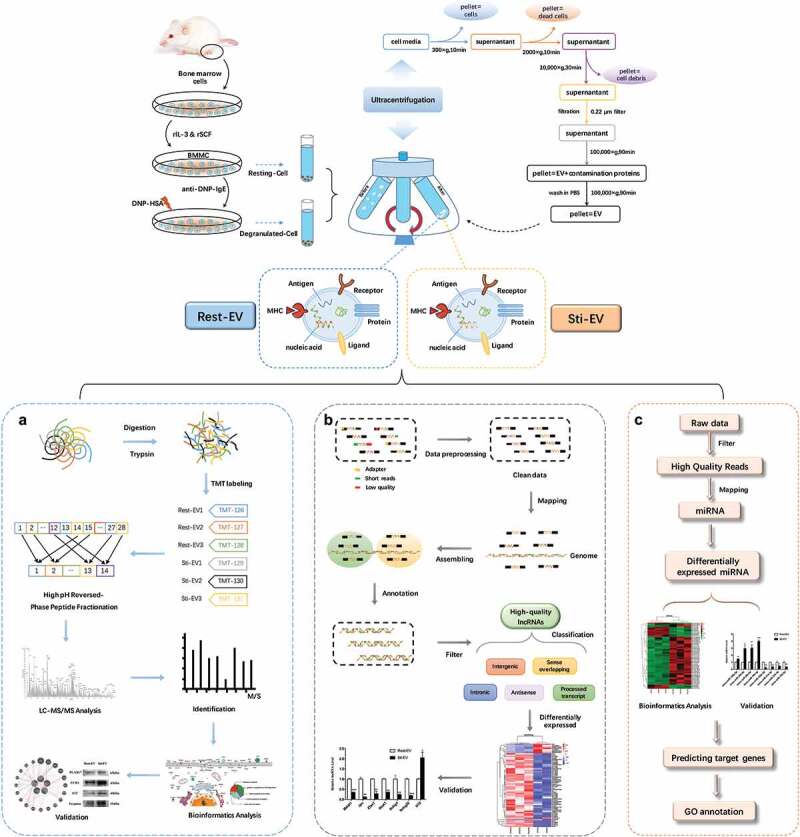
Schematic representation of BMMC-derived EVs isolation, and characterization. The TMT-labelling strategy elucidates the enrichment of proteins encapsulated in MC-derived EVs and RNA-seq to identify the expression profiles of lncRNAs and miRNAs. Murine bone marrow cells were induced to differentiate into MCs by rIL-3 and SCF in vitro. EVs released by resting and degranulated BMMCs (Rest-EV and Sti-EV) were isolated by successive differential centrifugation. (a) Flowchart of the TMT-labelling quantitative proteomic analysis of Rest-EV and Sti-EV. Proteins were extracted and digested by filter-aided sample preparation (FASP). The peptides were labelled with six-plex tandem mass tags (TMT) and analysed using EASY-nano-LC−MS/MS in MaxQuant software. The differentially expressed proteins (DEPs) were further analysed by bioinformatics tools and followed by biological validation using Western blotting. (b) Overview of the comprehensive scheme for the systematic identification of lncRNAs in Rest-EV and Sti-EV. The quality control of the RNA sequences was performed by the FastQC software. High-quality lncRNAs were obtained by a series of steps, such as mapping, assembling, annotation, and filtering. The assembled putative lncRNAs were classified into five categories, including antisense lncRNAs, intergenic lncRNAs (lincRNAs), processed transcript lncRNAs, sense intronic lncRNAs, and sense overlapping lncRNAs. The differentially expressed (DE) lncRNAs were further analysed by bioinformatics tools and followed by biological validation using qRT-PCR. (c) Flowchart of small RNA-seq data analysis. Preprocessing of the reads was accomplished through mapper.pl script of miRDeep2 software. Bowtie software was used to trim and align generated sequence reads; and mapping of the reads to miRBase was included. The DE miRNAs were investigated by the Bioconductor R packages and followed by biological validation using qRT-PCR. The miRTarBase database was used to analyse miRNA target interactions. Analysis of gene ontology annotation was performed by applying the DAVID functional annotation tool.