

Abstract
Yeast enolase serves as a prototype for metalloenzymes with labile, catalytic active site metal ions and is important for glycolysis and fermentation processes. Herein, microsecond molecular dynamics simulations of the protein-substrate and protein-product complexes are conducted to elucidate the mechanism of the opening of catalytically important active site loops. These simulations indicate that conversion of substrate to product is accompanied by diminished metal coordination and hydrogen-bonding interactions, as well as enhanced loop flexibility. Moreover, free energy simulations show that the loop opening is endergonic when substrate is bound but exergonic when product is bound. Thus, the conversion to product weakens the association of the loop with the ligand and binding site, thereby facilitating the loop opening after catalysis and enabling product release. These insights about active site loop motions in enzyme catalysis may be useful in guiding enzyme design efforts.
Keywords: metalloenzymes, enolase, catalysis, loop motions, hydrogen bond
Graphical Abstract
Metalloenzymes play significant and diverse roles in a wide range of biochemical processes.1–2 Yeast enolase serves as a prototype for metalloenzymes with labile metal ions that play a key role in the catalytic turnover,3 as indicated by the proposed catalytic cycle shown in Figure 1A.3 Enolases catalyze the reversible conversion of 2-phosphoglyceric acid (2PG) to phosphoenolpyruvic acid (PEP) in glycolysis (Figure 1C).4 The active site of yeast enolase contains two Mg2+ ions, which are denoted MgA2+ and MgB2+ in the present work. In the catalytic cycle, MgA2+ binds to the active site first, followed by the binding of substrate and then the binding of MgB2+. After substrate to product conversion, MgB2+ is released, followed by product release.
Figure 1.

Schematic depictions of (A) proposed catalytic cycle, (B) comparison between the closed and open loop states, and (C) proposed catalytic mechanism for yeast enolase. For part B, the open loop structure (green) was obtained from the PDB entry 1EBH, which corresponds to the E-MgA2+ state, and the closed loop structure (orange) was obtained from the PDB entry 1ONE, which corresponds to the E-MgA2+-S/P-MgB2+ state. The structures were superimposed by aligning the backbone Cα atoms in the protein dimer. Only chain A is shown for illustrative purposes.
Active site loops are common motifs in enzymes5 and can play significant roles, such as preventing side reactions and stabilizing reaction intermediates.6–9 Three loops, which correspond to Ser36-His43, Val153-Phe169, and Asp255-Asn266 (herein denoted L1, L2, and L3, respectively, as shown in Figure 1B), play significant roles in the catalytic cycle of yeast enolase.10–11 L1 binds to MgB2+ through the backbone and side chain oxygen atoms of Ser39 and also binds to the substrate through hydrogen-bonding interactions.12 L2 binds to the substrate through the hydrogen bond between His159 and the substrate phosphate group.13 L3 does not interact with the active site directly but is hydrogen bonded to L2. Crystallographic data indicate that these loops can adopt different conformations when the enzyme is in different states.14–16 For example, the three loops are open in the E-MgA2+ state14 but closed in the E-MgA2+-S-MgB2+ state,15 and they have been proposed to reopen for the release of MgB2+ and product (Figure 1).3 Crystallographic data also indicate differences in loop conformations when the enzyme is bound to 2PG versus PEP.16 Several computational studies of yeast enolase or homologous enzymes have been conducted in efforts to understand the structure and catalytic mechanism.17–23 Mixed quantum mechanical/molecular mechanical (QM/MM) calculations have been performed to study the dehydration reaction of yeast enolase.17–18 Molecular dynamics (MD) simulations have been performed to investigate the structural flexibility and protein interactions.19–21, 23
Despite the substantial number of experimental and theoretical studies, the mechanism of the loop closing and opening in enolase is still not fully understood. Herein, microsecond MD trajectories of the protein-2PG and protein-PEP systems are propagated to obtain a deeper understanding of this mechanism and its relation to catalysis. In particular, the impact of substrate conversion to product on the metal coordination, hydrogen-bonding interactions, and loop flexibility are examined. Moreover, the free energy surfaces associated with the loop closing and opening processes are also investigated by microsecond umbrella sampling simulations. This study provides fundamental insights about the potential roles of active site loops in enzymes, and the resulting knowledge may be useful in guiding enzyme design efforts.
The initial coordinates of the protein-2PG and protein-PEP systems were obtained from the PDB entry 1ONE, which provides coordinates for the E-MgA2+-S-MgB2+ and E-MgA2+-P-MgB2+ states.15 The ff14SB force field24 was used to model the protein system, and the TIP3P water model was employed to represent the solvent.25 Na+ and Cl− counterions were added to the system to provide a salt concentration of ~0.15 M. For each of the two systems, a 1.02 μs production MD trajectory was propagated with configurations saved every 10 ps. The pmemd.cuda program26 in the AMBER software package27 was used for these MD simulations. Additional details of the system preparation, force field parameters, and MD simulations are provided in the Supporting Information (SI).
Because of the compact packing and ordered hydrogen-bond network in the active site of yeast enolase, small perturbations of the ligand may significantly impact its structure and motions. The active site contains many charged residues (Figure S1 and Table S2), and a variety of hydrogen bonds can be formed between the enzyme and the substrate, especially between the enzyme and the phosphate group of the substrate (Figure S2 and Table S3). Similar patterns are observed in enzymes such as triosephosphate isomerase and glycerol 3-phosphate dehydrogenase, which have a “phosphate gripper” loop to anchor the phosphate group of the substrate.7 In addition, the OA atom of the substrate can potentially form hydrogen bonds with both L1 and L2 (Figure S2 and Table S3), suggesting its significant role in the loop closing and opening processes, as illustrated below.
The substrate to product conversion for yeast enolase is proposed to involve a dehydration reaction concurrent with protonation of Lys345 and deprotonation of Glu211 (Figure 1C).4 The appropriate protonation states of these two residues shown in Figure 1C have been used in our MD simulations of the protein-2PG and protein-PEP complexes to account for the dehydration reaction. To investigate the influence of the substrate to product conversion on the active site structure, we analyzed the MD trajectories of the protein-2PG and protein-PEP systems (i.e., the substrate-bound and product-bound systems, respectively). The substrate and product binding modes are similar for 2PG and PEP in the crystal structure and throughout the MD trajectories, with minor differences shown in Figures S3 and S4. However, we observed significant differences in the metal ion coordination and the hydrogen-bond network within the active site (Figure 2). In the protein-2PG system, the bridging carboxylate oxygen retains its coordination to both MgA2+ and MgB2+, whereas in the protein-PEP system, this oxygen atom is only coordinated to MgB2+. This coordination change is associated with a ~1 Å greater average distance between the two Mg2+ ions for the protein-PEP system compared to the protein-2PG system (Table S4). In addition, MgB2+ is coordinated to OC in the protein-2PG system and to OA in the protein-PEP system (Figure 2 and Table S4).
Figure 2.

Representative structures of the active site in (A) the protein-2PG system, and (B) the protein-PEP system. Note that Lys345 is protonated in (B) but not in (A) based on the proposed dehydration reaction mechanism.4 The hydrogen bonds between the phosphate group of 2PG or PEP and the protein, as well as between the hydroxyl group of 2PG and the protein, are depicted with dashed lines. These structures were obtained from the configurations at 360 ns in the production MD trajectories.
The hydrogen-bonding interactions between the protein and the ligand also exhibit significant differences for the protein-2PG versus the protein-PEP system (Figure 2 and Table S5). The hydroxyl group of 2PG is hydrogen bonded to the sidechains of Glu168 and Glu211, but these hydrogen bonds are not present in the protein-PEP system because of the removal of this hydroxyl group (Figure S2B and Table S5). The hydrogen bonding interaction between the ligand phosphate group and the protein residues also differs significantly for these two ligands. For example, the OC atom in PEP has an additional strong hydrogen bond with the Lys345 residue (Figure 2 and Table S5). Specifically, the average distance between Nζ of Lys345 and OC is 0.6 – 1.2 Å smaller for the protein-PEP system than for the protein-2PG system in our simulations (Table S4). This observation is consistent with the crystal structures 1ONE and 2ONE, which indicate that this distance is shorter by 0.3 and 0.7 Å, respectively, with bound PEP compared to bound 2PG.15–16 Additionally, a similar trend is observed in the crystal structure 2AKM, which corresponds to phosphate bonded to human neuronal enolase.28 In this structure, the chain with two Mg2+ ions has nearly identical hydrogen-bonding interactions between the protein and phosphate group as the protein-2PG system, while the chain with only one Mg2+ ion has a smaller distance between Nζ of Lys342 (which corresponds to Lys345 in yeast enolase) and the closest phosphate oxygen, as well as a rotated phosphate group (Figures S2 and S5). We propose that the protonation of Lys345 during the dehydration reaction4 may play a role in rotating the phosphate group, perturbing the hydrogen bonding between the protein and ligand, and inducing the loop opening in enolase.
In addition, the average number of hydrogen bonds between OA and the protein is ~3 for the protein-2PG system and less than 1 for the protein-PEP system (Table S5), illustrating the attenuated hydrogen-bonding interactions between the phosphate group and the active site loops L1 and L2 for the product compared to the substrate. For example, our microsecond MD simulations indicate a strong, persistent hydrogen bond between His159 and the phosphate group of 2PG. This hydrogen bond is not as persistent in the protein-PEP system (Table S5). This observation is consistent with the crystallographic data illustrating that a water separates His159 from the ligand when PEP is bound.20 We also observed significant flexibility of L2 in both the protein-2PG and protein-PEP systems; for each system, the L2 loop opens in one of the monomers during the microsecond MD trajectory. This observation is consistent with the diversity in the crystal structures, where the E-MgA2+-S, E-MgA2+-S-MgB2+, E-MgA2+-I-MgB2+ (I is a model for the enol intermediate), and E-MgA2+-P-MgB2+ states exhibit both open and closed L2 loops. A survey of the loop conformations in available crystal structures is provided in Table S6. Moreover, comparison of the computed partial charges for the substrate and enol intermediate indicates that the additional negative charge is delocalized onto the phosphate oxygen atoms as well as the carboxylate atoms (Table S1), enabling stronger hydrogen-bonding interactions between the ligand phosphate group and the active site loops. This observation is consistent with prior work indicating that loops L1 and L2 play a role in stabilizing the transition states along the catalytic reaction pathway by yeast enolase.18
To investigate the influence of substrate conversion to product on the flexibility of the active site loops, we calculated the root-mean-square fluctuations (RMSFs) of the Cα atoms in the three loops for both systems. The RMSFs of these Cα atoms in the protein-PEP system are approximately twice as large as those in the protein-2PG system (Figure 3), indicating that substrate conversion to product significantly enhances the flexibility of the loops. The crystal structure 2ONE also exhibits a partially disordered Ser39 and larger B factors for Cα atoms in residues 35–49, 157–163, and 245–265 when PEP is bound,16 implying greater flexibility of the relevant regions of the loops.
Figure 3.

The RMSF values of the Cα atoms in the three loops of the protein-2PG (black) and protein-PEP (red) systems. The solid and dashed lines correspond to Chains A and B, respectively.
To further study the influence of substrate conversion on the loop motions, we performed free energy simulations corresponding to the opening of loop L1 for each system. The two reaction coordinates used to represent this process are illustrated in Figure 4. The choice of the first reaction coordinate, which is the distance between MgB2+ and the Cα atom of Ser39, was straightforward. The choice of the second reaction coordinate, which is the distance between the backbone O of Ala38 and the backbone N of Glu47, was based on the observation that the open loop structure (PDB entry 1EBH,14 corresponding to the E-MgA2+ state) exhibits a hydrogen bond between the backbone O of Ala38 and the backbone N of Glu47, whereas this distance is large in the closed loop structure (PDB entry 1ONE,15 corresponding to the E-MgA2+-S/P-MgB2+ state). The free energy pathways connecting the closed and open loop conformations for the E-MgA2+-S-MgB2+ state and the E-MgA2+-P-MgB2+ state were calculated using these two reaction coordinates. To estimate the values of the two reaction coordinates for the open loop conformation, the open loop structure of the E-MgA2+ state (PDB entry 1EBH) was superimposed on the closed loop structure of the E-MgA2+-S/P-MgB2+ state (PDB entry 1ONE), as depicted in Figure 4. After performing 2.8 μs of umbrella sampling along these two reaction coordinates for each system, the potentials of mean force (also denoted the free energy surfaces) were generated by unbiasing the data using the weighted histogram analysis method (WHAM).29 Additional details of these free energy simulations are provided in the SI.
Figure 4.

Reaction coordinates in (A) the closed loop structure of the E-MgA2+-S-MgB2+ state (PDB entry 1ONE), and (B) the open loop structure of the E-MgA2+ state (PDB entry 1EBH). The reaction coordinates are represented by the dashed lines. The metal ions and loop colored orange are from the closed loop structure, while the loop colored green is from the open loop structure. The two structures were superimposed by aligning the Cα atoms of the protein backbone. The two reaction coordinates are as follows: (1) the distance between MgB2+ and the Cα atom of Ser39 and (2) the distance between the backbone O of Ala38 and the backbone N of Glu47.
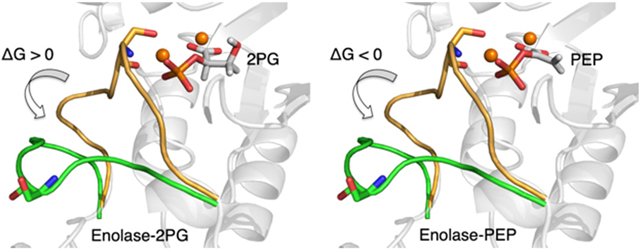
The two-dimensional free energy surfaces illustrated in Figure 5 highlight significant differences between the two systems. The loop opening process is substantially more thermodynamically favorable for the protein-PEP system than for the protein-2PG system. In particular, the opening process is endergonic, with a free energy increase of 3 kcal/mol, for the protein-2PG system and is slightly exergonic, with a free energy decrease of 5 kcal/mol, for the protein-PEP system. These results are consistent with the observation of the closed loop in the E-MgA2+-S-MgB2+ crystal structure15 and the proposal that the loop reopens for the release of MgB2+ and the product. Furthermore, Figure 5B indicates that the protein-PEP system has an intermediate state located between the closed and open loop states. This structure is similar to the crystal structure of the S39A mutant of yeast enolase (PDB entry 1L8P),11 in which loop L1 is in a semi-closed state due to diminished interaction between Ser39Oγ, which is in loop L1, and the active site (Figure S6).
Figure 5.

Two-dimensional free energy profiles for the loop closing and opening processes in (A) the protein-2PG system and (B) the protein-PEP system. For illustrative purposes, only free energies within 20 kcal/mol of the lowest free energy are shown. The two reaction coordinates are illustrated in Figure 4.
To better understand the difference in the free energy change associated with the loop opening process for the protein-2PG and protein-PEP systems, we performed hydrogen-bonding analyses for loop L1 and the ligands (Table S7). This analysis indicates that the average number of hydrogen-bonding interactions involving loop L1 is the same for both ligands, and in both cases loop L1 forms ~2 more hydrogen bonds in the open loop conformation than in the closed loop conformation (Table S7), suggesting that these interactions are not responsible for the free energy differences. However, the average number of hydrogen-bonding interactions involving the ligand is greater for the protein-2PG system than for the protein-PEP system, mainly due to the hydroxyl group on 2PG, although the number of hydrogen-bonding interactions remains the same for the open and closed loop states in both cases. Interestingly, the hydroxyl group in 2PG hydrogen bonds to Glu168 and Glu211 in the closed loop conformation but exhibits an internal hydrogen bond to the ligand phosphate group that apparently prevents hydrogen bonding to Glu168 in the open loop conformation (Table S8 and Figure S7). This internal hydrogen bond, which only forms for 2PG in the open loop conformation and prevents other stabilizing hydrogen-bonding interactions, may be one of the reasons that the loop opening process is not thermodynamically favorable in the protein-2PG system but is thermodynamically favorable in the protein-PEP system.
In summary, our MD simulations indicate that the substrate to product conversion in yeast enolase alters the metal coordination in the active site and perturbs the hydrogen-bonding interactions between the protein and the ligand, enhancing the thermal fluctuations and flexibility of the active site loops in the product state. According to our free energy simulations, the loop opening is significantly more thermodynamically favorable for the product state, thereby facilitating the loop opening after catalysis, subsequently also favoring product release. This increased understanding of the catalytic mechanism of yeast enolase may be used to mutate this enzyme to alter its catalytic activity or specificity. To influence the loop opening, our analysis suggests that mutating Gly37 to a larger residue may facilitate the opening of L1 through steric effects, whereas mutating Ala38 to Ser may inhibit the opening of L1 through enhanced hydrogen-bonding interaction with the substrate phosphate group. In addition to playing an essential role in glycolysis, gluconeogenesis, and fermentation processes, enolase is also a promising therapeutic target30 and a potential biomarker of tumors.31–32 Hence, these insights could also be useful for drug design efforts. More generally, highlighting the significant role of active site loop motions in enzyme catalysis may serve as a guide for enzyme design in a wide range of applications.
Supplementary Material
Acknowledgments
We thank Feng Pan for helping to choose the reaction coordinates. We appreciate the helpful suggestions from Clorice Reinhardt and the reviewers. We are grateful for the financial support from the National Institute of Health (grant number GM056207). We acknowledge computational support from the Extreme Science and Engineering Discovery Environment (XSEDE),33 which is supported by the National Science Foundation (grant number ACI-1548562). Specifically, this work utilized the GPU resources of Comet at the San Diego Supercomputer Center (allocation number TG-MCB120097).
Funding Sources
National Institutes of Health Grant GM056207
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Details of the system preparation, molecular dynamics simulations, and umbrella sampling; force field parameters; supporting figures and tables (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Thomson AJ; Gray HB, Bio-Inorganic Chemistry. Curr. Opin. Chem. Biol 1998, 2, 155–158. [DOI] [PubMed] [Google Scholar]
- 2.Andreini C; Bertini I; Cavallaro G; Holliday G; Thornton J, Metal Ions in Biological Catalysis: From Enzyme Databases to General Principles. J. Biol. Inorg. Chem 2008, 13, 1205–1218. [DOI] [PubMed] [Google Scholar]
- 3.Weston JF, Designing Models for Metalloenzymes In Advances in Mathematical Chemistry and Applications, Elsevier: Netherlands; United Kingdom; MA, USA, 2015; pp 243–264. [Google Scholar]
- 4.Reed GH; Poyner RR; Larsen TM; Wedekind JE; Rayment I, Structural and Mechanistic Studies of Enolase. Curr. Opin. Struct. Biol 1996, 6, 736–743. [DOI] [PubMed] [Google Scholar]
- 5.Brändén C-I; Tooze J, Introduction to Protein Structure, Second Edition Taylor & Francis: New York, NY, 1999. [Google Scholar]
- 6.Pompliano DL; Peyman A; Knowles JR, Stabilization of a Reaction Intermediate as a Catalytic Device: Definition of the Functional Role of the Flexible Loop in Triosephosphate Isomerase. Biochemistry 1990, 29, 3186–3194. [DOI] [PubMed] [Google Scholar]
- 7.Malabanan MM; Amyes TL; Richard JP, A Role for Flexible Loops in Enzyme Catalysis. Curr. Opin. Struct. Biol 2010, 20, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knowles JR, Enzyme Catalysis: Not Different, Just Better. Nature 1991, 350, 121–124. [DOI] [PubMed] [Google Scholar]
- 9.Fan Y; Cembran A; Ma S; Gao J, Connecting Protein Conformational Dynamics with Catalytic Function as Illustrated in Dihydrofolate Reductase. Biochemistry 2013, 52, 2036–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brewer JM; Glover CV; Holland MJ; Lebioda L, Significance of the Enzymatic Properties of Yeast S39a Enolase to the Catalytic Mechanism. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1998, 1383, 351–355. [DOI] [PubMed] [Google Scholar]
- 11.Poyner RR; Larsen TM; Wong S-W; Reed GH, Functional and Structural Changes Due to a Serine to Alanine Mutation in the Active-Site Flap of Enolase. Arch. Biochem. Biophys 2002, 401, 155–163. [DOI] [PubMed] [Google Scholar]
- 12.Wedekind JE; Poyner RR; Reed GH; Rayment I, Chelation of Serine 39 to Mg2+ Latches a Gate at the Active Site of Enolase: Structure of the Bis (Mg2+) Complex of Yeast Enolase and the Intermediate Analog Phosphonoacetohydroxamate at 2.1-. Ang. Resolution. Biochemistry 1994, 33, 9333–9342. [DOI] [PubMed] [Google Scholar]
- 13.Brewer JM; Glover CV; Holland MJ; Lebioda L, Enzymatic Function of Loop Movement in Enolase: Preparation and Some Properties of H159n, H159a, H159f, and N207a Enolases. J. Protein Chem 2003, 22, 353–361. [DOI] [PubMed] [Google Scholar]
- 14.Wedekind JE; Reed GH; Rayment I, Octahedral Coordination at the High-Affinity Metal Site in Enolase: Crystallographic Analysis of the Mgii-Enzyme Complex from Yeast at 1.9. Ang. Resolution. Biochemistry 1995, 34, 4325–4330. [DOI] [PubMed] [Google Scholar]
- 15.Larsen TM; Wedekind JE; Rayment I; Reed GH, A Carboxylate Oxygen of the Substrate Bridges the Magnesium Ions at the Active Site of Enolase: Structure of the Yeast Enzyme Complexed with the Equilibrium Mixture of 2-Phosphoglycerate and Phosphoenolpyruvate at 1.8 Å Resolution. Biochemistry 1996, 35, 4349–4358. [DOI] [PubMed] [Google Scholar]
- 16.Zhang E; Brewer JM; Minor W; Carreira LA; Lebioda L, Mechanism of Enolase: The Crystal Structure of Asymmetric Dimer Enolase– 2-Phospho-D-Glycerate/Enolase– Phosphoenolpyruvate at 2.0 Å Resolution. Biochemistry 1997, 36, 12526–12534. [DOI] [PubMed] [Google Scholar]
- 17.Alhambra C; Gao J; Corchado JC; Villa J; Truhlar DG, Quantum Mechanical Dynamical Effects in an Enzyme-Catalyzed Proton Transfer Reaction. J. Am. Chem. Soc 1999, 121, 2253–2258. [Google Scholar]
- 18.Liu H; Zhang Y; Yang W, How Is the Active Site of Enolase Organized to Catalyze Two Different Reaction Steps? J. Am. Chem. Soc 2000, 122, 6560–6570. [Google Scholar]
- 19.Hakobyan D; Nazaryan K, Investigation of Interaction between Enolase and Phosphoglycerate Mutase Using Molecular Dynamics Simulation. J. Biomol. Struct. Dyn 2006, 23, 625–633. [DOI] [PubMed] [Google Scholar]
- 20.Marcos V; Dias SMG; Mello LV; da Silva Giotto MT; Gavalda S; Blonski C; Garratt RC; Rigden DJ, Structural Flexibility in Trypanosoma Brucei Enolase Revealed by X-Ray Crystallography and Molecular Dynamics. FEBS J. 2007, 274, 5077–5089. [DOI] [PubMed] [Google Scholar]
- 21.Hakobyan D; Nazaryan K, Molecular Dynamics Study of Interaction and Substrate Channeling between Neuron-Specific Enolase and B-Type Phosphoglycerate Mutase. Proteins 2010, 78, 1691–1704. [DOI] [PubMed] [Google Scholar]
- 22.Mutlu O; Yakarsonmez S; Sariyer E; Danis O; Yuce-Dursun B; Topuzogullari M; Akbulut E; Turgut-Balik D, Comprehensive Structural Analysis of the Open and Closed Conformations of Theileria Annulata Enolase by Molecular Modelling and Docking. Comput. Biol. Chem 2016, 64, 134–144. [DOI] [PubMed] [Google Scholar]
- 23.Sariyer E; Yakarsonmez S; Danis O; Turgut-Balik D, A Study of Bos Taurus Muscle Specific Enolase; Biochemical Characterization, Homology Modelling and Investigation of Molecular Interaction Using Molecular Docking and Dynamics Simulations. Int. J. Biol. Macromol 2018, 120, 2346–2353. [DOI] [PubMed] [Google Scholar]
- 24.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, Ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99sb. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 26.Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC, Routine Microsecond Molecular Dynamics Simulations with Amber on Gpus. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- 27.Case DA; Cheatham TE; Darden T; Gohlke H; Luo R; Merz KM Jr; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber Biomolecular Simulation Programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin J; Chai G; Brewer JM; Lovelace LL; Lebioda L, Fluoride Inhibition of Enolase: Crystal Structure and Thermodynamics. Biochemistry 2006, 45, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grossfield A, “Wham: The Weighted Histogram Analysis Method”, Version 2.0.9.1, Http://Membrane.Urmc.Rochester.Edu/Wordpress/?Page_Id=126 (Accessed Dec 20, 2017).
- 30.Avilán L; Gualdrón-López M; Quiñones W; González-González L; Hannaert V; Michels PA; Concepción J-L, Enolase: A Key Player in the Metabolism and a Probable Virulence Factor of Trypanosomatid Parasites—Perspectives for Its Use as a Therapeutic Target. Enzyme Res. 2011, 2011, Article ID 932549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isgrò MA; Bottoni P; Scatena R, Neuron-Specific Enolase as a Biomarker: Biochemical and Clinical Aspects In Advances in Cancer Biomarkers, Springer: Dordrecht, 2015; pp 125–143. [DOI] [PubMed] [Google Scholar]
- 32.Vizin T; Kos J, Gamma-Enolase: A Well-Known Tumour Marker, with a Less-Known Role in Cancer. Radiol. Oncol 2015, 49, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD, Xsede: Accelerating Scientific Discovery. Comput. Sci. Eng 2014, 16, 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.