

Short abstract
Background
Poly (ADP-ribose) polymerase 1 (PARP-1) plays pivotal roles in immune and inflammatory responses. Accumulating evidence suggests PARP-1 as a promising target for immunomodulation in multiple sclerosis and natalizumab-associated progressive multifocal leukoencephalopathy.
Objective
This study explores expression of PARP-1 and downstream effectors in multiple sclerosis and during natalizumab treatment.
Methods
Transcriptional expressions were studied by real-time reverse transcriptase polymerase chain reaction on CD4+T/CD8+T/CD14+/B cells and peripheral blood mononuclear cells from healthy volunteers, untreated and natalizumab-treated non-progressive multifocal leukoencephalopathy and progressive multifocal leukoencephalopathy multiple sclerosis patients.
Results
PARP-1 expression was higher in CD4+T, CD8+T and B cells from untreated patients compared to healthy volunteers. Natalizumab treatment restored deregulated PARP-1 expression in T cells but not in B cells. Sustained upregulation of PARP-1 was associated with decreased expression of downstream PARP-1 factors such as TGFBR1/TGFBR2/BCL6 in B cells. Notably, a higher expression of PARP-1 was detected in progressive multifocal leukoencephalopathy patients.
Conclusions
Given the importance of PARP-1 in inflammatory processes, its upregulation in multiple sclerosis lymphocyte populations suggests a potential role in the immune pathogenesis of multiple sclerosis. Strikingly higher PARP-1 expression in progressive multifocal leukoencephalopathy cases suggests its involvement in progressive multifocal leukoencephalopathy disease pathomechanisms. These results further support the value of PARP-1 inhibitors as a potential novel therapeutic strategy for multiple sclerosis and natalizumab-associated progressive multifocal leukoencephalopathy.
Keywords: PARP-1, natalizumab, multiple sclerosis, progressive multifocal leukoencephalopathy (PML), JCV
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) resulting from an autoimmune attack targeting myelin sheets in the CNS, leading to demyelination, axonal and neuronal injury.1 Natalizumab (Tysabri, Biogen), a recombinant humanised monoclonal antibody that targets α4β1 and α4β7 integrins on the surface of leukocytes is regarded as an effective disease-modifying therapy for relapsing–remitting multiple sclerosis (RRMS) that prevents invasion of the CNS through the blood–brain barrier, thus reducing inflammation and preventing the formation of new focal lesions. These effects translate into a significant reduction of relapse rates and disability progression.2 However, treatment with natalizumab has been associated with the development of progressive multifocal leukoencephalopathy (PML), a devastating opportunistic lytic infection of oligodendrocytes in the CNS that is caused by reactivation of the latent human polyomavirus JC virus (JCV).3 JCV seropositivity, longer treatment duration, especially beyond 2 years, and prior treatment with immunosuppressants has been identified as risk factors and are used for clinical guidance.4
Poly (ADP-ribose) polymerase 1 (PARP-1) is the most abundant and well-characterised member of the PARP nuclear enzyme superfamily that catalyses the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD+) to a broad panel of acceptor proteins such as histones and transcription factors.5 PARP-1 is involved in a wide range of cellular processes including DNA repair, cell proliferation and death signalling, transcriptional regulation and inflammation.6,7 Diverse studies conducted in the murine experimental autoimmune encephalomyelitis model of MS suggested a potential role for PARP-1 in the pathogenesis of MS,8–11 triggering the development of PARP-1 inhibitors as promising approaches for immunomodulation in MS.12 Besides their potential value for MS treatment, PARP-1 inhibitors have also been suggested as novel therapeutic drugs for PML.13
Here, we examined PARP-1 expression in various lymphocyte subpopulations from untreated and natalizumab-treated MS patients and in patients with natalizumab-associated PML. We report the differential expression of PARP-1 and downstream effectors in T and B cells, together with deregulated PARP-1 expression in patients with PML.
Methods
Subjects
Patient characteristics and cohorts are depicted in Table 1. Samples were collected during visits of the patients in the years 2008–2015 and for PML cases in the years 2008–2012. Five different and heterogenous cohorts (except monocyte and B cell cohorts that were homogeneous) were used for the study. Considering the duration of natalizumab treatment as a risk factor for developing PML, we divided our cohorts of natalizumab-treated patients into two groups: treatment duration of 3–24 months and longer than 24 months. A group of 15 natalizumab-treated patients who developed PML was also included in the peripheral blood mononuclear cell (PBMC) cohort. Samples were drawn after PML diagnosis. The JCV serostatus was available from 57 out of 58 natalizumab-treated patients of the PBMC cohort. PML patients were all JCV seropositive (15/15); 10 short-term treated non-PML patients (3–24 months, 10/21) and 10 long-term treated non-PML patients (>24 months, 10/22) were JCV seropositive. All untreated patients had no immunomodulation in the 6 months before or during the study.
Table 1.
Characteristics of patients.
| Group | N | Gender F/M | Age (years) mean ± SD | No. of natalizumab infusions mean ± SD | EDSS median (IQR) | No. of relapses in the past 6 months mean ± SEM |
|---|---|---|---|---|---|---|
| Cohort for B cell analysis | ||||||
| • Healthy volunteers | 12 | 10/2 | 41.0 ± 4.09 | NA | NA | NA |
| • Untreated RRMS | 12 | 8/4 | 55.7 ± 3.13 | NA | 3.0 (2.5–3.8) | 0.16 ± 0.11 |
| • Nat 3–24 months | 12 | 10/2 | 40.5 ± 3.64 | 14.0 ± 1.97 | 3.25 (1.6–3.8) | 0.25 ± 0.17 |
| • Nat >24 months | 12 | 9/3 | 39.2 ± 2.32 | 66.7 ± 6.60 | 2.5 (2.0–3.5) | 0 |
| Cohort for CD4+T cell analysis | ||||||
| • Healthy volunteers | 12 | 8/4 | 36.1 ± 2.78 | NA | NA | NA |
| • Untreated RRMS | 12 | 9/3 | 46.8 ± 2.81 | NA | 2.25 (1.5–3.8) | 0.08 ± 0.08 |
| • Nat 3–24 months | 12 | 8/2 | 42.6 ± 3.93 | 12.8 ± 2.35 | 3.0 (2.0–4.3) | 0.33 ± 0.14 |
| • Nat >24 months | 12 | 10/2 | 39.8 ± 2.23 | 39.6 ± 1.67 | 3.25 (3.0–3.8) | 0 |
| Cohort for CD8+T cell analysis | ||||||
| • Healthy volunteers | 12 | 8/4 | 41.4 ± 2.93 | NA | NA | NA |
| • Untreated RRMS | 12 | 9/3 | 47.4 ± 2.98 | NA | 2.0 (1.5–3.2) | 0.16 ± 0.11 |
| • Nat 3–24 months | 10 | 8/2 | 39.8 ± 3.73 | 8.7 ± 2.70 | 2.5 (1.3–4.0) | 0.60 ± 0.26 |
| • Nat >24 months | 12 | 10/2 | 41.0 ± 2.80 | 51.1 ± 4.55 | 3.0 (2.1–4.5) | 0.16 ± 0.16 |
| Cohort for monocyte analysis | ||||||
| • Healthy volunteers | 11 | 9/2 | 39.4 ± 4.12 | NA | NA | NA |
| • Untreated RRMS | 12 | 8/4 | 55.7 ± 3.13 | NA | 3.0 (2.5–3.8) | 0.16 ± 0.11 |
| • Nat 3–24 months | 12 | 10/2 | 40.5 ± 3.64 | 14.0 ± 1.97 | 3.25 (1.6–3.8) | 0.25 ± 0.17 |
| • Nat >24 months | 11 | 8/3 | 40.2 ± 2.29 | 69.0 ± 6.81 | 2.5 (2.0–3.5) | 0 |
| Cohort for PBMC analysis | ||||||
| • Healthy volunteers | 14 | 8/6 | 46.0 ± 2.93 | NA | NA | NA |
| • Untreated RRMS | 20 | 14/6 | 52.1 ± 1.88 | NA | 2.0 (1.7–2.5) | 0.05 ± 0.05 |
| • Nat 3–24 months | 21 | 16/5 | 40.0 ± 2.79 | 14.0 ± 1.74 | 3.5 (2.7–4.2) | 0.35 ± 0.10 |
| • Nat >24 months | 22 | 17/5 | 40.6 ± 2.20 | 49.0 ± 3.62 | 3.25 (2.5–4.0) | 0 |
| • PML | 15 | 12/3 | 45.9 ± 2.22 | 33.5 ± 3.34 | – | – |
IQR: interquartile range; EDSS: Expanded Disability Status Scale; RRMS: relapsing–remitting multiple sclerosis; Nat: natalizumab; PBMC: peripheral blood mononuclear cell; PML: progressive multifocal leukoencephalopathy.
Standard protocol approvals, registrations and patient consents
Written informed consent was obtained from all patients and healthy donors spontaneously recruited. The study was approved by the cantonal institutional review board of Basel City and Basel Country.
Cell separation
PBMCs were isolated from ethylenediamine tetraacetic acid (EDTA) anticoagulated venous blood by density gradient centrifugation (Lymphoprep; Axon Lab, Switzerland). CD4+T/CD8+T/CD14+T and B cell subpopulations were separated from PBMCs using MACS technology (positive isolation using CD4, CD8, and CD14 microbeads, human, B-cell negative enrichment kit II; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Purity of isolated CD4+T (97.9% ± 0.21), CD8+T (96.4% ± 0.42), CD14+ (95.9% ± 0.6) and B cells (97.7% ± 0.36) was analysed with an Attune focusing flow cytometer (Applied Biosystems, Darmstadt, Germany).
RNA isolation
QIAzol (QIAgen AG, Hombrechtikon, Switzerland) was used to lyse PBMCs and isolated cell subpopulations. Total RNA (including miRNA) was extracted using an miRNeasy mini kit (QIAgen) according to the manufacturer’s protocol.
Messenger RNA expression analysis
Total RNA was reverse-transcribed using qScript XLT cDNA SuperMix (Quantabio) according to the manufacturer’s protocol. cDNA was used as a template for the real-time reverse transcriptase polymerase chain reaction (RT-PCR) based on the 5′ nuclease chemistry with ABI PRISM 7500 sequence detection system (Applied Biosystems, Switzerland), using the following assay-on-demand reagents (Applied Biosystems): PARP1 (Hs 00242302_m1); TGFBR1 (Hs 00610318_m1); TGFBR2 (Hs 00234253_m1); BCL6 (Hs 00153368_m1). As previously reported,14 PUM1 (Hs 00472881_m1) was used as a reference for normalisation and relative expression analyses. The comparative cycle threshold method (Applied Biosystems) was used for calculations of relative quantitation of targets.
Data and statistical analysis
GraphPad Prism software (La Jolla, CA, USA) was used for statistical analysis. For the various group comparisons the non-parametric Mann–Whitney test was applied. P < 0.05 was considered significant. Correlations between expressions were assessed using Spearman’s non-parametric correlations.
Results
Natalizumab restores deregulated PARP-1 expression in T-cell subsets but not in B cells
PARP-1 mRNA expression was assessed in CD4+T, CD8+T, B cells and CD14+ monocytes from different cohorts of controls and study patients. Interestingly, PARP-1 was significantly upregulated in untreated RRMS patients compared to healthy volunteers in all tested T and B-cell subsets (Figure 1(a–c)). In contrast, PARP-1 expression was downregulated in CD4+T (Figure 1(a)) and CD8+T (Figure 1(b)) cells of natalizumab-treated patients. Downregulation was observed already after short-term treatment. In B cells, however, sustained upregulation of PARP-1 was observed under natalizumab therapy irrespective of treatment duration (Figure 1(c)). In monocytes, only a trend for upregulation of PARP-1 expression in untreated RRMS patients compared to healthy volunteers was found (Figure 1(d)).
Figure 1.

Expression of poly (ADP-ribose) polymerase 1 (PARP-1) mRNA in CD4+T, CD8+T, B cells and monocytes. Transcriptional expression of PARP-1 was analysed with real-time reverse transcriptase polymerase chain reaction in CD4+T (a), CD8+T cells (b), B cells (c) and monocytes (d) from healthy volunteer, untreated patients and natalizumab-treated patients (3–24 months or >24 months). Relative expression levels (median with interquartile range) are depicted. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
Deregulated TGFBR expression in CD4+T and B cells
PARP-1 has been suggested to regulate TGFBR expression negatively in T cells.15 We explored TGFBR1 and TGFBR2 expression in the T and B-cell subsets of our cohorts of patients and controls. In CD4+T cells, no significant differences were observed in TGFBR1 mRNA expression levels between the four groups (Figure 2(a), left panel). Interestingly, PARP-1 upregulation correlated with significant downregulation of TGFBR2 in CD4+T cells from untreated MS patiens versus healthy volunteers (Figure 2(a), right panel) (r = –0.60; P = 0.002). However, in contrast to the restored PARP-1 mRNA levels in patients treated with natalizumab, sustained downregulation of TGFBR2 was observed under treatment, although significance was lost in the long-term treated group, suggesting a possible delay in the restoration of TGFBR2 mRNA levels. In B cells, PARP-1 upregulation was associated with both TGFBR1 and TGFBR2 downregulation (P = 0.051 and P = 0.038, respectively) in untreated patients versus healthy volunteers (Figure 2(b)). Long-term treatment with natalizumab increased TGFBR1 expression significantly (Figure 2(b), left panel) contrasting to TGFBR2 mRNA expression levels that remained downregulated under therapy (Figure 2(b), right panel). No significant differential expression of TGFBR1 and TGFBR2 was detected in CD8+T cells (data not shown).
Figure 2.

Expression of TGFBR1 and TGFBR2 mRNA in CD4+T and B cells. Transcriptional expressions of TGFBR1 (left panels) and TGFBR2 (right panels) were analysed with real-time reverse transcriptase polymerase chain reaction in CD4+T (a) and B cells (b) from healthy volunteers, untreated and natalizumab-treated patients (3–24 months; >24 months). Relative expression levels (median with interquartile range) are depicted. ***P < 0.001; **P < 0.01, *P < 0.05; ns: not significant.
Differential expression of B-cell lymphoma 6 (BCL6) in B and CD8+T cells from untreated and natalizumab-treated patients
PARP-1 has been proposed to play an important role in switching off BCL6 transcription in B cells.16 In our cohort, in parallel to the uncovered PARP-1 upregulation, a trend for downregulation of BCL6 was observed in untreated and both groups of natalizumab-treated patients (Figure 3(a)). Notably, significant lower expression of BCL6 mRNA was detected in samples from patients with long-term therapy. We further extended our analyses to T-cell subsets. In CD4+T cells, where BCL6 was shown to play a critical role for regulatory T cell (Treg)-mediated control of T helper type 2 inflammation,17 no significant difference was found in our cohort of patients (data not shown). In contrast, in CD8+T cells, BCL6 expression levels correlated with PARP-1 expression, with significant downregulation in untreated patients compared to healthy volunteers (r = –0.51; P = 0.021) and restored BCL6 expression in natalizumab-treated patients (r = –0.61; P = 0.002) (Figure 3(b)).
Figure 3.
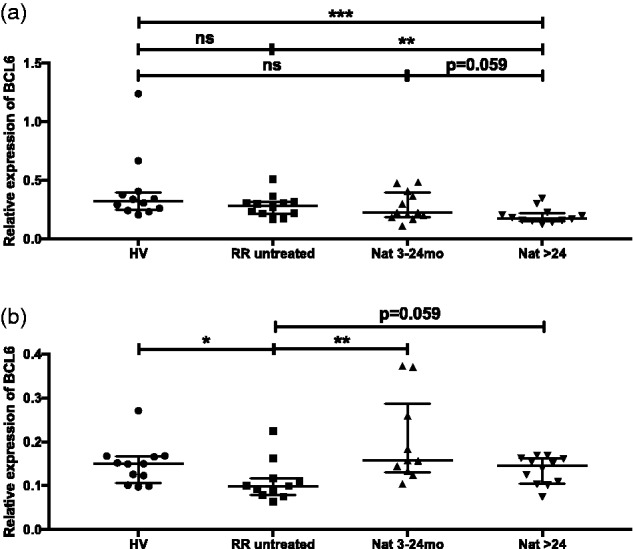
Expression of BCL6 mRNA in B cells and CD8+T cells. Transcriptional expression of BCL6 was analysed with real-time reverse transcriptase polymerase chain reaction in B cells (a) and CD8+T cells (b) from healthy volunteers, untreated and natalizumab-treated patients (3–24 months; >24 months). Relative expression levels (median with interquartile range) are depicted. ***P < 0.001; **P < 0.01; *P < 0.05; ns: not significant.
PARP1/TGFBR deregulation in PML
Regarding the reported suppressive effect of a PARP-1 inhibitor on JCV replication,13 we further expanded our investigation of PARP-1 expression to samples from patients with natalizumab-associated PML. The analysis was performed on PBMCs due to the limitation of sample volume. A total of 15 PML patients whose natalizumab treatment duration varied from 17 to 62 months were included in the study. In contrast to the MS-related upregulation of PARP-1 in B and T-cell subsets, no significant difference was found in PBMCs of untreated and control groups, probably reflecting the heterogeneity of the PBMC mixture (Figure 4(a)). Also, natural killer cells, which were not studied, may contribute to the expression in PBMCs. The upregulation of PARP-1 mRNA expression was also detected in long-term natalizumab-treated patients. Importantly, a higher expression of PARP-1 was detected in patients with PML. TGFBR1 and TGFBR2 were downregulated in long-term treated patients, in parallel with the expression levels of PARP-1 (Figure 4(b), left and right panel, respectively). Strikingly lower expressions of TGFBR1 and TGFBR2 were found in PML patients, thereby being in line with the high PARP-1 expression levels.
Figure 4.

Expression of poly (ADP-ribose) polymerase 1 (PARP-1), TGFBR1 and TGFBR2 mRNA in peripheral blood mononuclear cells (PBMCs). Transcriptional expression of PARP-1 (a), TGFBR1 ((b), left panel) and TGFBR2 ((b), right panel) were analysed with real-time reverse transcriptase polymerase chain reaction in PBMCs from healthy volunteers, untreated, natalizumab-treated patients (3–24 months; >24 months) and natalizumab-associated progressive multifocal leukoencephalopathy (PML) patients. Relative expression levels (median with interquartile range) are depicted. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns: not significant.
Discussion
Growing evidence supports a significant role for PARP-1 in neurological diseases.18 In the field of neuroimmunology, most data were acquired from experimental animal models.12 In contrast, only few studies have been conducted using human MS samples. PARP-1 activation has been detected in apoptotic oligodendrocytes in MS lesions.19 In a study performed on peripheral blood monocytes of RRMS patients and secondary progressive multiple sclerosis (SPMS), Farez and colleagues showed that PARP-1 enzymatic activity was significantly higher in patients with SPMS.10 Here, we report for the first time higher expression of PARP-1 in lymphocyte subsets from RRMS patients compared to healthy volunteers. Noteworthy, higher PARP-1 expression was detected in CD4+T, CD8+T and B cells, whereas no significant increase was found in CD14+ monocytes, suggesting a possible specific effect on common lymphoid progenitors.
PARP-1 appears as an important regulator of TGFBR expression and transforming growth factor beta (TGF-β) signalling. In fact, increased expression of TGFBR1 and TGFBR2 were found in CD4+ T cells from PARP-1–/– mice.15 In our cohort of human CD4+T cell samples, increased PARP-1 expression in untreated patients correlated with decreased expression of TGFBR2 but not TGFBR1. Zhang and colleagues proposed distinct underlying mechanisms of negative regulation of TGFBR1 and TGFBR2 expressions by PARP-1, involving on one side a selective binding of PARP-1 to the promoter of TGFBR2 and on the other side an inhibition of TGBFR1 expression mediated by PARP-1 enzymatic activity.15 Such divergent or additional/compensatory mechanisms might be involved in the differential effect of PARP-1 on the expression of TGFBR1 and TGFBR2 in our CD4+T samples. Significant downregulation of TGFBR2 expression in untreated RRMS patients compared to healthy volunteers was previously found in another study from our lab using different CD4+T cell samples.20 These findings may contribute to a better understanding of the pivotal role of TGF-β signalling in the regulation of immune responses. Hence, Li and colleagues reported lethal inflammation associated with T-cell activation in mice with T-cell-specific deletion of TGFBR2.21 Importantly, deregulation of TGF-β signalling, a critical regulator of the development and function of Tregs, might contribute to the impaired immunoregulatory function of Tregs in MS.22,23 In parallel, growing evidence is supporting an important role for PARP-1 in the regulation of Treg differentiation and function. Hence, Nasta and colleagues reported increased Foxp3+ regulatory T cells in PARP-1 deficiency.24 More recently, Luo and colleagues suggested that PARP-1 negatively regulates the immunosuppressive function of Treg cells at the post-translational level by way of FOXP3 poly (ADP-ribosyl)ation.25 Collectively, these data suggest a potential important impact of the deregulation of the PARP-1/TGFBR axis on T-cell function in MS.
In B cells, which are also central players of the immune cascades triggering CNS inflammation,26,27 deregulation of PARP-1/TGFBR signalling might also contribute to MS disease activity. In fact, using PARP-1-deficient mice, Ambrose et al. unravelled an essential role of PARP-1 in normal T-cell-dependent antibody responses and the regulation of isotype expression.28 In mice, B-cell-specific deletion of TGFBR2 highlighted the important role of TGFBR signalling in controlling B-cell homeostasis and responses, notably IgG production.29 In patients’ B cells, unlike in T cells, deregulation of PARP-1/TGFBR signalling was observed both in untreated patients and in natalizumab-treated patients. Considering the proposed critical regulatory role of B cells in the immune response that controls JCV infection,30 these data might be of importance in the setting of natalizumab-induced PML development where strikingly overexpressed PARP-1/TGFBR signalling was detected.
Deregulated expression of BCL6 in B cells might also contribute to MS disease pathomechanisms. In fact, BCL6 functions as a transcriptional repressor that impairs premature B-cell activation/differentiation31 and plays a critical role in the development of a diverse primary B-cell repertoire.32 In CD8+T cells, considering the critical role of BCL6 in the generation and maintenance of activated memory CD8+T cells,33 the downregulation of BCL6 in untreated patients could possibly be involved in the reported deficiency of CD8+T effector memory T cells in MS.34 BCL6 was also shown to control granzyme B expression in murine effector CD8+T cells.35 In our patient CD8+T cells, no significant change in the expression of granzyme B was detected (data not shown).
Conclusions
Our data support a potential impact of PARP-1 deregulation in MS and treatment-associated PML pathomechanisms, therefore strengthening further the proposed PARP-1 inhibition approach as a novel therapy for MS. However, considering the primary function of PARP-1 in DNA damage detection and repair, the use of PARP-1 inhibitors as therapy for inflammatory diseases requires caution due to the risk of genomic instability.
Acknowledgements
The author(s) would like to thank Mrs Nadège Lagarde for excellent technical assistance.
Contributor Information
Maria Meira, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Claudia Sievers, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Francine Hoffmann, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Heidi Bodmer, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Tobias Derfuss, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Jens Kuhle, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Aiden Haghikia, Department of Neurology, Ruhr-University Bochum, Germany.
Ludwig Kappos, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Raija LP Lindberg, Departments of Biomedicine and Neurology, University Hospital Basel, Switzerland.
Conflict of Interests
The author(s) have no conflicts of interest to declare.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Swiss National Science Foundation (320030_160168).
ORCID iDs
Maria Meira https://orcid.org/0000-0001-8695-1226
Tobias Derfuss https://orcid.org/0000-0001-8431-8769
Ludwig Kappos https://orcid.org/0000-0003-4175-5509
References
- 1.Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med 2018; 378: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kappos L, O’Connor PW, Polman CH, et al. Clinical effects of natalizumab on multiple sclerosis appear early in treatment course. J Neurol 2013; 260: 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mills EA, Mao-Draayer Y. Understanding progressive multifocal leukoencephalopathy risk in multiple sclerosis patients treated with immunomodulatory therapies: a bird's eye view. Front Immunol 2018; 9: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger JR. Classifying PML risk with disease modifying therapies. Mult Scler Relat Disord 2017; 12: 59–63. [DOI] [PubMed] [Google Scholar]
- 5.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 2017; 31: 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bock FJ, Todorova TT, Chang P. RNA regulation by poly(ADP-ribose) polymerases. Mol Cell 2015; 58: 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosado MM, Bennici E, Novelli F, et al. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013; 139: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiarugi A. Inhibitors of poly(ADP-ribose) polymerase-1 suppress transcriptional activation in lymphocytes and ameliorate autoimmune encephalomyelitis in rats. Br J Pharmacol 2002; 137: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott GS, Kean RB, Mikheeva T, et al. The therapeutic effects of PJ34 [N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide.HCl], a selective inhibitor of poly(ADP-ribose) polymerase, in experimental allergic encephalomyelitis are associated with immunomodulation. J Pharmacol Exp Ther 2004; 310: 1053–1061. [DOI] [PubMed] [Google Scholar]
- 10.Farez MF, Quintana FJ, Gandhi R, et al. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol 2009; 10: 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavone L, Aldinucci A, Ballerini C, et al. PARP-1 inhibition prevents CNS migration of dendritic cells during EAE, suppressing the encephalitogenic response and relapse severity. Mult Scler 2011; 17: 794–807. [DOI] [PubMed] [Google Scholar]
- 12.Cavone L, Chiarugi A. Targeting poly(ADP-ribose) polymerase-1 as a promising approach for immunomodulation in multiple sclerosis? Trends Mol Med 2012; 18: 92–100. [DOI] [PubMed] [Google Scholar]
- 13.Nukuzuma S, Kameoka M, Sugiura S, et al. Suppressive effect of PARP-1 inhibitor on JC virus replication in vitro. J Med Virol 2013; 85: 132–137. [DOI] [PubMed] [Google Scholar]
- 14.Meira M, Sievers C, Hoffmann F, et al. Natalizumab-induced POU2AF1/Spi-B upregulation: a possible route for PML development. Neurol Neuroimmunol Neuroinflamm 2016; 3: e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P, Nakatsukasa H, Tu E, et al. PARP-1 regulates expression of TGF-beta receptors in T cells. Blood 2013; 122: 2224–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambrose HE, Papadopoulou V, Beswick RW, et al. Poly-(ADP-ribose) polymerase-1 (Parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene 2007; 26: 6244–6252. [DOI] [PubMed] [Google Scholar]
- 17.Sawant DV, Sehra S, Nguyen ET, et al. Bcl6 controls the Th2 inflammatory activity of regulatory T cells by repressing Gata3 function. J Immunol 2012; 189: 4759–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sriram CS, Jangra A, Madhana RM, et al. Multiple facets of poly(ADP-ribose) polymerase-1 in neurological diseases. Neural Regen Res 2015; 10: 49–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veto S, Acs P, Bauer J, et al. Inhibiting poly(ADP-ribose) polymerase: a potential therapy against oligodendrocyte death. Brain 2010; 133: 822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meira M, Sievers C, Hoffmann F, et al. Unraveling natalizumab effects on deregulated miR-17 expression in CD4+ T cells of patients with relapsing-remitting multiple sclerosis. J Immunol Res 2014. ; 2014: 897249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006; 25: 455–471. [DOI] [PubMed] [Google Scholar]
- 22.Viglietta V, Baecher-Allan C, Weiner HL, et al. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004; 199: 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee PW, Severin ME, Lovett-Racke AE. TGF-beta regulation of encephalitogenic and regulatory T cells in multiple sclerosis. Eur J Immunol 2017; 47: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasta F, Laudisi F, Sambucci M, et al. Increased Foxp3+ regulatory T cells in poly(ADP-ribose) polymerase-1 deficiency. J Immunol 2010; 184: 3470–3477. [DOI] [PubMed] [Google Scholar]
- 25.Luo X, Nie J, Wang S, et al. Poly(ADP-ribosyl)ation of FOXP3 protein mediated by PARP-1 protein regulates the function of regulatory T cells. J Biol Chem 2015; 290: 28675–28682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michel L, Touil H, Pikor NB, et al. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol 2015; 6: 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Probstel AK, Sanderson NS, Derfuss T. B cells and autoantibodies in multiple sclerosis. Int J Mol Sci 2015; 16: 16576–16592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambrose HE, Willimott S, Beswick RW, et al. Poly(ADP-ribose) polymerase-1 (Parp-1)-deficient mice demonstrate abnormal antibody responses. Immunology 2009; 127: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 2000; 13: 443–451. [DOI] [PubMed] [Google Scholar]
- 30.Durali D, de Goer de Herve MG, Gasnault J, et al. B cells and progressive multifocal leukoencephalopathy: search for the missing link. Front Immunol 2015; 6: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev 2012; 247: 172–183. [DOI] [PubMed] [Google Scholar]
- 32.Duy C, Yu JJ, Nahar R, et al. BCL6 is critical for the development of a diverse primary B cell repertoire. J Exp Med 2010; 207: 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichii H, Sakamoto A, Hatano M, et al. Role for Bcl-6 in the generation and maintenance of memory CD8+ T cells. Nat Immunol 2002; 3: 558–563. [DOI] [PubMed] [Google Scholar]
- 34.Pender MP, Csurhes PA, Pfluger CM, et al. Deficiency of CD8+ effector memory T cells is an early and persistent feature of multiple sclerosis. Mult Scler 2014; 20: 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida K, Sakamoto A, Yamashita K, et al. Bcl6 controls granzyme B expression in effector CD8+ T cells. Eur J Immunol 2006; 36: 3146–3156. [DOI] [PubMed] [Google Scholar]