

Abstract
The type-1 parathyroid hormone receptor (PTHR1), which regulates calcium homeostasis and tissue development, has two native agonists, parathyroid hormone (PTH) and PTH-related protein (PTHrP). PTH forms a complex with the PTHR1 that is rapidly internalized and induces prolonged cAMP production from endosomes. In contrast, PTHrP induces only transient cAMP production, which primarily arises from receptors on the cell surface. We show that backbone modification of PTH(1-34)-NH2 and abaloparatide (a PTHrP derivative) with a single homologous β-amino acid residue can generate biased agonists inducing prolonged cAMP production from receptors at the cell surface. This unique spatio-temporal profile could be useful for distinguishing effects associated with the duration of cAMP production from effects associated with the site of cAMP production.
Graphical Abstract
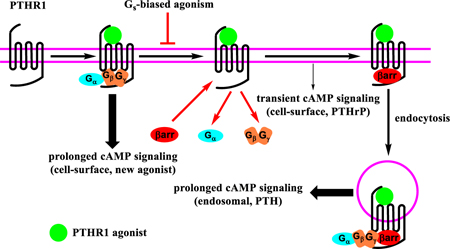
Information transfer at the molecular level is critical in living systems, and many natural messages are encoded in amino acid sequences. Evolutionary modulation of these messages is focused on side chains of proteins and peptides. Chemical synthesis enables an expansion in polypeptide language via backbone modification, which can be implemented without changing side chains. We have begun to explore this approach in the context of polypeptide hormones that activate B family GPCRs, many of which are targets of therapeutic interest.1–4 Previously, β-amino acids have been used to modulate proteolytic stability, receptor-subtype selectivity, and signaling profiles of peptidic GPCR agonists.5–9 The present study introduces a new capability of α→β substitution, modulation of the spatiotemporal characteristics of signal transduction.
Agonist binding to a GPCR induces conformational changes that are sensed by cytosolic binding partners. Different intracellular partners, such as Gs or β-arrestins, are predicted to engage distinct receptor conformations.10 Biased agonism11 is observed when a ligand favors a receptor conformation attractive to one partner relative to others. Our group previously discovered that replacing multiple α amino acid residues of glucagon-like peptide-1 (GLP-1) with cyclic β amino acid residues generated biased agonists favoring receptor interaction with β-arrestins relative to Gs. However, these α/β peptides were much less potent agonists than the original α peptide (GLP-1).9 Here we show that minimal backbone alteration enabled by the use of homologous β residues can generate potent agonists with high signaling selectivity at the PTHR1 and an altered spatiotemporal signaling profile relative to α-peptide prototypes.
The PTHR1 is an important target for the treatment of osteoporosis and hypoparathyroidism. Activation of the PTHR1 by its two native ligands, PTH and PTHrP, initiates Gs/cAMP/PKA signaling and β arrestin recruitment.12, 13 PTH and PTHrP differ, however, in their spatio-temporal signaling profiles.14 PTHrP causes transient cAMP production from receptors on the cell surface, while PTH triggers prolonged cAMP production after the receptor-agonist complex has translocated to endosomes.15–17
Signaling duration is an important determinant of the physiological effects exerted by PTHR1 agonists,18 but the subcellular location of GPCR signaling can also influence physiological outcome.19 Therefore, molecular tools that enable dissection of the spatial and temporal dimensions of PTHR1 signaling would be useful for elucidating the physiological roles of this receptor.20, 21 We asked whether backbone modification could provide a PTHR1 agonist with a unique spatio-temporal signaling profile relative to natural hormones, specifically, an agonist that would induce prolonged receptor activation that occurs only at the cell surface.
Because binding of β-arrestin to the PTHR1 is necessary for receptor internalization and attenuates cell-surface Gs/cAMP signaling,22 PTHR1 engaged by a Gs-biased agonist might persist at the cell surface and cause prolonged cAMP production from that location, if the agonist had sufficient affinity. Previous approaches to Gs-biased agonists of the PTHR1 have involved amino acid side chain modification and achieved only limited success.13, 23, 24 We pursued α→β backbone modification5, 8, 25, 26 with PTH(1-34) and in PTHrP analogue abaloparatide (ABL), both of which are used to treat osteoporosis.27, 28
The N-terminal region of peptidic PTHR1 agonists inserts deeply into the transmembrane domain (PTHR1-TMD) and causes conformational changes to which transducers respond.29 The PTHR1 conformation required for Gs activation appears to differ from that needed for recruitment of β-arrestins.30 We hypothesized that backbone modification in the N-terminal region of PTH(1-34) might induce PTHR1 conformations that differentially affect Gs activation and β-arrestin recruitment. Our search for Gs-biased agonists based on PTH(1-34)-NH2 began with a set of previously described analogues generated by replacing each of the first eight residues, individually, with the (S*)-β3, the (S*)-β2 or the (R*)-β2 homologue (S* and R* defined in SI).8 Several of these backbone-modified analogues (Figure S1) were indistinguishable from PTH(1-34)-NH2 in stimulating cAMP production, a marker of Gs activation, as evaluated in HEK-293 cells stably expressing human PTHR1 and the GloSensor for cAMP detection (Table S1). In new studies, we evaluated these analogues for their ability to recruit β-arrestins to the PTHR1 via bioluminescence resonance energy transfer (BRET) measurements using CHO-FlpIn cells stably transfected with PTHR1-Rluc8 and β-arrestin-1-Venus or β-arrestin-2-Venus (Table S1). Considerable variation in recruitment of β-arrestins to the PTHR1 was observed. Incorporating the (S*)-β2 homologue at position 5 or the (R*)-β2 homologue at position 7 or 8 (peptides 1-3) caused the most significant BRET reductions (Figure 2a and S6a).
Figure 2.

Activity profiles of PTH and α/β Gs-biased PTHR1 agonists. a.b. Concentration-response effects of PTH and peptides 1-6 for cAMP production and β-arrestin-1 recruitment (β-arrestin-2 data in SI). Data represent mean ± s.e.m from n ≥ 6 independent experiments. c. Calculated bias factors of 1-6 relative to PTH(1-34)-NH2. *, #Statistically significant difference from PTH using one-way ANOVA (* P < 0.0001; # P < 0.001).
With PTH(1-34)-NH2 as the reference agonist, we quantified the signaling bias of 1-3 based on the Black-Leff operational model (Figure 2c), in which ΔΔlog(τ/KA) denotes the bias factor.31, 32 This quantification method minimizes the dependence of bias factor on cell type.31 Both 2 and 3 were ~10-fold biased toward the Gs-mediated signaling pathway vs. β-arrestin recruitment, relative to PTH(1-34)-NH2. Peptide 1 displayed a more moderate bias. We found that previously described peptides containing a single side-chain modification,24 Tyr1-PTH(1-34)-NH2 and Trp1-PTHrP(1-36)-NH2, are defective for β-arrestin recruitment, as reported; however, both were ~5-fold less potent than PTH(1-34)-NH2 in stimulating cAMP production (Figure S2). Neither of these side chain-modified peptides displayed the level of signal bias observed for backbone-modified peptides 1-3.
ABL shows a significant bias toward cAMP production relative to PTH(1-34)-NH2 (Figure S3). Despite this bias, ABL is relatively effective at recruiting β-arrestins to the PTHR1 (Figure S4), which motivated us to use ABL to evaluate the generality of the α→β modification strategy. Initial modifications of ABL involved positions 7 and 8, based on observations with PTH(1-34)-NH2 analogues. However, most of these modifications caused substantial declines in stimulation of cAMP production (Figure S5). The differential tolerance of β homologue incorporation at positions 7 and 8 suggests that the PTHR1-TMD-binding modes of PTH(1-34)-NH2 and ABL are different, which is consistent with the discovery that the N-terminal regions of PTH(1-34) and PTHrP(1-34) are not functionally equivalent for receptor binding.33 More interesting results emerged from α→β substitutions at positions 1 and 2 of ABL. Peptides 4-6 were very effective at stimulating cAMP production but significantly less effective than ABL or PTH(1-34)-NH2 at recruiting β-arrestins (Figures 2b, S6b and S7).
The signaling duration of Gs-biased agonists 1-6 was evaluated with “washout” assays in which cells were stimulated with an agonist for a defined period, and then the agonist was washed away, and cAMP production was monitored (“ligand-off phase”).34, 35 For 2, 3 and 5, cAMP production was more transient than for PTH(1-34)-NH2 (Figure S8). In contrast, 1 induced more prolonged signaling relative to PTH(1-34)-NH2 (Figure 3a). While ABL was more short-acting than PTH(1-34)-NH2 at their equiactive concentrations,36 both 4 and 6 performed similarly to PTH(1-34)-NH2 in this assay at the tested concentration (Figures 3b and S9).
Figure 3.

Effect of competitive antagonist and BA1 on signal duration of PTHR1 agonists. a.b. Post-washout cAMP production mediated by 1 nM PTH, 1, 4 or 6 in HEK293 cells stably expressing hPTHR1. c. Washout assay data from part a and b presented as area under curve (AUC); data for measurements with BA1 are included. Data represent mean ± s.e.m of n = 4 (PTH(1-34)-NH2 and 1), n = 3 (4 and 6) independent experiments. All the ligand activities are normalized to that of PTH(1-34)-NH2 at 1 nM. Statistically significant difference using one-way ANOVA (**** P < 0.0001, *** P < 0.001, ** P < 0.01).
To evaluate the location of the cAMP production stimulated by 1, 4 or 6, we introduced an excess of PTHR1 antagonist (D)Trp12,Tyr34-bPTH(7-34) during the washout assay. We hypothesized that this peptidic antagonist should compete with agonist molecules for PTHR1 remaining on the cell surface, but the antagonist should not reach internalized agonist-receptor complexes.22, 37, 38 For 1, 4, and 6, the presence of antagonist after washout caused a profound drop in the duration and extent of cAMP production (Figures 3 and S10). In contrast, only a modest drop was observed after washout with antagonist for PTH(1-34)-NH2, which is known to continue stimulating cAMP production from endosomes.15 These observations support the conclusion that most cAMP production induced by 1, 4 or 6 involves PTHR1 at the cell surface.
Both the PTHR1 and β-arrestin are overexpressed in the cells used for the BRET assay, while the washout assay was performed in HEK293 cells expressing β-arrestin at endogenous levels. We therefore evaluated β-arrestin recruitment induced by 1, 4 or 6 via co-immunoprecipitation (co-IP) of endogenous β-arrestin and the PTHR1 in HEK293 cells stably expressing hemagglutinin (HA)-tagged PTHR1.13 As shown in Figures 4 and S11, PTH(1-34)-NH2 effectively induced co-IP of the PTHR1 and β-arrestin, while agonists 1, 4 and 6 induced very little co-IP. Peptide 6 was further studied via confocal microscopy in HEK293 cells expressing SNAP-tagged PTHR1 and GFP-tagged β and β-arrestin, 6 did not induce observ-arrestin-2. While PTH(1-34)-NH2 effectively triggered the co-localization of the PTHR1 able β-arrestin recruitment to the PTHR1 (Figure S12). These observations support the conclusion that biased agonists 1, 4 and 6 are relatively ineffective at inducing β-arrestin recruitment to the PTHR1, which causes receptors activated by these peptides to remain at the cell surface.
Figure 4.

Recruitment of endogenous β-arrestin to PTHR1 detected by co-immunoprecipitation. HEK293 cells stably expressing HA-tagged PTHR1 were challenged with PTH(1-34)-NH2 and peptides 1-6. The left and right panels of b are from the same blot with unrelated data omitted from the center
Blocking endosomal acidification was shown to prolong cAMP production of PTH.17 We therefore conducted additional washout experiments with bafilomycin A1 (BA1), an inhibitor of endosomal acidification,39 to gain further insight on the signaling profiles of 1, 4 and 6. After agonist washout, cells were incubated in a solution containing both BA1 and the antagonist. Inclusion of BA1 after washout had very little effect on the extent of post-washout cAMP production induced by 4 or 6 (Figures 3c and S13a), but BA1 caused a substantial increase in post-washout cAMP production induced by 1, relative to the level observed with the antagonist alone. This observation suggests that the single α➞β2 replacement at position 5 of PTH(1-34)-NH2 (to generate 1) exerts dual effects on the agonist profile by diminishing β-arrestin recruitment induced by 1 and rendering 1 susceptible to acidification-induced dissociation from the receptor within endosomes. These two effects could be synergistic in restricting the cAMP production induced by 1 to receptors on the cell surface in the absence of BA1, although the endosomal dissociation effect is presumably minor. When tested at the same ligand concentration, ABL also displayed more sustained post-washout cAMP production in the presence of BA1 (Figure S13b), suggesting that endosomal acidification induces ABL dissociation from the receptor. In contrast to the agonist activity of 1 or ABL, the spatial restriction of cAMP production induced by 4 or 6 seems to arise predominantly from their inability to induce receptor internalization.
We have shown that introducing a single extra CH2 unit into the backbone of a long polypeptide agonist can substantially change the signaling profile that results from engagement of a class B GPCR. Moreover, altering the position of the extra CH2 along the backbone can lead to different signaling outcomes. (In the SI, we interpret the behavior of our biased agonists in terms of recent advances in GPCR structural analysis.) Because this backbone-focused approach maintains the native side chain sequence, and because the backbone-modified peptides can be prepared via conventional solid-phase synthesis, designing and generating a “test set” of β-containing analogues based on a known polypeptide agonist is straightforward (in contrast to exploring a potentially infinite set of analogues containing unnatural side chains40). While displaying high potency and efficacy in stimulating cAMP production via activation of the PTHR1, β-residue-containing peptides 1, 4 and 6 are highly defective for β-arrestin recruitment. When compared to the α-peptide prototypes at equiactive concentrations, these three α/β peptides induce longer duration of cAMP production that emanates predominantly from receptors at the cell surface. There is a long-standing belief that sustained cAMP production from PTHR1 stimulation will lead to RANKL activation and bone resorption.[NEED REF!] However, it is unclear at present whether the spatial aspect of cAMP production contributes to the RANKL activation. The highly Gs-biased agonists 4 and 6 could be useful tools to explore questions in this vein.
Supplementary Material
Figure 1.

α/β PTHR1 agonists displaying significant Gs-biased agonism relative to PTH(1-34)-NH2. See the supporting information for the definition of R* and S*. nL denotes norleucine. U denotes 2-aminoisobutyric acid.
ACKNOWLEDGMENT
This research was supported in part by NIH Grant R01 GM056414 (to S.H.G), R01 DK111427 (to J.P.V.), R01 DK116780 (to J.P.V.), T32-GM008424 (to A.D.W.), P01 DK11794 (to T.J.G.), and P30 AR066261 (to T.J.G)
Footnotes
Supporting Information
Protocols for peptide synthesis, functional cell assays, co-immunoprecipitation, and confocal microscopy, method for the quantification of bias factors, and analytical data for all the peptides.
S.H.G is a cofounder of Longevity Biotech, Inc., which is pursuing biomedical applications of α/β-peptides
REFERENCES
- 1.Broichhagen J; Podewin T; Meyer-Berg H; von Ohlen Y; Johnston NR; Jones BJ; Bloom SR; Rutter GA; Hoffmann-Roder A; Hodson DJ; Trauner D, Optical Control of Insulin Secretion Using an Incretin Switch. Angew. Chem. Int. Ed 2015, 54, 15565–15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fremaux J; Venin C; Mauran L; Zimmer RH; Guichard G; Goudreau SR, Peptide-oligourea hybrids analogue of GLP-1 with improved action in vivo. Nat. Commun 2019, 10, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroll C; Mansi R; Braun F; Dobitz S; Maecke HR; Wennemers H, Hybrid Bombesin Analogues: Combining an Agonist and an Antagonist in Defined Distances for Optimized Tumor Targeting. J. Am. Chem. Soc 2013, 135, 16793–16796. [DOI] [PubMed] [Google Scholar]
- 4.Liu YT; Cai YY; Liu W; Li XH; Rhoades E; Yan ECY, Triblock peptide-linker-lipid molecular design improves potency of peptide ligands targeting family B G protein-coupled receptors. Chem. Commun 2015, 51, 6157–6160. [DOI] [PubMed] [Google Scholar]
- 5.Cheloha RW; Maeda A; Dean T; Gardella TJ; Gellman SH, Backbone modification of a polypeptide drug alters duration of action in vivo. Nat. Biotechnol 2014, 32, 653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheloha RW; Chen BM; Kumar NN; Watanabe T; Thorne RG; Li LJ; Gardella TJ; Gellman SH, Development of Potent, Protease-Resistant Agonists of the Parathyroid Hormone Receptor with Broad beta Residue Distribution. J. Med. Chem 2017, 60, 8816–8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson LM; Barrick S; Hager MV; McFedries A; Homan EA; Rabaglia ME; Keller MP; Attie AD; Saghatelian A; Bisello A; Gellman SH, A Potent alpha/beta-Peptide Analogue of GLP-1 with Prolonged Action in Vivo. J. Am. Chem. Soc 2014, 136, 12848–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S; Cheloha RW; Watanabe T; Gardella TJ; Gellman SH, Receptor selectivity from minimal backbone modification of a polypeptide agonist. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 12383–12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hager MV; Johnson LM; Wootten D; Sexton PM; Gellman SH, beta-Arrestin-Biased Agonists of the GLP-1 Receptor from beta-Amino Acid Residue Incorporation into GLP-1 Analogues. J. Am. Chem. Soc 2016, 138, 14970–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mary S; Damian M; Louet M; Floquet N; Fehrentz JA; Marie J; Martinez J; Baneres JL, Ligands and signaling proteins govern the conformational landscape explored by a G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 8304–8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopal S; Rajagopal K; Lefkowitz RJ, Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 2010, 9, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abousamra AB; Juppner H; Force T; Freeman MW; Kong XF; Schipani E; Urena P; Richards J; Bonventre JV; Potts JT; Kronenberg HM; Segre GV, Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat os-teoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 2732–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gesty-Palmer D; Chen MY; Reiter E; Ahn S; Nelson CD; Wang ST; Eckhardt AE; Cowan CL; Spurney RF; Luttrell LM; Lefkowitz RJ, Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem 2006, 281, 10856–10864. [DOI] [PubMed] [Google Scholar]
- 14.Cheloha RW; Gellman SH; Vilardaga JP; Gardella TJ, PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol 2015, 11, 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrandon S; Feinstein TN; Castro M; Wang B; Bouley R; Potts JT; Gardella TJ; Vilardaga JP, Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol 2009, 5, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vilardaga JP; Jean-Alphonse FG; Gardella TJ, Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol 2014, 10, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gidon A; Al-Bataineh MM; Jean-Alphonse FG; Stevenson HP; Watanabe T; Louet C; Khatri A; Calero G; Pastor-Soler NM; Gardella TJ; Vilardaga JP, Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat. Chem. Biol 2014, 10, 707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okazaki M; Ferrandon S; Vilardaga JP; Bouxsein ML; Potts JT; Gardella TJ, Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 16525–16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsvetanova NG; von Zastrow M, Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol 2014, 10, 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horwitz MJ; Tedesco MB; Sereika SM; Syed MA; Garcia-Ocana A; Bisello A; Hollis BW; Rosen CJ; Wysolmerski JJ; Dann P; Gundberg C; Stewart AF, Continuous PTH and PTHrP infusion causes suppression of bone formation and discordant effects on 1,25(OH)(2) vitamin D. J. Bone Miner. Res 2005, 20, 1792–1803. [DOI] [PubMed] [Google Scholar]
- 21.Ricarte FR; Le Henaff C; Kolupaeva VG; Gardella TJ; Partridge NC, Parathyroid hormone(1-34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A-CRTC3 signaling. J. Biol. Chem 2018, 293, 20200–20213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrari SL; Behar V; Chorev M; Rosenblatt M; Bisello A, Endocytosis of ligand-human parathyroid hormone receptor 1 complexes is protein kinase C-dependent and involves beta-arrestin2-Real-time monitoring by fluorescence microscopy. J. Biol. Chem 1999, 274, 29968–29975. [DOI] [PubMed] [Google Scholar]
- 23.Bisello A; Chorev M; Rosenblatt M; Monticelli L; Mierke DF; Ferrari SL, Selective ligand-induced stabilization of active and desensitized parathyroid hormone type 1 receptor conformations. J. Biol. Chem 2002, 277, 38524–38530. [DOI] [PubMed] [Google Scholar]
- 24.Cupp ME; Nayak SK; Adem AS; Thomsen WJ, Parathyroid Hormone (PTH) and PTH-Related Peptide Domains Contributing to Activation of Different PTH Receptor-Mediated Signaling Pathways. J. Pharmacol. Exp Ther 2013, 345, 404–418. [DOI] [PubMed] [Google Scholar]
- 25.Peggion E; Mammi S; Schievano E; Silvestri L; Schiebler L; Bisello A; Rosenblatt M; Chorev M, Structure-function studies of analogues of parathyroid hormone (PTH)-1-34 containing beta-amino acid residues in positions 11-13. Biochemistry 2002, 41, 8162–8175. [DOI] [PubMed] [Google Scholar]
- 26.Schievano E; Mammi S; Carretta E; Fiori N; Corich M; Bisello A; Rosenblatt M; Chorev M; Peggion E, Conformational and biological characterization of human parathyroid hormone hPTH(1-34) analogues containing beta-amino acid residues in positions 17-19. Biopolymers 2003, 70, 534–547. [DOI] [PubMed] [Google Scholar]
- 27.Berg C; Neumeyer K; Kirkpatrick P, Teriparatide. Nat Rev Drug Discov 2003, 2, 257. [DOI] [PubMed] [Google Scholar]
- 28.Shirley M, Abaloparatide: First Global Approval. Drugs 2017, 77, 1363–1368. [DOI] [PubMed] [Google Scholar]
- 29.Zhao LH; Ma SS; Sutkeviciute I; Shen DD; Zhou XE; de Waal PW; Li CY; Kang YY; Clark LJ; Jean-Alphonse FG; White AD; Yang DH; Dai AT; Cai XQ; Chen J; Li C; Jiang Y; Watanabe T; Gardella TJ; Melcher K; Wang MW; Vilardaga JP; Xu HE; Zhang Y, Structure and dynamics of the active human parathyroid hormone receptor-1. Science 2019, 364, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vilardaga JP; Frank M; Krasel C; Dees C; Nissenson RA; Lohse MJ, Differential conformational requirements for activation of G proteins and the regulatory proteins arrestin and G protein-coupled receptor kinase in the G protein-coupled receptor for parathyroid hormone (PTH)/PTH-related protein. J. Biol. Chem 2001, 276, 33435–33443. [DOI] [PubMed] [Google Scholar]
- 31.Kenakin T; Watson C; Muniz-Medina V; Christopoulos A; Novick S, A Simple Method for Quantifying Functional Selectivity and Agonist Bias. ACS Chem Neurosci 2012, 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Black JW; Leff P, Operational Models of Pharmacological Agonism. Proc. R. Soc. Lond., B, Biol. Sci 1983, 220, 141–162. [DOI] [PubMed] [Google Scholar]
- 33.Gardella TJ; Luck MD; Wilson AK; Keutmann HT; Nussbaum SR; Potts JT; Kronenberg HM, Parathyroid Hormone (PTH)-PTH-related Peptide Hybrid Peptides Reveal Functional Interactions between the 1-14 and 15-34 Domains of the Ligand. J. Biol. Chem 1995, 270, 6584–6588. [DOI] [PubMed] [Google Scholar]
- 34.Dean T; Vilardaga JP; Potts JT; Gardella TJ, Altered selectivity of parathyroid hormone (PTH) and PTH-Related protein (PTHrP) for distinct conformations of the PTH/PTHrP receptor. Mol. Endocrinol 2008, 22, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura T; Noda H; Joyashiki E; Hoshino M; Watanabe T; Kinosaki M; Nishimura Y; Esaki T; Ogawa K; Miyake T; Arai S; Shimizu M; Kitamura H; Sato H; Kawabe Y, Identification of an orally active small-molecule PTHR1 agonist for the treatment of hypoparathyroidism. Nat. Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hattersley G; Dean T; Corbin BA; Bahar H; Gardella TJ, Binding Selectivity of Abaloparatide for PTH-Type-1-Receptor Conformations and Effects on Downstream Signaling. Endocrinology 2016, 157, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldman ME; McKee RL; Caulfield MP; Reagan JE; Levy JJ; Gay CT; Dehaven PA; Rosenblatt M; Chorev M, A new highly potent parathyroid-hormone antagonist:[D-Trp12,Tyr34]bPTH-(7-34)NH2. Endocrinology 1988, 123, 2597–2599. [DOI] [PubMed] [Google Scholar]
- 38.Thomsen ARB; Plouffe B; Cahill TJ; Shukla AK; Tarrasch JT; Dosey AM; Kahsai AW; Strachan RT; Pani B; Mahoney JP; Huang LY; Breton B; Heydenreich FM; Sunahara RK; Skiniotis G; Bouvier M; Lefkowitz RJ, GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshimori T; Yamamoto A; Moriyama Y; Futai M; Tashiro Y, Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhib-its acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem 1991, 266, 17707–17712. [PubMed] [Google Scholar]
- 40.Zhang HK; Sturchler E; Zhu J; Nieto A; Cistrone PA; Xie J; He LL; Yea K; Jones T; Turn R; Di Stefano PS; Griffin PR; Dawson PE; McDonald PH; Lerner RA, Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat. Commun 2015, 6, 8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.