
Figure 1.
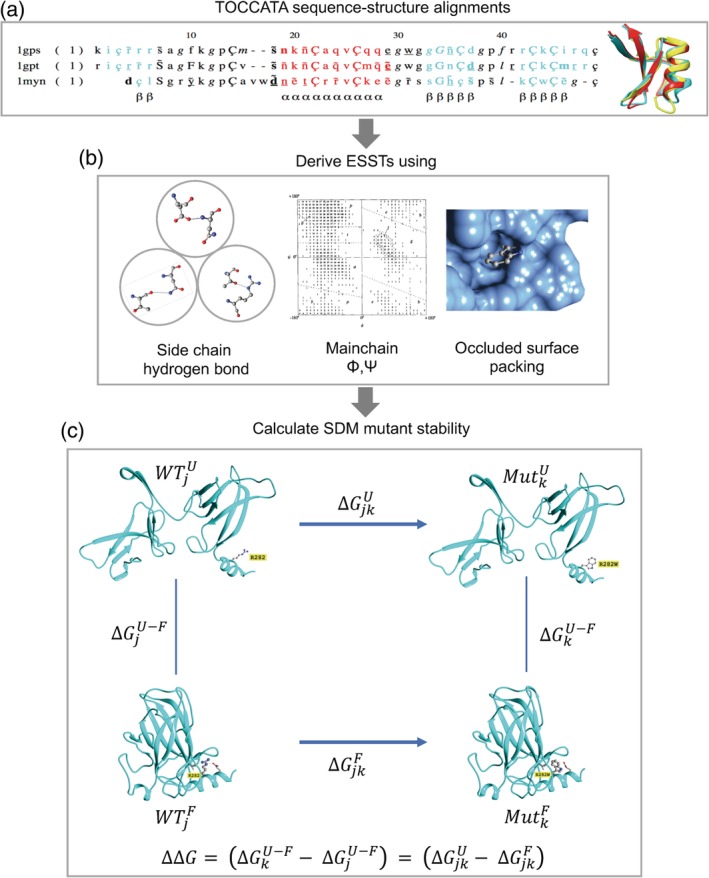
Site‐directed mutator (SDM) method. (a) Sequence‐structure alignments from TOCCATA for protein families are used to calculate the ESSTs. (b) 216 ESSTs are calculated using a combination of eight side‐chain hydrogen bonding patterns, nine main‐chain conformations based on ϕ and ψ dihedral angles, and three residue‐occluded surface packings. (c) The folding–unfolding free energy diagram represented as a thermodynamic cycle for site‐directed mutagenesis. The difference in free energy of unfolding of the wild‐type residue j, and mutant residue k, is related to the free energy changes associated with the mutation in the unfolded and folded states. Using ESSTs, the difference in stability score upon mutation [as shown in Equation (2)] is calculated analogously with Equation (1). As an example, the structure of p53 (PDB http://firstglance.jmol.org/fg.htm?mol=2OCJ) is used to illustrate the mutation at residue position 282 from arginine to tryptophan. Parts of the figure were derived from the graphical abstract taken from Ref. [27]