
Figure 2.
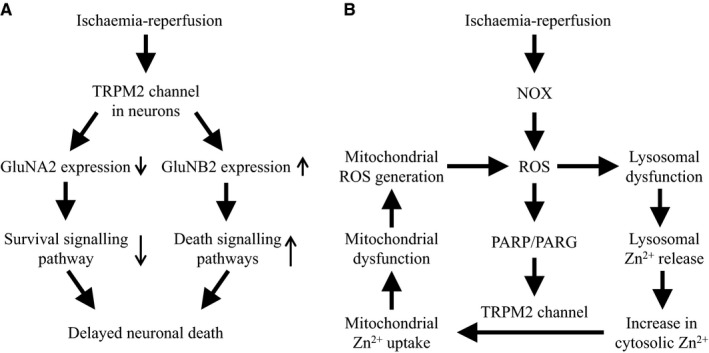
TRPM2‐dependent molecular mechanisms for delayed neuronal death. Two distinctive TRPM2‐mediated molecular mechanisms for delayed neuronal death leading to ischaemia‐reperfusion brain damage have been proposed. A, Elevated generation of reactive oxygen species (ROS) during ischaemia‐reperfusion and subsequent activation of the TRPM2 channel in hippocampal neurons induce down‐regulation of the GluNA2‐containing NMDAR‐mediated survival signalling pathway and up‐regulation of the GluNB2‐containing NMDAR‐mediated death‐promoting signalling pathways, resulting in neuronal death. B, Elevated ROS during reperfusion following transient ischaemia stimulates NADPH‐dependent oxidases (NOX)‐mediated ROS generation. ROS causes lysosomal loss and dysfunction and release of Zn2+, elevating the cytosolic Zn2+ level. ROS also induces activation of the TRPM2 channel in the mitochondria as well as on the cell surface via promoting ADPR generation catalysed by poly(ADPR) polymerase (PARP) and poly(ADPR) glycohydrolase (PARG) in the nucleus. Activation of the TRPM2 channel in the mitochondria increases mitochondrial uptake of Zn2+ that triggers mitochondrial loss and dysfunction and mitochondrial ROS generation. Therefore, activation of the TRPM2 channel sets in motion a positive feedback mechanism ultimately drives lysosomal and mitochondrial dysfunction and neuronal death