

Abstract
Mitochondrial genomes can sustain mutations that are simultaneously detrimental to individual fitness and yet, can proliferate within individuals owing to a replicative advantage. We analysed the fitness effects and population dynamics of a mitochondrial genome containing a novel 499 bp deletion in the cytochrome b(1) (ctb-1) gene (Δctb-1) encoding the cytochrome b of complex III in Caenorhabditis elegans. Δctb-1 reached a high heteroplasmic frequency of 96% in one experimental line during a mutation accumulation experiment and was linked to additional spontaneous mutations in nd5 and tRNA-Asn. The Δctb-1 mutant mitotype imposed a significant fitness cost including a 65% and 52% reduction in productivity and competitive fitness, respectively, relative to individuals bearing wild-type (WT) mitochondria. Deletion-bearing worms were rapidly purged within a few generations when competed against WT mitochondrial DNA (mtDNA) bearing worms in experimental populations. By contrast, the Δctb-1 mitotype was able to persist in large populations comprising heteroplasmic individuals only, although the average intracellular frequency of Δctb-1 exhibited a slow decline owing to competition among individuals bearing different frequencies of the heteroplasmy. Within experimental lines subjected to severe population bottlenecks (n = 1), the relative intracellular frequency of Δctb-1 increased, which is a hallmark of selfish drive. A positive correlation between Δctb-1 and WT mtDNA copy-number suggests a mechanism that increases total mtDNA per se, and does not discern the Δctb-1 mitotype from the WT mtDNA. This study demonstrates the selfish nature of the Δctb-1 mitotype, given its transmission advantage and substantial fitness load for the host, and highlights the importance of population size for the population dynamics of selfish mtDNA.
This article is part of the theme issue ‘Linking the mitochondrial genotype to phenotype: a complex endeavour’.
Keywords: mitochondrial deletion, selfish genetic element, genomic conflict, fitness, selection, heteroplasmy
1. Introduction
Across the eukaryotic branches on the tree of life, there exists significant variation in the number of protein-coding genes in the genomes of mitochondria, ranging from three to 66 [1,2]. However, most animal species typically contain the same 13 protein-coding mitochondrial genes, a small fraction of their complete protein-coding capacity. In spite of this limited protein-coding capacity, mitochondria may have had disproportionate effects on eukaryotes, starting with their origin and influencing major aspects of their subsequent evolution, from speciation [3–5] and evolution of sex [6,7] to adaptation to different environments [8–10] and senescence [11,12].
The population biology of mitochondria can be described as nested hierarchies, and competition, random genetic drift and natural selection can operate at different levels of organization [13]. Thus, competition can take place between species, between populations and individuals of the same species (intraspecific), between the cells of an individual and between molecules of mitochondrial DNA (mtDNA) within a cell. Eukaryotic cells, depending on the species and the stage of development, can harbour from one to hundreds of thousands of mitochondria, and each mitochondrion can contain multiple copies of the mitochondrial genome. Hence, a cell with multiple copies of mtDNA can possess different genotypes (mitotypes or haplotypes) owing to spontaneously occurring mtDNA mutations or partial biparental transmission, a condition referred to as heteroplasmy [14–17]. While the ratio of different types of mtDNAs in a heteroplasmic population may fluctuate, usually one mitotype predominates in frequency with other mitotypes observed at low frequencies. Hence, a mutant mitochondrial genome may have minimal effect on the physiology of the cell and the individual when its copy-number is low in the cell. In such instances, the organismal phenotype is thought to be determined by the predominant mtDNA variant [15]. The forces of genetic drift and selection operating within cells can increase the copy-number of a mutant mitotype to a degree that it starts affecting the physiology and fitness of the cell and the individual, which are usually detrimental and likely to result in selection against the mutation. While most detrimental heteroplasmic mutations are removed from populations either by genetic drift, selection or by cells targeting defective mitochondria for degradation (mitophagy) [18], they sometimes increase in frequency and reach fixation. Indeed, heteroplasmy has been implicated in mitochondrial diseases associated with senescence, infertility and cancer in animals [16,19–24].
Because selection can operate on different levels of organization, it can also operate in opposing directions at different levels. In addition to the creation of a heteroplasmic state, mtDNA mutations may also give rise to variants that can be described as ‘selfish’ or ‘parasitic’ mtDNA molecules that possess a transmission advantage over their wild-type (WT, henceforth) counterparts despite being neutral or detrimental to organismal fitness [25–27]. A mitochondrial mutation can increase the fitness of a DNA molecule within cells and yet be detrimental at the level of individuals, contributing to genetic conflict. The potential of mitochondria to incur mutations that result in such genetic conflict is well recognized [28–30]. Not only can there be selection within cells that is opposed by selection between individuals, but because mitochondria are usually uniparentally inherited, mutations can arise that are beneficial to the sex that transmits its mitochondria to the next generation to the disadvantage of the opposite sex. Examples of the latter category include the multiple varieties of cytoplasmic male sterility mutations that are commonly observed in the mitochondrial genomes of flowering plants [31]. Such selfish mtDNA molecules, originating owing to mutations, commence at a low frequency but can rapidly exceed the frequency of the WT mtDNA within the cytoplasm, if their spread is left unabated.
Recent research has demonstrated that deletion-bearing mtDNA molecules often bear the signature of selfish genetic elements. Large-deletion bearing mtDNA molecules resulting in a petite mutant phenotype in small experimental populations of the yeast Saccharomyces cerevisiae were among the first to be described as selfish mtDNA elements, and were often observed to replicate along with the WT mtDNA in a state of heteroplasmy [32–34]. These petite mutations in yeast are one of the best characterized examples of within-cell selection for deleterious mitochondrial mutations, and are thought to gain a transmission advantage owing to faster replication. Additionally, germline mtDNA deletions that are transmitted to progeny have also been reported in humans [35] and nematodes [36,37]. Deletions of mitochondrial genes in the nematode genus Caenorhabditis exhibit patterns that are consistent with selfishness in that they are strongly detrimental to the individual, and yet, reach high intracellular frequency and persist over long periods in populations [26,36,38,39]. In the first documented case of a selfish mitochondrial deletion (uaDf5) in Caenorhabditis elegans, close to 25% (11 genes) of the mitochondrial genome was eliminated during mutagenesis, including atp6, ctb-1, nd1 and nd2 [36]. The uaDf5 deletion persisted in laboratory populations despite significant negative consequences for fitness-related traits. Additionally, natural populations of Caenorhabditis briggsae were found to harbour a heteroplasmic mitochondrial deletion that included a portion of nd5 [26,37]. The nd5 deletion-bearing mitochondrial genomes exist in a heteroplasmic state at 30–40% frequency within C. briggsae. As was the case of the large deletion in C. elegans, these deletions are detrimental to fitness. In both C. elegans and C. briggsae, the negative effects on individual fitness are countered by transmission advantage of the mitochondria that harbour the deletions. mtDNA deletions are thought to be functionally recessive, with a phenotype manifesting beyond a high threshold level of heteroplasmy (greater than or equal to 60%) [40,41], although there can be considerable variation in the phenotypic onset of mtDNA genetic defects [42]. Intriguingly, a small percentage of normal mtDNA (2–27%) can have a disproportionately large ameliorative effect on the phenotype despite a high frequency of the heteroplasmic mtDNA deletion [43].
The reduced ability of populations to rid themselves of strongly deleterious mitochondrial mutations by natural selection can give rise to compensatory mutations, either in nuclear or other mitochondrial genes, that mitigate the effects of the original mutations. An important mechanistic question regarding large-scale heteroplasmic mtDNA deletions is whether they are indeed propagating as selfish genetic elements, a designation that is met if the said elements fulfil two key criteria, namely: (i) possess a transmission advantage relative to other DNA encoded in the genome of the organism, and (ii) are neutral or detrimental with respect to organismal fitness [25]. Caenorhabditis elegans provides an attractive multicellular system to understand the evolutionary dynamics of mitochondrial mutations in real time and the mechanisms by which deleterious mutations are maintained in populations. To that end, we have begun to investigate mitochondrial mutations, including mtDNA deletions, that arose spontaneously and reached high frequencies in a mutation accumulation (MA) experiment in C. elegans [44]. One MA line (line 1G) was found to harbour five heteroplasmic mtDNA mutations relative to the ancestral control, of which three exceeded frequencies of 50%. Of these, a 499 bp frameshift deletion in the cytochrome b(1) (ctb-1, henceforth) gene had reached a frequency of 96%. For simplicity, we refer to this mutant mtDNA genome as Δctb-1-bearing mtDNA given that (i) the large ctb-1 deletion had the highest frequency among all of the detected mtDNA heteroplasmies within this line, and (ii) the ctb-1 deletion serves as a convenient, easily detectable marker for mutant mtDNA within this experimental line. Herein we analyse the fitness consequences of this mutant Δctb-1 mtDNA genome, the timing of its origin and its population dynamics that enable its evolutionary persistence in populations despite a significant fitness cost.
2. Methods
(a). Identification of Δctb-1-bearing mtDNA in a spontaneous mutation accumulation experiment
A 499 bp deletion in the mitochondrial ctb-1 gene originated in one replicate line (1G) of a long-term C. elegans spontaneous MA experiment with varying population sizes [44–46]. The MA experiment was conducted over 409 consecutive MA generations. Line 1G was one of twenty replicate MA lines that were propagated via single hermaphrodite descent each generation (n = 1) and had reached MA generation 346 when submitted for genome sequencing. The lower MA generation number (generation 346) was owing to frequent backups during the course of the MA experiment, presumably because of the build-up of a mutation load leading to reduced fitness relative to the ancestor. Following the termination of the MA experiment, whole-genome sequencing of all 35 MA lines and their ancestral pre-MA control was conducted via Illumina paired-ends sequencing and all mtDNA mutations across the 35 MA lines were identified [44]. MA line 1G was found to harbour five heteroplasmic mtDNA mutations relative to the ancestral control: (i) a G → T substitution in the tRNA-Asn gene (frequency 13%), (ii) a 499 bp frameshift deletion in the ctb-1 gene (frequency 96%), (iii) a single T insertion in a homopolymeric run within the nd5 gene [(T)8 → (T)9; frequency 3%], (iv) an insertion of a T pair in a homopolymeric run within the nd5 gene [(T)8 → (T)10; frequency 70%], and (v) a C → T nonsynonymous substitution (Thr → Ile) in the nd5 gene (frequency 94%). Owing to the presence of three high-frequency (greater than or equal to 70%) heteroplasmic mtDNA mutations in line 1G, it is likely that the majority of mtDNA molecules within this lineage harbour more than one of these heteroplasmic mtDNA variants. Given that the ctb-1 deletion is the heteroplasmy with the highest frequency and is easily genotyped by polymerase chain reaction (PCR), we refer to the mutant mitotypes as Δctb-1-bearing mtDNA or Δctb-1 mitotype for the sake of simplicity.
(b). Timing the spontaneous origins of multiple heteroplasmies in mutation accumulation line 1G
An immense advantage of C. elegans as a model system for experimental evolution studies is the species' ability to survive long-term cryogenic storage at −86°C. During the course of the spontaneous MA experiment, the MA lines had been cryogenically preserved at regular time-intervals. In addition to the pre-MA ancestral control, we thawed stocks of line 1G frozen at nine additional time-intervals, namely MA generations 25, 51, 72, 96, 137, 157, 221, 300 and 346, and isolated genomic DNA from L4 larvae using a previously described supplemental nematode protocol with a Puregene Genomic DNA Tissue Kit [44]. These genomic DNA samples were initially screened for the frequency of Δctb-1 via droplet digital PCR (ddPCR, henceforth). The sequence context of the other mtDNA mutations in line 1G were not suitable for designing reliable ddPCR probes and the frequency of these mutations was estimated from peak heights in chromatograms generated by Sanger sequencing.
(c). Droplet digital polymerase chain reaction to estimate the frequency of Δctb-1 and mtDNA copy-number
We used ddPCR to determine the frequency of the Δctb-1 mitotype, and the relative copy-number of Δctb-1 and WT mtDNA. We refer to the mitochondrial alleles present in the ancestral control of our MA lines as WT alleles in this study. Bio-Rad ddPCR probes targeted a sequence within the ctb-1 deletion, a region of cox-1, and a conserved region of the second exon of actin-2 (electronic supplementary material, table S1). To determine the degree of heteroplasmy, a single L4 worm was lysed in 5 µl worm lysis buffer (90 µl 10× PCR buffer + 10 µl 10 mg ml−1 proteinase K). The single worm lysate was diluted 1 : 5 in molecular grade water and 2.2 µl of the diluted worm lysate was added to 19.8 µl of ddPCR master mix, which included both ctb-1 and cox-1 Bio-Rad Digital PCR assays in a single multiplexed PCR reaction. In an adjacent well, the actin-2 Bio-Rad Digital PCR assay was performed on the same single worm lysate to measure the concentration of nuclear genomes in the sample. The fluorescin (FAM) probe in the ctb-1 assay only hybridizes to the WT mtDNA whereas the hexachlorofluorescin (HEX) probe in the cox-1 assay hybridizes to both Δctb-1 and WT mtDNA in the sample. Heteroplasmic frequency was determined by subtracting the concentration (copies μl−1) of WT mtDNA in the sample from the total mtDNA concentration and dividing it by the total (f(ΔmtDNA) = [total mtDNA concentration − WT mtDNA concentration]/total mtDNA concentration). This quantity was then compared to the nuclear DNA concentration to determine relative mtDNA copy-number per cell. Droplets were generated using the BioRad Automated Droplet Generator. Droplet generation was considered successful if more than 7000 droplets were generated. Subsequently, the PCR droplets were read on a BioRad QX200 Droplet Reader and droplet distribution was analysed by the automated threshold setting in Quantisoft and inspected visually. The FAM probe used for the ctb-1 WT is slightly more efficient than the HEX probe used for cox-1. As a result, when C. elegans N2 DNA is used as a template for these ddPCR reactions, the heteroplasmic Δctb-1 are negative (−0.099, s.d. = 0.028). This is owing to the fact that the heteroplasmy frequency was calculated by subtracting the frequency of ctb-1 WT from that of cox-1.
(d). Estimating the frequencies of heteroplasmic mutations in nd5 and tRNA-Asn
The intracellular frequencies of nd5 and tRNA-Asn mutations were determined from Sanger sequencing. The following primers were used to sequence the region of nd5 that contained three previously detected mutations: forward, 5′-TCATCTTCATCTTGGGAGGATTT-3′ and reverse, 5′-GTGTCCTCAAGGCTACCACC-3′. The peak heights at heteroplasmic sites were measured from sequencing chromatograms using ImageJ. For the nd5 nonsynonymous mutation at mtDNA:12 304, the height of the mutant allele peak Tmut and the height of the WT allele peak Cwt were used to estimate the frequency of mtDNA containing the nonsynonymous mutation as follows:
The same approach was used to calculate the frequency of the tRNA-Asn base substitution at mtDNA:1679.
For the T8 → T9 frameshift mutation at mtDNA:11 778, the heteroplasmy frequency was determined from the average of two sites. The sequence context at the boundaries at the end of the homopolymeric run and the downstream nucleotides is TAG and we used the first T + A and A + G in the chromatograms to determine the frequency as follows:
where T9 is the T in the first double peak, A1 is the A in the first double peak, A2 is the A in the second double peak and G1 is the G in the second double peak. When the (T)8 → (T)10 insertion is present in addition to the T8 → T9 insertion, the following calculations were performed to calculate the frequencies of both (T)8 → (T)9 and (T)8 → (T)10 mutations as follows:
and
where A, G and T10 are the corresponding bases in the first triple peak downstream of the insertion.
A forward primer (5′-GGTGTTACAGGGGCAACATT-3′) within the region that is deleted in ctb-1 and a reverse primer (5′-GTGTCCTCAAGGCTACCACC-3′) downstream of the nd5 missense mutation amplified a 7470 bp region of the non-deletion bearing mtDNA to test if any of the nd5 mutations were associated with the WT ctb-1. PCR was performed using Promega GoTaq Long PCR reagents. Gel purified (QIAEXII Gel Extraction Kit) PCR products were used to reamplify the nd5 region (forward primer: 5′-TCATCTTCATCTTGGGAGGATTT-3′, reverse primer: 5′-GTGTCCTCAAGGCTACCACC-3′) for sequencing. Similarly, a forward primer (5′-ATCAATTGCCCAAAGGGGAGT-3′) upstream of the tRNA-Asn mutation and a reverse primer (5′-TGGCCCTCAAATTGGAATAA-3′) within the ctb-1 deletion amplified a 3435 bp region to test if the tRNA-Asn mutation was associated with the WT ctb-1. Gel purified PCR products were sequenced using the tRNA-Asn forward primer and a tRNA-Asn reverse primer. DNA sequencing of nd5 and tRNA-Asn was done on 1G worms sampled at every time point during the original MA experiment, and at the beginning and termination (generation 75) of the experimental evolution in large populations. Sanger sequencing was performed by Eton Biosciences.
(e). Sequestering the Δctb-1-bearing mtDNA in a wild-type background
In order to assess the fitness effects and population dynamics of Δctb-1-bearing mtDNA relative to WT mtDNA, we first isolated the mitochondria of line 1G in a WT nuclear background, free of other nuclear mutations that arose in line 1G during the MA procedure. This was accomplished by crossing 1G hermaphrodites to males belonging to a line bearing a mutation in the nuclear fog-2 gene. In WT C. elegans, hermaphrodites have two X chromosomes and males have one X chromosome. The fog-2 gene functions in a pathway responsible for sperm production in hermaphrodites. Hence, a fog-2 loss-of-function [fog-2(lf)] mutation turns XX individuals that would have been hermaphrodites into functional females only capable of reproduction via outcrossing with males. After two generations of crosses to males of the mutant fog-2 strain, XX individuals were rendered homozygous for the fog-2(lf) and therefore incapable of self-fertilizing. This conversion of XX selfing hermaphrodites into obligate outcrossing females facilitated subsequent outcrossing with males bearing a WT nuclear genome. Commencing with the F3 generation, females with Δctb-1-bearing mtDNA were subjected to 11 additional generations of backcrosses to fog-2(lf) males with a WT (non-MA) nuclear background during which the contribution of the nuclear DNA of the MA line was reduced by half in each generation (electronic supplementary material, figure S1). Finally, backcrossed females with Δctb-1-bearing mtDNA were backcrossed with WT N2 males in the last two generations (generations 14 and 15), thereby replacing the WT fog-2 allele in the females and restoring them to functional hermaphrodites with the ability to self-fertilize. After 15 generations, the proportion of the nuclear DNA from the original 1G line is estimated to be 0.515 of the total nuclear DNA, or approximately 3 × 10−5, which virtually removes all other nuclear mutations that arose during MA. This backcrossing experiment yielded six replicate lines comprising worms with the Δctb-1-bearing mtDNA from line 1G in a WT nuclear background. These six lines (1G.C, 1G.L, 1G.M, 1G.N, 1G.T and 1G.U) were cryogenically preserved at −80°C.
(f). Assays for four fitness-related traits
The fitness effects of Δctb-1-bearing mtDNA from line 1G were assayed in the six 1G lines harbouring the mutant DNA in a WT nuclear background relative to WT N2 controls. Frozen stocks of the six experimental WT backcrossed 1G lines (1G.C, 1G.L, 1G.M, 1G.N, 1G.T and 1G.U) and the N2 control line were thawed and one and three individuals, respectively, were selected and expanded into 15 replicates to establish within-line replication (n = 90 and 45 worms for the 1G and N2 lines, respectively). The 15 Δctb-1-bearing hermaphrodites from each of the generated 1G lines (n = 90) were assayed for four fitness-related traits (productivity, survivorship to adulthood, developmental time and longevity) and compared to WT N2 nematodes (n = 45) using standard C. elegans protocols [45–47]. These 135 lines (90 Δctb-1-bearing and 45 N2 control) were then maintained by transferring single L4 (fourth larval stage) hermaphrodite individuals for two additional generations to remove environmental effects (maternal and grandmaternal). A single third-generation descendant from each thawed replicate was subsequently employed in the actual fitness assay. For logistic purposes, the fitness assays were performed in three rounds, each with two of the Δctb-1-bearing bearing lines (n = 30) and an ancestral N2 control (n = 15). All assays were performed under benign laboratory conditions at 20°C, an optimal temperature for C. elegans growth. A schematic of the design of the four fitness assays is presented in the electronic supplementary material, figure S2.
To assay survivorship to adulthood (also referred to as survivorship), 10 L1 (first larval stage) progeny of each third-generation hermaphrodite were randomly selected upon hatching and isolated on a separate 35 mm nematode growth medium (NGM) Petri dish seeded with an Escherichia coli OP50 lawn; then 36 h after isolation, the number of individuals surviving past the L4 larval stage to reach adulthood were quantified. Survivorship values can range from 0 to 1, and were calculated by dividing the number of adult worms by the number of L1 larvae originally sequestered [45,46].
To measure productivity, a single worm, a full sibling of the 10 individuals assayed for survivorship, was randomly selected as an L4 larva and transferred to a new 35 mm NGM Petri dish seeded with an E. coli OP50 lawn. Every 24 h ± 30 min thereafter, each hermaphrodite being assayed was transferred to a fresh Petri dish over 8 days after reaching the L4 (last) larval stage. Transfers were terminated if the worm was found dead prior to the completion of these 8 days. Following each daily transfer of the assayed individuals, plates with eggs were placed at 20°C for an additional 24 h period to enable hatching. To enable progeny counts, the plates were then stored at 4°C to kill the progeny larvae. Progeny were counted by staining the agar pad and E. coli lawn with a 0.075% water dilution of toluidine blue, which rendered the dead worms transparent and visible on the contrasting purple background for the approximately 2–5 min period required for counting. Productivity was calculated as the total number of progeny produced; non-reproductive individuals were scored as having zero productivity [45,46].
The single worm assayed for productivity above was additionally used to measure developmental time. Following its sequestration as a lone L4 larva onto a seeded Petri dish, the worm was visually inspected under a dissecting scope every 2 h. The hermaphrodite was determined to be an adult hermaphrodite if it had: (i) undergone its L4 larval moult, (ii) had a developed vulva, and (iii) displayed at least one developed egg in the uterus. Developmental time was calculated as the number of hours it took a newly-hatched L1 larva to reach adulthood [47].
We measured longevity (number of days) on the same worm that was assayed for productivity and developmental time. After day 8 of egg laying, the worms were transferred to a new 35 mm NGM plate seeded with OP50 for a longevity assay. Each day thereafter, these nematodes that had largely ceased egg production were visualized under the dissecting scope until mortality. A nematode was determined to be dead if it was no longer locomoting, did not respond to gently tapping the plate or agar, did not respond to gentle prodding of the tail with a pick and had no pharyngeal activity. Longevity was measured as the total number of days from a newly-hatched L1 larva to mortality.
Relative fitness values for three traits (productivity, survivorship to adulthood and longevity) were generated by assigning the mean absolute fitness value of the WT N2 control a value of 1 (n = 45). Fitness measures for these three traits were standardizing by dividing a replicate line's observed fitness value by the mean fitness value of the WT N2 control to yield relative fitness values. Developmental rate was expressed as the inverse of the relative developmental time.
(g). Competition experiments
Stocks of five experimental replicate C. elegans lines bearing the Δctb-1 mtDNA in a WT nuclear background (1G.C, 1G.L, 1G.N, 1G.T and 1G.U) were thawed along with WT N2 worms. Worms were picked from each thaw and placed on separate 60 mm NGM plates with E. coli OP50 seed. A single worm from the progeny of each thaw was transferred for two generations to remove potential freezer effects. The F3 generation worms were used to initiate an experimental evolution study to investigate the population dynamics of Δctb-1-bearing lines under competitive and non-competitive conditions.
For each of the five experimental lines (1G.C, 1G.L, 1G.N, 1G.T and 1G.U), we established two populations, one competitive and one non-competed control. Hence, the competition experiment comprised 10 populations in total (five competition and five non-competed controls) (electronic supplementary material, figure S3). Each competition population was initiated by adding 50 L4 hermaphrodites from a Δctb-1-bearing experimental line and 50 L4 worms from the WT N2 stock onto a 100 mm NGM plate seeded with an E. coli OP50 lawn (1 ml), for a total population size of n = 100 worms with Δctb-1 and WT mtDNA bearing worms in equal frequencies. In addition, we established five non-competed control populations by transferring 50 L4 hermaphrodites from a Δctb-1 bearing experimental line in isolation onto a fresh 100 mm NGM plate. In summary, the first generation (generation 0 in electronic supplementary material, figure S3) of the competition lines comprised an equal ratio of Δctb-1 mtDNA: WT mtDNA worms whereas the non-competed control lines were established with 100% Δctb-1 mtDNA worms. A standard C. elegans bleaching protocol was used to synchronize the evolving populations at the first larval stage (L1) of each generation and ensure that adult worms from a previous generation did not continue to contribute to the gene pool across multiple generations. Four days after L4 worms were placed onto a fresh plate to establish a new generation, each plate was bleached with a 30% bleach and 15% 5M NaOH solution. Bleaching killed living worms but rendered eggs unharmed. The eggs that survived the bleach were re-plated onto new 100 mm NGM plates with OP50 to establish the next generation. The competition experiment was conducted over 16 consecutive generations (electronic supplementary material, figure S3).
To determine the population frequencies of the Δctb-1-bearing and WT mtDNA haplotypes on each plate in each generation, we randomly selected 30 L4 worms for genotyping from each of the 10 experimental plates (five competition and five non-competed controls). The worms were added to worm lysis buffer (10× PCR buffer and proteinase K) and the digests were used in single-worm PCR with cytb-1 primers to assess the presence/absence of the Δctb-1 mitotype. The ctb-1 forward and reverse primers are 5′-TAGCATTTTCAACAGTGCAG-3′ and 5′-CGCAAAATTGCATAACTCAAAT-3′, respectively. The expected WT and Δctb-1 PCR products are 661 and 162 bp in length, respectively. The PCR products were run on a 1% agarose gel (100 ml 1× tris-acetate-EDTA; 1 g RA agarose; 1 µl GelRed) at 105 V for 30 min.
In addition, large populations that were established from replicate lines 1G.C, 1G.N, 1G.T and 1G.U were maintained on 100 mm NGM plates seeded with an E. coli OP50 lawn (1 ml) for 75 generations and maintained in the same manner as that outlined for the competed and non-competed lines. At generations 0, 30 and 75, a random sample of 15 individuals were used to determine the frequency of Δctb-1 and relative mtDNA copy-number in individual worms by ddPCR. The frequency results were analysed in a two-way ANOVA after arcsine transformation. The Pearson correlation coefficient between relative copy-numbers of Δctb-1 and WT ctb-1 were calculated for these 15 individuals per line at each of the three time-points.
(h). Estimating the relative fitness of Δctb-1 bearing mtDNA from competition experiments
The composite relative fitness, w, of the Δctb-1 mitotype was calculated from its average frequency pooled across the competition experiments comprising the five replicate lines across generations. First, the log of the ratio (p/q) in each generation t was calculated, where, p equals the average frequency of the Δctb-1 mitotype, and q equals the average frequency of the WT ctb-1 allele. The log ratio of p/q was mapped as a function of generation and linear regression was performed on the data. The slope of the regression line, b, equals log(w). Hence, the relative fitness, w, was calculated as 10b where b represents the slope of the log(p/q)t over t generations.
(i). Role of selfish drive versus genetic drift in the proliferation of the Δctb-1 mitotype within individuals
To test if the Δctb-1 mitotype's rapid increase in frequency was owing to selfish drive or genetic drift, we first performed directional selection for a lower frequency of Δctb-1. From each of the populations at generation 75 (1G.C, 1G.N, 1G.T and 1G.U), five L4 hermaphrodites were transferred to individual 35 mm NGM plates seeded with OP50. After allowing 2 days for egg-laying, these adult (parent) nematodes were subsequently genotyped by single-worm PCR using primers for ctb-1. Five progeny of one parent per line that produced the strongest WT PCR product were used to establish the next generation. After five rounds of selection for a Δctb-1 PCR product of reduced intensity, the offspring of a single individual from line 1G.T with a low frequency of the Δctb-1 mitotype (37%) were used to establish 25 lines that were propagated by picking a single L4 hermaphrodite at random in each generation to establish the next generation. After six generations of propagating 25 lines by single-progeny descent, single-worm ddPCR was used to determine the frequency of the Δctb-1 mitotype.
3. Results
(a). Appearance and proliferation of the Δctb-1 mitotype as a heteroplasmy during mutation accumulation
A 499 bp frameshift deletion in ctb-1 arose during a spontaneous MA experiment in C. elegans and was detected via whole-genome sequencing at the termination of the MA experiment (figure 1a) [44]. The frequency of the deletion within individuals of this particular MA line (1G) was estimated to be 96%. The deletion encompassed approximately 45% of the ctb-1 gene (figure 1b). Hence, not only does this Δctb-1 allele have the potential to result in a truncated protein product with no residual function, it may also generate frameshifted transcripts leading to altered proteins. Specifically, this Δctb-1 allele is predicted to produce a 106 aa peptide, whereas the original WT CTB-1 protein comprised 370 aa, owing to the introduction of a stop codon downstream of the deleted segment.
Figure 1.
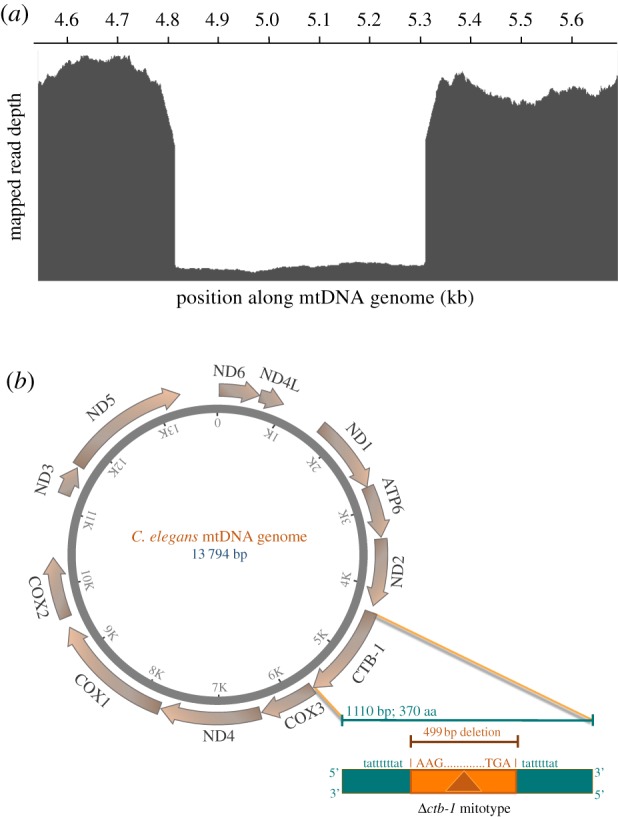
Identification and location of the ctb-1 deletion in MA line 1G. (a) A 499 bp frameshift mtDNA deletion in ctb-1 (Δctb-1) occurred spontaneously in a C. elegans spontaneous mutation accumulation line, 1G, and was detected via whole-genome sequencing [44]. The figure shows the read depth from Illumina whole genome sequencing mapped to the ctb-1 region of the C. elegans mitochondrial genome in line 1G. The read depth inside the deletion is only 4% of the read depth of the sequences immediately flanking the deletion, suggesting that the frequency of the Δctb-1 mitotype is 96% within this MA line. (b) A map of the C. elegans mitochondrial genome (adapted from [48]), illustrating the location, extent and the sequence context of Δctb-1. The protein-coding sequence of the ctb-1 gene spans 1110 bp (protein length 370 aa). (Online version in colour.)
We screened previously cryopreserved stocks of MA line 1G at several time-intervals of the MA experiment via PCR and ddPCR to determine: (i) the approximate timing of origin of the Δctb-1 mitotype, and (ii) the change in its frequency within line 1G during the course of the MA experiment. The Δctb-1 mitotype was not detected in worms at MA generation 25, but was present at 7% frequency by MA generation 51. Its frequency rose rapidly until it had reached 90% by MA generation 91. Following MA generation 91 and until MA generation 364, its frequency remained high, fluctuating between 80% and 98% (figure 2).
Figure 2.
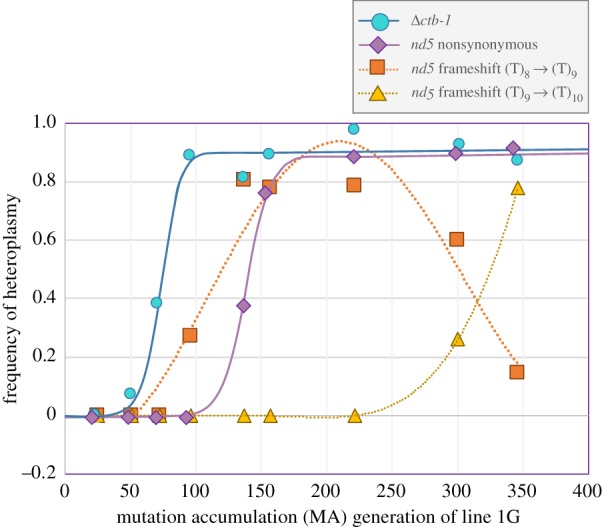
Frequency trajectories of four spontaneous mtDNA mutations that arose in MA line 1G during the course of the MA experiment. The vertical axis represents the intracellular frequency of individual mtDNA mutations as estimated by ddPCR (Δctb-1) or Sanger sequencing. The horizontal axis represents the number of generations since the start of the MA experiment. (Online version in colour.)
(b). Additional high-frequency mtDNA mutations in mutation accumulation line 1G originated after Δctb-1
Following the origin of the Δctb-1 allele in line 1G, an insertion of a single T nucleotide in nd5 was detected in 26% frequency in a run of 8Ts at location 11 778 by MA generation 90 and subsequently reaching a frequency of 81% by MA generation 137 (figure 2). This single T insertion in nd5 remained in high frequency at MA generations 157 and 221, with an estimated frequency of 78% at both time-points. However, when a second T insertion had appeared at the same site by MA generation 300, the single T mutation declined in frequency until it had been largely replaced by the double T insertion. The combined frequencies of the single and double T insertion at this site were 86% and 92% in MA generations 300 and 346, respectively. In addition to these insertions, a nonsynonymous mutation in nd5 had reached a frequency of 38% by generation 137 (figure 2). This mutation also reached high frequency and fluctuated around 90% from generation 222 until the end of the MA experiment. Based on their high frequencies, it seemed likely that these mutations were on the same molecule as the Δctb-1 mutant allele and not linked to the low frequency ctb-1 WT mtDNA. We tested this assumption by first performing PCR with one primer within the deleted ctb-1 sequence and a reverse primer downstream of the nd5 mutations, followed by Sanger sequencing of nd5. The PCR product only contained sequence from the ctb-1 WT allele. At each time-point analysed in the MA experiment, and in each experimental population established with the mtDNA from MA line 1G, the ctb-1 WT allele is associated with the nd5 WT allele. Similarly, the nd5 mutations were associated with the Δctb-1 mutant allele and we found no evidence of recombination between the ctb-1 WT and Δctb-1 molecules. In addition to these mutations that persisted in the mtDNA genome of line 1G until the end of the MA experiment, a T insertion at site 11 722 within the nd5 gene was detected at 41% frequency in MA generation 137 but subsequently went extinct (figure 2). A single nucleotide insertion at this site occurred in multiple independent lines during the original MA experiment, suggesting that it is a mutational hotspot [44]. The intracellular frequencies of the nd5 mutations in this study were primarily assessed via Sanger sequencing, but their frequencies can be compared to those previously obtained by whole-genome Illumina sequencing from the end of the MA experiment [44]. The single and double T insertions were estimated to be at 14% and 78% frequency by Sanger sequencing, respectively, but at 3% and 70% by Illumina sequencing. The nonsynonymous nd5 mutant allele was estimated at 91% and 94% frequency by Sanger and Illumina sequencing, respectively. In addition to mutations in ctb-1 and nd5, whole-genome sequencing of MA lines had identified a tRNA-Asn mutation with 13% frequency in line 1G [44]. Based on Sanger sequencing, this mutation was in 8% frequency at the end of the MA experiment, and at 5% at MA generation 300. There is no evidence of this mutation prior to MA generation 300. Sanger sequencing of PCR products from the WT ctb-1 molecule, which used a reverse primer to the deleted region in ctb-1 and a forward primer upstream of tRNA-Asn, only yielded a WT tRNA-Asn sequence. We conclude that the Δctb-1 mutant allele is linked to other mutations in nd5 and tRNA-Asn, which arose sequentially after the original ctb-1 deletion, and there is no evidence of recombination between any of these mutations in the mitochondrial genome of line 1G or its derivatives.
(c). Δctb-1-bearing mtDNA engender a significant fitness cost
In order to test the fitness effects of Δctb-1-bearing mtDNA relative to WT mtDNA, the mutant mitochondria were sequestered in a WT N2 genetic background, free of other nuclear mutations that arose during MA within line 1G (electronic supplementary material, figure S1). Four fitness traits, namely productivity, survivorship to adulthood, longevity and developmental time were measured in six lines (1G.C, 1G.L, 1G.M, 1G.N, 1G.T, 1G.U) harbouring the Δctb-1 mtDNA from line 1G in a WT N2 nuclear background (electronic supplementary material, figure S2; table 1). The Δctb-1 bearing worms varied significantly from the WT mtDNA lines with respect to all of the four fitness traits assayed (figure 3; electronic supplementary material, table S2).
Table 1.
Fitness of Δctb-1-bearing mtDNA in a wild-type (WT) nuclear background relative to control worms of the laboratory strain, N2, bearing WT mtDNA. (Fifteen replicates each of three N2 control and six experimental Δctb-1-bearing (1G.C–1G.U) lines were assayed. Estimates of the mean phenotype for four fitness traits are provided for the WT N2 control (; three lines each with 15 replicates, n = 45) and the six 1G-derived, Δctb-1 bearing lines (; n = 90). Mean fitness values across n = 15 replicates for individual control and experimental lines are also provided (three N2 control and six Δctb-1-bearing lines).)
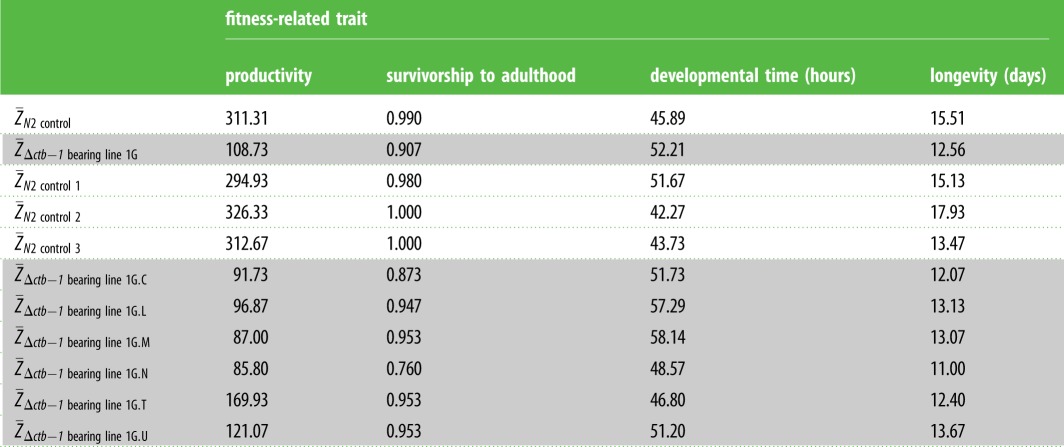 |
Figure 3.
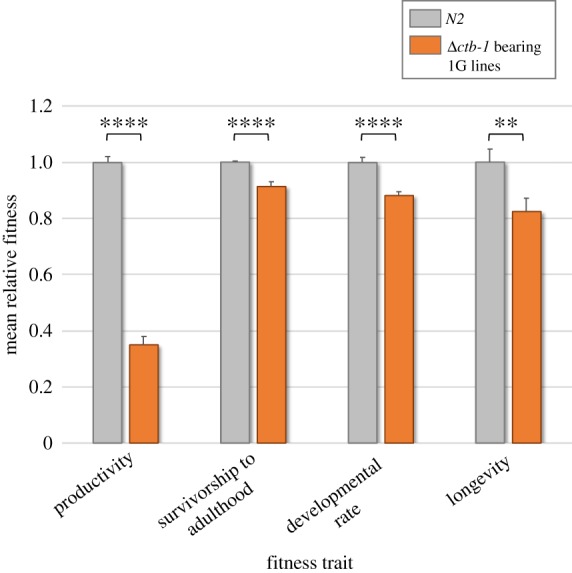
Relative trait means of Δctb-1 bearing 1G replicate lines and the WT N2 control. Mean fitness values for each of the four traits were measured across six 1G lines (orange), each with 15 replicates where possible (n = 87 or 90) and three N2 control lines (grey), each with 15 replicates (n = 45). Phenotypic assays were conducted for four fitness-related traits, namely productivity, survivorship to adulthood, developmental rate and longevity. For simplicity, the mean relative fitness value for each of the four traits in the WT N2 control was scaled to a value of 1. All of the lines bearing the Δctb-1 mtDNA perform significantly worse than the WT N2 line. **p ≤ 0.01, ****p ≤ 0.0001. Error bars represent one standard error. (Online version in colour.)
Δctb-1 bearing lines exhibited a significant decline in productivity relative to the WT N2 control (figure 3; Wilcoxon rank sum, z = 8.88, d.f. = 1, p < 0.0001). The mean productivity of 109 offspring per hermaphrodite in Δctb-1 bearing lines is only 35% of the 311 offspring produced by the WT N2 control. Mean relative productivity of the Δctb-1 bearing lines ranged from 28% (lines 1G.M and 1G.N) to 55% (line 1G.T) of the average WT productivity. There was no significant difference in the mean productivity of the six individual 1G lines bearing the Δctb-1 mitotype (table 1; Wilcoxon/Kruskal–Wallis, χ25 = 8.47, p = 0.13).
Δctb-1 bearing lines exhibited a significant decline in survivorship to adulthood relative to the N2 control (figure 3; Wilcoxon rank sum, z = 4.11, d.f. = 1, p < 0.0001). The mean survivorship of Δctb-1 bearing and N2 control worms is 90% and 99%, respectively. Hence, Δctb-1 bearing worms have a relative mean survivorship of 92% (8% mean decline) compared to WT worms. Mean survivorship of the Δctb-1 bearing lines ranged from 77% (line 1G.N) to 96% (lines 1G.L, 1G.M, 1G.T and 1G.U) of the average WT survivorship. There was a significant difference in the mean survivorship of the six individual 1G lines bearing the Δctb-1 mitotype (table 1; Wilcoxon/Kruskal–Wallis, χ25 = 12.79, p = 0.026), with lines 1G.C and 1G.N in particular having lowered survivorship of 77–88% relative to the N2 control.
There was a significant reduction in the developmental rate of Δctb-1 bearing lines versus the N2 control (figure 3; Wilcoxon rank sum, z = 5.1, d.f. = 1, p < 0.0001). Δctb-1 bearing worms took, on average, 6.3 h longer than WT to lay their first fertilized egg, representing an average 13.7% increase in the time taken to reach reproductive maturity. Δctb-1 bearing lines took 2–27% longer relative to the N2 control to reach reproductive maturity. There was a significant difference in the mean developmental time of the six individual 1G lines bearing the Δctb-1 mitotype (table 1; Wilcoxon/Kruskal–Wallis, χ25 = 34.19, p < 0.0001).
The longevity of Δctb-1 bearing lines was significantly lower than that of the N2 control (figure 3; Wilcoxon rank sum, z = 2.54, d.f. = 1, p = 0.011). On average, the lifespan of Δctb-1 bearing worms was approximately 3 days shorter relative to the N2 control, representing an average 19% decrease in the former. The mean longevity of Δctb-1 bearing lines ranged from 71–88% of the N2 control's mean longevity. However, there was no significant difference in the mean longevity of the six individual 1G lines bearing the Δctb-1 mitotype (table 1; Wilcoxon/Kruskal–Wallis, χ25 = 1.42, p = 0.92).
(d). Rapid extinction of Δctb-1 when competed with wild-type mtDNA
A competition assay was performed to test the fitness of the Δctb-1 mitotype in the replicate 1G lines when in competition with the WT N2 mtDNA. Five replicate 1G lines with the Δctb-1-bearing mtDNA in a WT nuclear background (1G.C, 1G.L, 1G.N, 1G.T, 1G.U) were competed against WT N2 worms in a mixed population commencing with equal frequencies of deletion-bearing and WT mtDNA worms (electronic supplementary material, figure S3). All five competition populations showed a steep decline in the frequency and rapid extinction of Δctb-1 mtDNA across successive generations of competition with worms bearing WT mitochondria (figure 4a). The frequency of Δctb-1 bearing worms had fallen below levels of detection in all populations by the eighth generation (figure 4a). The competition experiment was continued until generation 16, with 30 individuals sampled from each population in every generation, but no individuals were detected as harbouring the Δctb-1 mitotype from generations 9–16.
Figure 4.
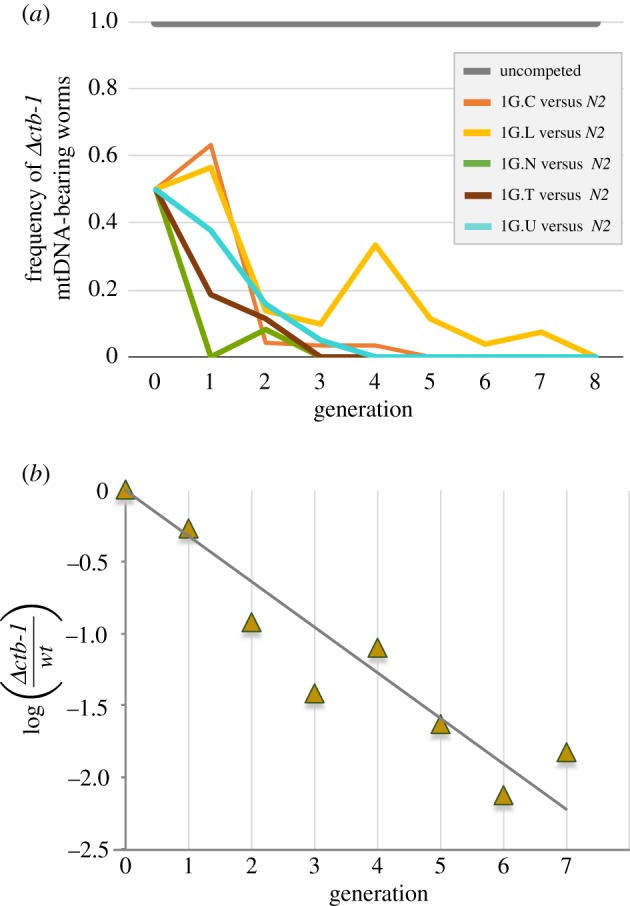
Evolutionary dynamics of Δctb-1 mtDNA under competitive conditions. (a) Population-level frequencies of the Δctb-1 mtDNA heteroplasmy in five 1G replicate lines under non-competed (grey lines) versus competitive conditions (coloured lines). In competitive assay populations established with equal ratios of Δctb-1 and WT mtDNA bearing worms (50 Δctb-1 bearing 1G hermaphrodites+50 WT mtDNA-bearing N2 hermaphrodites), the population dynamics of the Δctb-1 mitotype displays a steep decline in frequency with time. The Δctb-1 mtDNA heteroplasmy remains in high frequency (approx. 1.0) within each replicate line across generations under noncompetitive (control) conditions. In general, there is a significant reduction in the frequency of the Δctb-1 mitotype from 0.5 to less than 0.2 within two generations under competitive conditions, and eventual loss (undetectable via PCR) from the population between generations 3–8. The Δctb-1 mitotype remained undetected between generations 9–16. (b) A linear regression of the change in log[f(Δctb-1/WT mtDNA)] with time (generations). The single data point for each generation represents the average values of the Δctb-1 mutant mitotype and WT mtDNA across five independent replicates of line 1G (1G.C, 1G.L, 1G.N, 1G.T and 1G.U). The relative fitness, w, of the Δctb-1 mutant mitotype was calculated from the slope of the regression line and estimated to be 0.48, implicating a large deleterious fitness cost of the ctb-1 deletion bearing mtDNA and its gradual eradication in large competitive populations via purifying selection. (Online version in colour.)
The five replicate Δctb-1 bearing 1G lines were also used to establish large populations that were maintained in parallel to the competition experiment, but without any WT N2 worms. In these non-competed populations, the Δctb-1 was never observed to go extinct (figure 4a). This suggests that the decline in the population frequency and eventual loss of Δctb-1 under competitive conditions was owing to the substantially lower fitness of Δctb-1 bearing worms relative to WT, and not because of the loss of the Δctb-1 mitotype within worms that contained the deletion at the beginning of the experiment.
The relative fitness reduction associated with the Δctb-1 mitotype was also calculated from its average frequency pooled across the five replicate competition populations across generations. First, the log of the ratio (p/q) in each generation t was calculated, where, p and q equal the average frequency of the Δctb-1 and WT ctb-1 mitotype, respectively. The log ratio of p/q was mapped as a function of generation and linear regression was performed on the data (figure 4b). The slope of the regression line, b, was determined to be −0.3174, which yielded a relative fitness (w) value of 0.4815. Hence, the overall competitive fitness of Δctb-1 bearing worms is only 48% of worms with WT mitochondria. A negative selection coefficient of approximately 0.52 (52%) represents an extremely significant fitness cost for Δctb-1 bearing worms.
(e). Frequency of Δctb-1 declines with time in large populations
Four Δctb-1 bearing lines (1G.C., 1G.N, 1G.T and 1G.U) were maintained as large populations for 75 generations. The initial average frequency of the heteroplasmic Δctb-1 in these worms was 93.4% (figure 5). There was a slight, but significant, difference in the frequency of the heteroplasmic Δctb-1 between the lines at the beginning of their evolution in large populations (ANOVA, F = 11.07, p < 0.0001). At the lower end of the spectrum, 1G.U and 1G.T had the Δctb-1 mitotype in 91 and 92% frequency, respectively. At the higher end of the spectrum, the Δctb-1 mitotype in 1G.C and 1G.N was at 95 and 96% frequency, respectively. We additionally determined the frequencies of four additional heteroplasmies in these four 1G-derived lines after their mutant mtDNA genomes were sequestered in a WT nuclear background. The single T insertion heteroplasmy in nd5 in these experimental lines at the start of this experiment ranged from 13% to 24% with an average frequency of 20%. The frequency of the double T insertion heteroplasmy in nd5 in these experimental lines ranged from 67% to 74% with an average frequency of 70%. The combined frequencies the single and double T insertions at this site ranged from 87% to 93% with an average of 90%. The frequency of the nonsynonymous mutation heteroplasmy in nd5 ranged from 83% to 91% with an average frequency of 86%. Lastly, the base substitution in tRNA-Asn ranged from 0% to 17% with an average of 11% across the 1G-derived lines.
Figure 5.

A box plot of the Δctb-1 frequency across time in non-competed large populations of lines 1G.C, 1G.N, 1G.T and 1G.U. The average frequency of Δctb-1 is 93.4%, 93.2% and 74.0% at generation 0, 30 and 75, respectively. (Online version in colour.)
In the absence of worms homoplasmic for WT mtDNA, the frequency of Δctb-1 within lines initially appeared to be stable at high frequency. After 30 generations at large population size, the average frequency in all four lines remained above 90%, with an average of 93%. However, by generation 75, the average frequency of the heteroplasmic Δctb-1 mitotype had declined to 74% (two-way ANOVA, generation effect: F = 85.49, p < 0.0001). In addition to the decline in the average frequency of the heteroplasmic Δctb-1 mitotype in these populations, there was an increase in the variation in frequency of Δctb-1 between individuals within lines. The standard deviation in the frequency of Δctb-1 within the four populations at the beginning (generation 0) ranged from 1.3% to 3.8%, with an average of 2.7%. By generation 30, the average within-population standard deviation had increased slightly to 4.8% but was 14.8% by generation 75. The gradual increase in the within-population variation might explain why the frequency of the strongly deleterious Δctb-1 mitotype declines very slowly at first. The efficiency of selection was probably impeded in the early generations of these populations because there was insufficient variation in the frequency of Δctb-1 between individuals and hence, insufficient variation in fitness. As the population-level variation increased with time, so did the variation in fitness and subsequently, the reduction of Δctb-1 mitotypes.
The frequencies of the three additional heteroplasmies in nd5 (single T insertion, double T insertion and nonsynonymous mutation) showed similar decline as the Δctb-1 mutant after propagation at large population sizes over 75 generations. The average frequency of the nonsynonymous mutation declined to 70% and the combined frequency of the single and double T insertions was estimated to be at 77%. Interestingly, the frequency of the double TT insertion exhibited the greatest decline to 57%, whereas the frequency of the single T insertion was virtually unchanged at 20%. In addition, the low frequency tRNA-Asn mutant went extinct in each of the four large populations.
(f). mtDNA copy-number was elevated in some, but not all Δctb-1 bearing lines
Previous work has shown that the mtDNA copy-number is frequently elevated in individuals that are heteroplasmic for a mitochondrial gene deletion [36,49,50]. This increase in mtDNA copy-number is probably the result of compensatory mechanisms that cells resort to when their respirational demands are not being met [50]. After backcrossing the Δctb-1 mitochondrial genome into a WT nuclear background, four independently backcrossed lines were tested for mtDNA copy-number. Immediately following the backcrossing regime, two of these lines, 1G.C and 1G.N, had elevated mtDNA copies compared to control lines with WT mitochondria (generation 0; figure 6; one-way ANOVA: F = 6.25, p = 0.0003). The average mtDNA copy-number increase for lines 1G.C and 1G.N was 60% and 47%, respectively. By contrast, the total mtDNA copy-number was not elevated in lines 1G.T and 1G.U relative to WT mitochondria. Lines 1G.T and 1G.U also had a slightly lower frequency of Δctb-1, but perhaps more importantly, they had almost two times (1.93×) higher frequency of the WT mitochondrial genome. On average, lines 1G.T and 1G.U also appeared to perform better in the fitness-related traits assays (table 1).
Figure 6.

A box plot of the relative mtDNA copy-number across time in non-competed large populations of lines 1G.C, 1G.N, 1G.T and 1G.U as determined by ddPCR. The relative mtDNA copy-number for WT N2 mtDNA is displayed for reference. (Online version in colour.)
There was no significant change in mtDNA copy-number in the large populations by generation 30 (two-way ANOVA, generation effect: F = 1.17, p = 0.28). However, by generation 75, the total mtDNA copy-number in lines 1G.C and 1G.N had declined to WT levels. At that stage in the experiment, all four populations that were established as independently WT-backcrossed replicates of the original 1G line had total mtDNA copy-numbers indistinguishable from N2 worms with WT mitochondrial genomes (Tukey–Kramer HSD: ancestral control versus 1G.C, p = 0.27; ancestral control versus 1G.N, p = 1.0; ancestral control versus 1G.T, p = 0.64; ancestral control versus 1G.U, p = 1.0). The reduction in total mtDNA copy-number within these four replicate 1G lines with time could be an adaptation if the cost of increasing the total number of mitochondria is greater than the benefit from increasing the copy-number of WT mtDNA.
(g). Strength of correlation between Δctb-1 and wild-type mtDNA copy-number weakened with time
There is a significant positive correlation between Δctb-1 and WT mtDNA copy-number (r = 0.42, p = 0.001; figure 7a). This correlation may stem from two principal sources. The first source of this correlation could be the compensatory processes that increase mtDNA copy-number in response to mitochondrial dysfunction. In the event of a severe reduction of mitochondrial functions, such as would accompany a loss-of-function mutation in the mitochondrial genome, the cells might increase the replication of mtDNA indiscriminately, resulting in an increased copy-number of both normal and defective mtDNA genomes. The second potential source is experimental variation that could affect the numbers of mutant and WT mtDNA in the same manner. Although we made every effort to sample individual worms at the same stage of development, it is possible that there existed some experimental variation in the developmental stage of the worms at the time of analysis, which in turn resulted in variation in mtDNA copy-number and a positive correlation between mutant and WT mtDNA copy-number. The coefficient of variation (CV) for Δctb-1 and WT mtDNA copy-number across both populations and generations was 0.62 and 1.15, respectively. Calculating the CV separately within particular generations or populations does not change this conclusion. This suggests that the variation in Δctb-1 mtDNA copy-number was no greater than expected when its higher average copy-number was taken into account. Furthermore, we find no evidence that the variation in Δctb-1 mtDNA contributes disproportionally to the variation in total mtDNA copy-number as has been previously suggested [49].
Figure 7.

Relationship between the copy-number of Δctb-1 and WT mtDNA. (a) A scatterplot of the relationship between relative copy-number of Δctb-1 and WT mtDNA at generation 0 in large populations. The plot combines ddPCR results for lines 1G.C, 1G.N, 1G.T and 1G.U. There is a significant correlation between Δctb-1 and WT mtDNA copy-number at generation 0 (r = 0.42, p = 0.001). (b) Individual correlation coefficients between Δctb-1 and WT mtDNA copy-number as a function of time (number of generations) for non-competed populations of 1G.C, 1G.N, 1G.T and 1G.U. The strength of the correlation between Δctb-1 and WT mtDNA copy number declines with time (r = −0.69, p = 0.013). (Online version in colour.)
Within the four Δctb-1 bearing lines that were evolved at large population size for 75 generations, the strength of the correlation between Δctb-1 and WT copy-number declined with time (r = −0.69, p = 0.013) and had all but disappeared by generation 75 (figure 7b). If the correlation between Δctb-1 and WT mtDNA copy-number is primarily an artefact of an asynchronous developmental stage of the worm (second explanation suggested in the preceding paragraph), it should exert a similar effect at both generations 0 and 75, which was not observed. The weakening of the correlation with time is, however, consistent with our first hypothesis in the preceding paragraph that the reduction in total mtDNA copy-number is owing to adaptive mutations, presumably in the nuclear genome, that disable or reduce the compensatory increase in mtDNA replication in response to mitochondrial dysfunction. Alternatively, by generation 75, the frequency of Δctb-1 may have fallen below the levels that would trigger a compensatory increase in mtDNA copy-number. However, at that stage in the evolution of these heteroplasmic Δctb-1 populations, the Δctb-1 mitotype was still present in high frequency, 74% on average, and the number of WT mtDNA genomes was only a quarter of the numbers observed in homoplasmic WT worms.
(h). Δctb-1 is selfish
We conducted five generations of artificial selection for a reduction in the frequency of Δctb-1. The average heteroplasmy levels in eight worms that were descended from population 1G.T was 40%, with a range from 23 to 65%. We chose the descendants of an individual with a Δctb-1 frequency of 37%, which was the closest to the mean, to establish 25 independent replicate lines. These 25 lines were maintained by single-progeny descent each generation to maximize the strength of genetic drift and minimize the efficacy of natural selection at the inter-individual level. If the intra-individual population dynamics of the Δctb-1 mitotype are dominated by genetic drift, the changes in its frequency should be random. If there is selection against the Δctb-1 mitotype, its frequency should decline. Conversely, if the Δctb-1 mitotype has a transmission advantage despite its severe fitness cost at the individual level, its frequency should increase. After only six generations of single individual bottlenecks, there existed considerable variation between the 25 lines, with the frequency of Δctb-1 ranging from 23 to 81% (figure 8). However, only one of the 25 lines (4%) had a reduced Δctb-1 frequency. The average frequency of Δctb-1 rose to 54%, a significant increase from the original 37% (Student's: t = 5.9, p < 0.0001). Assuming that the change in Δctb-1 frequency over time stems from changes in the germline frequency of heteroplasmy rather than fluctuations in Δctb-1 in the somatic tissue, we conclude that Δctb-1 has a transmission advantage over WT mitochondria despite engendering a severe fitness cost at the individual level.
Figure 8.

The distribution of the frequency of Δctb-1 mtDNA in 25 lines after six generations of bottlenecking via single progeny descent. All 25 lines were descended from an individual with a Δctb-1 frequency of 37%, indicated by the dashed vertical line. The frequency of Δctb-1 mtDNA exhibited a rapid and significant increase (average 54%) under this regime of minimal selection between individuals.
4. Discussion
A spontaneous MA study in C. elegans with varying effective population sizes revealed that mitochondrial mutations predicted to be strongly deleterious reached high frequency within individual MA lines [44]. The focal mitochondrial mutations reached a higher frequency in MA lines with the most severe population bottlenecks (n = 1 individual) relative to larger-sized MA lines (n = 10 and 100 individuals), consistent with their detrimental effects on fitness. Specifically, two n = 1 MA lines (of a total 20 lines) possessed high-frequency mtDNA heteroplasmies comprising deletions of protein-coding or tRNA genes. In general, random genetic drift is assumed to be the major evolutionary force enabling the increase in frequency and/or fixation of deleterious mutations. This is especially pertinent in MA experiments, which typically involve passaging populations through severe genetic bottlenecks in order to maximize the effects of genetic drift. In the case of mitochondrial deletions, there is evidence that they can have a transmission advantage despite their serious consequences for the reproductive success of their hosts [33,36,37,51–53]. However, the mechanisms that maintain deleterious mitochondrial mutations in populations are still poorly understood.
In this study, we focused on one of several previously identified spontaneous mitochondrial deletions in our C. elegans MA lines [44], namely a high-frequency deletion in the ctb-1 gene of MA line 1G referred to as the Δctb-1 mitotype. As previously mentioned, the mtDNA genomes of line 1G also possessed two additional high-frequency heteroplasmies in addition to Δctb-1, namely a nonsynonymous base substitution and a frameshift mutation in the mtDNA gene, nd5. The primary objective of this study was to determine if this mutant mitotype, which contains all three high-frequency heteroplasmic mutations including Δctb-1 in addition to two low frequency mutations, is a selfish genetic element. We found no evidence for recombination between any of the mutations, given that the WT ctb-1 was always associated with the WT alleles of both nd5 and tRNA-Asn. For reasons explained earlier, we refer to this mutant mitotype as Δctb-1 or Δctb-1-bearing mtDNA. To qualify as a selfish genetic element, a particular mitotype should have negative or neutral effects on fitness in conjunction with a transmission advantage over other mitochondrial genotypes. A previous whole genome sequencing analysis of MA line 1G (in which the Δctb-1 deletion originated) suggested that at the termination of the MA experiment, an unprecedented 96% of the mtDNA genomes within individuals of this line contained this deletion [44]. This mutation frequency seemed extraordinarily high and is likely to be severely deleterious given that approximately 50% of the coding sequence was deleted while simultaneously altering the reading frame of the coding sequence downstream of the deletion. For comparison, two other mitochondrial deletions, uaDf5 and mptDf1, that were discovered in laboratory strains of C. elegans, are frequently in 60–80% frequency [36,49]. Herein, we demonstrate that in high frequency, the Δctb-1 mitotype is indeed severely deleterious for each of four assayed fitness-related traits. In this respect, the Δctb-1 mitotype is similar to deletions uaDf5 and mptDf1 that have been previously described in C. elegans, resulting in delayed development, reduced productivity, shorter lifespan and lowered survivorship to adulthood [36,38,49]. Furthermore, Δctb-1 bearing worms are rapidly driven to extinction when competed against WT worms. However, we are unable to delineate the specific contribution of each of the five linked mutations comprising the Δctb-1 mitotype to the composite 52% fitness decline. Indeed, one or more mutations may in fact be compensatory in nature. Irrespective, this compensatory role, if present, does not preclude or offset the substantial fitness cost to the worms bearing this mitotype.
Given that C. elegans is amenable to long-term cryopreservation, we were offered a unique opportunity to retroactively screen stocks of line 1G frozen at several time-intervals during the progression of the MA experiment and thereby determine the approximate timing of the origin of Δctb-1 and reconstruct its population trajectory over approximately 364 generations of experimental evolution. Δctb-1 first appeared between MA generations 25 and 50, and already existed at a 7% frequency by generation 51. Its rapid proliferation within line 1G is evident by its occurrence at 90% frequency by generation 90. Subsequently, mutations at two different sites in nd5 also reach high frequency. Given the strongly negative fitness effects associated with this mitotype, combined with the extreme genetic drift that dominates in MA experiments, such a rapid rise could, in principle, be owing to either random drift within the germline of this MA line, or a transmission advantage of the mutant mitochondrial genome. Although the frequency of Δctb-1 declined under directional selection, its frequency increased once again when selection for its reduction was relaxed. If genetic drift within the germline was the primary reason for the initial rapid proliferation of the Δctb-1 mitotype in the original MA experiment, we expect the frequency of Δctb-1 to change at random, with no significant net increase or decrease after we relaxed selection for lower abundance. Instead, our results suggest that the Δctb-1 mitotype has a replication or transmission advantage over WT mitochondria. Hence, our data demonstrating (i) strongly deleterious fitness consequences of the Δctb-1 mitotype, and (ii) its transmission advantage over WT mitochondria, together qualify it as a selfish genetic element.
The importance of large population size for the containment of selfish mtDNA has been noted in different species, including yeast [33] and C. briggsae [54]. In large populations, selection between individuals results in lower abundance of selfish mtDNA. Conversely, at small population size, selection within individuals results in greater abundance of selfish mtDNA. In our experiments comprising large, non-competed populations of the derivatives of line 1G over the course of 75 generations, the frequency of Δctb-1 initially exhibited a very slight decline but the rate of decline accelerated with time. In the initial stages of the experiment, when the frequency of Δctb-1 was near its maximum whereas that of the WT was near its minimum, the variation in the numbers of mutant and WT mitochondria generated each generation probably did not translate into large differences in fitness between individuals. Increasing variation in the frequencies of Δctb-1 and WT mtDNA with time concomitantly increases variation in fitness, resulting in greater efficiency of selection for individuals with reduced abundance of Δctb-1. Furthermore, as the frequencies of Δctb-1 and WT become more equal, random partitioning of mitochondria into eggs generates greater variation in the numbers of Δctb-1 and WT within individuals than when Δctb-1 was high. Subsequently, this engenders greater variation in fitness. It is currently unclear if Δctb-1 would have reached an equilibrium frequency owing to the opposing forces of individual and within-individual selection, or if it would continue to decline until the Δctb-1 mitotype became extinct. If there is a real threshold level below which detrimental mitochondria are selectively neutral, Δctb-1 might also persist in the populations until it is eventually lost by genetic drift.
An upregulation of mtDNA copy-number has been suggested to play a role in the proliferation of mutant mitochondrial genomes [49,50]. Although we observe an increase in mtDNA copy-number in some replicate lines of 1G, this was not apparent in other lines that contained the same Δctb-1 deletion in high frequency. There was a correlation between mutant and WT mtDNA quantity, which can be interpreted as the result of a nonspecific increase in mtDNA. That is, the mechanisms responsible for an increase in mtDNA do not discriminate between Δctb-1 and WT and generically increase total mtDNA copy-number. This interpretation is also supported by comparing the variation in the copy-numbers of Δctb-1 and WT mtDNA. If the increase in mtDNA is preferentially for the Δctb-1 mtDNA while WT mtDNA was tightly regulated, we would expect greater variation in Δctb-1 mtDNA between individuals and lines, as Δctb-1 mtDNA would increase disproportionately. Although Δctb-1 mtDNA did indeed exhibit greater variation, we found no significant differences in the CV between Δctb-1 and WT mtDNA. It appears that this greater variation in Δctb-1 is a function of a greater average number of Δctb-1 than WT mtDNA per individual.
Within the large, non-competed populations of line 1G replicates evolved for 75 generations, there was selection for a lower frequency of the Δctb-1 mitotype, but also an opportunity for compensatory evolution to mitigate the deleterious consequences of Δctb-1. During evolution in these large populations, the total mtDNA copy-number dropped to levels that are typical of the WT N2 control. Furthermore, the correlations between Δctb-1 and WT mtDNA were no longer significant after 75 generations, even though the average frequency of Δctb-1 remained fairly high, at 74%. It is possible that the frequency of the Δctb-1 mitotype had dropped to levels that did not stimulate a compensatory increase in mtDNA copy-number. However, it is also possible that the lack of compensatory increase in mtDNA is itself adaptive. For example, the increased investment in mitochondria may be too costly and generate too little return on the investment, so that an increase in mtDNA copy-number instead contributes to the fitness cost of the Δctb-1 mitotype.
Mitochondrial mutations contribute to a multitude of age-related and degenerative diseases and understanding the persistence and proliferation of deleterious mtDNA variants could provide new ways to target and treat their debilitating effects [55–57]. Furthermore, the sources of genetic conflict between the nuclear and mitochondrial genomes also have implications for the evolution of organellar genomes and the evolutionary history of eukaryotes. A key question remaining to be answered pertains to the mechanism by which mitochondrial deletions can have a competitive advantage within cells. One distinct feature of mitochondria is that they have highly reduced genome sizes, perhaps in order to increase their replication rate [58–60]. It had been proposed that deletion-bearing mtDNA molecules gain a selfish transmission advantage because they replicate more rapidly owing to their smaller size [61]. The experimental evidence for the hypothesis that shorter mitochondrial genomes are faster replicators is mixed. Some studies have demonstrated that shorter mitochondrial genomes can replicate more rapidly than larger, WT mtDNA genomes within the same cell [62,63]. Likewise, in vivo experiments in mouse neurons suggested that mitochondrial genomes with large deletions accumulate faster than genomes with small deletions [64]. This proposed characteristic of mitochondrial genomes could provide a transmission advantage for the Δctb-1 mitotype because they replicate more rapidly owing to their smaller size. However, a recent study in C. elegans found no empirical support for this ‘small-genome’ hypothesis [49] given that mtDNA heteroplasmies involving point mutations and smaller deletions show clonal expansion at a rate similar to that of large deletion-bearing mitochondria [65]. Rather, recent work in C. elegans suggest that mutant mtDNA molecules ‘hijack’ endogenous pathways of mitonuclear signalling to upregulate the mitochondrial unfolded protein response pathway to facilitate an increase in their copy-number [49,50].
Levels of mitochondrial heteroplasmy are the product of mutation and selection at different levels of organization. To address questions about replicative advantage of different mtDNA variants within individuals, it is imperative that one eliminate sources of confounding factors, such as selection between individuals. Experiments on the population dynamics of selfish mtDNA have revealed that their abundance can be very sensitive to population size [54]. One of the advantages of using single individual bottlenecks to study the dynamics of mitochondrial mutations is the near absence of selection between individuals, which can reveal the relative contributions of selection and drift within the germline.
Supplementary Material
Acknowledgements
We thank the editors, Fabrizio Ghiselli and Liliana Milani for extending an invitation to contribute to this special issue and for their helpful suggestions. This manuscript greatly benefitted from comments provided by two anonymous reviewers. We are grateful to John Jaenike and Suzanne Estes for their insightful comments and to Anke Konrad for contributing a figure.
Data accessibility
Fitness data values for four fitness-related traits are provided in the electronic supplementary material.
Competing interests
The authors declare no competing financial or other interests.
Funding
This research was partially supported by National Science Foundation grant no. MCB-1330245 to V.K. U.B. and V.K. were additionally supported by start-up funds from the Department of Veterinary Integrative Biosciences, College of Veterinary Medicine and Biomedical Sciences at Texas A&M University.
References
- 1.Kairo A, Fairlamb AH, Gobright E, Nene V. 1994. A 7.1 kb linear DNA molecule of Theileria parva has scrambled rDNA sequences and open reading frames for mitochondrially encoded proteins. EMBO J. 13, 898–905. ( 10.1002/j.1460-2075.1994.tb06333.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray MW. 2018. Mitochondrial genomes. In Molecular life sciences (eds Wells RD, Bond JS, Klinman J, Masters BSS), pp. 695–709. New York, NY: Springer. [Google Scholar]
- 3.Burton RS, Pereira RJ, Barreto FS. 2013. Cytonuclear genomic interactions and hybrid breakdown. Annu. Rev. Ecol. Evol. Syst. 44, 281–302. ( 10.1146/annurev-ecolsys-110512-135758) [DOI] [Google Scholar]
- 4.Chang CC, Rodriguez J, Ross J. 2016. Mitochondrial–nuclear epistasis impacts fitness and mitochondrial physiology of interpopulation Caenorhabditis briggsae hybrids. G3 (Bethesda) 6, 209–219. ( 10.1534/g3.115.022970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Telschow A, Gadau J, Werren JH, Kobayashi Y. 2019. Genetic incompatibilities between mitochondria and nuclear genes: effect on gene flow and speciation. Front. Genet. 10, 62 ( 10.3389/fgene.2019.00062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Havird JC, Hall MD, Dowling DK. 2015. The evolution of sex: a new hypothesis based on mitochondrial mutational erosion. Bioessays 37, 951–958. ( 10.1002/bies.201500057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radzvilavicius AL, Blackstone NW. 2015. Conflict and cooperation in eukaryogenesis: implications for the timing of endosymbiosis and the evolution of sex. J. R. Soc. Interface 12, 20150584 ( 10.1098/rsif.2015.0584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das J. 2006. The role of mitochondrial respiration in physiological and evolutionary adaptation. Bioessays 28, 890–901. ( 10.1002/bies.20463) [DOI] [PubMed] [Google Scholar]
- 9.da Fonseca RR, Johnson WE, O'Brien SJ, Ramos MJ, Antunes A.. 2008. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9, 119 ( 10.1186/1471-2164-9-119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott GR, Schulte PM, Egginton S, Scott AL, Richards JG, Milsom WK. 2011. Molecular evolution of cytochrome c oxidase underlies high-altitude adaptation in the bar-headed goose. Mol. Biol. Evol. 28, 351–363. ( 10.1093/molbev/msq205) [DOI] [PubMed] [Google Scholar]
- 11.Correia-Melo C, Birch J, Passos JF. 2016. Powering senescence: the ugly side of mitochondria. Cell Cycle 15, 2541–2542. ( 10.1080/15384101.2016.1204852) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korolchuk VI, Miwa S, Carroll B, von Zglinicki T.. 2017. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine 21, 7–13. ( 10.1016/j.ebiom.2017.03.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rand DM. 2001. The units of selection on mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 32, 415–448. ( 10.1146/annurev.ecolsys.32.081501.114109) [DOI] [Google Scholar]
- 14.Harrison RG, Rand DM, Wheeler WC. 1985. Mitochondrial DNA size variation within individual crickets. Science 228, 1446–1448. ( 10.1126/science.228.4706.1446) [DOI] [PubMed] [Google Scholar]
- 15.Kmiec B, Woloszynska M, Janska H. 2006. Heteroplasmy as a common state of mitochondrial genetic information in plants and animals. Curr. Genet. 50, 149–159. ( 10.1007/s00294-006-0082-1) [DOI] [PubMed] [Google Scholar]
- 16.Stewart JB, Chinnery PF. 2015. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. ( 10.1038/nrg3966). [DOI] [PubMed] [Google Scholar]
- 17.Karavaeva IE, Golyshev SA, Smirnova EA, Sokolov SS, Severin FF, Knorre DA. 2017. Mitochondrial depolarization in yeast zygotes inhibits clonal expansion of selfish mtDNA. J. Cell Sci. 130, 1274–1284. ( 10.1242/jcs.197269) [DOI] [PubMed] [Google Scholar]
- 18.Knorre DA. 2019. Intracellular quality control of mitochondrial DNA: evidence and limitations. Phil. Trans. R. Soc. B 375, 20190176 ( 10.1098/rstb.2019.0176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeviani M, Antozzi C. 1997. Mitochondrial disorders. Mol. Hum. Reprod. 3, 133–148. ( 10.1093/molehr/3.2.133) [DOI] [PubMed] [Google Scholar]
- 20.Wallace DC. 1999. Mitochondrial diseases in man and mouse. Science 283, 1482–1488. ( 10.1126/science.283.5407.1482) [DOI] [PubMed] [Google Scholar]
- 21.Keogh M, Chinnery PF. 2013. Hereditary mtDNA heteroplasmy: a baseline for aging? Cell Metab. 18, 463–464. ( 10.1016/j.cmet.2013.09.015) [DOI] [PubMed] [Google Scholar]
- 22.Wallace DC, Chalkia D. 2013. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 5, a021220 ( 10.1101/cshperspect.a021220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greaves LC, et al. 2014. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 10, e1004620 ( 10.1371/journal.pgen.1004620) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakshmanan LN, Yee Z, Ng LF, Gunawan R, Halliwell B, Gruber J. 2018. Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals. Aging Cell 17, e12814 ( 10.1111/acel.12814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurst GD, Werren JH. 2001. The role of selfish genetic elements in eukaryotic evolution. Nat. Rev. Genet. 2, 597–606. ( 10.1038/35084545) [DOI] [PubMed] [Google Scholar]
- 26.Clark KA, Howe DK, Gafner K, Kusuma D, Ping S, Estes S, Denver DR. 2012. Selfish little circles: transmission bias and evolution of large deletion-bearing mitochondrial DNA in Caenorhabditis briggsae nematodes. PLoS ONE 7, e41433 ( 10.1371/journal.pone.0041433) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma H, O'Farrell PH. 2016. Selfish drive can trump function when animal mitochondrial genomes compete. Nat. Genet. 48, 798–802. ( 10.1038/ng.3587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eberhard WG. 1980. Evolutionary consequences of intracellular organelle competition. Q. Rev. Biol. 55, 231–249. ( 10.1086/411855) [DOI] [PubMed] [Google Scholar]
- 29.Cosmides LM, Tooby J. 1981. Cytoplasmic inheritance and intragenomic conflict. J. Theor. Biol. 89, 83–129. ( 10.1016/0022-5193(81)90181-8) [DOI] [PubMed] [Google Scholar]
- 30.Barr CM, Neiman M, Taylor DR. 2005. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 168, 39–50. ( 10.1111/j.1469-8137.2005.01492.x) [DOI] [PubMed] [Google Scholar]
- 31.Schnabel PS, Wise RP. 1998. The molecular basis of cytoplasmic make sterility and fertility restoration. Trends Plant Sci. 3, 175–180. ( 10.1016/S1360-1385(98)01235-7) [DOI] [Google Scholar]
- 32.MacAlpine DM, Kolesar J, Okamoto K, Butow RA, Perlman PS. 2001. Replication and preferential inheritance of hypersuppressive petite mitochondrial DNA. EMBO J. 20, 1807–1817. ( 10.1093/emboj/20.7.1807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor DR, Zeyl C, Cooke E. 2002. Conflicting levels of selection in the accumulation of mitochondrial defects in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 99, 3690–3694. ( 10.1073/pnas.072660299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jasmin JN, Zeyl C. 2014. Rapid evolution of cheating mitochondrial genomes in small yeast populations. Evolution 68, 269–275. ( 10.1111/evo.12228) [DOI] [PubMed] [Google Scholar]
- 35.Poulton J, Deadman ME, Ramacharan S, Gardiner RM. 1991. Germ-line deletions of mtDNA in mitochondrial myopathy. Am. J. Hum. Genet. 48, 649–653. [PMC free article] [PubMed] [Google Scholar]
- 36.Tsang WY, Lemire BD. 2002. Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem. Cell Biol. 80, 645–654. ( 10.1139/o02-135) [DOI] [PubMed] [Google Scholar]
- 37.Howe DK, Denver DR. 2008. Muller's Ratchet and compensatory mutation in Caenorhabditis briggsae mitochondrial genome evolution. BMC Evol. Biol. 8, 62 ( 10.1186/1471-2148-8-62) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liau WS, Gonzalez-Serricchio AS, Deshommes C, Chin K, LaMunyon CW. 2007. A persistent mitochondrial deletion reduces fitness and sperm performance in heteroplasmic populations of C. elegans. BMC Genet. 8, 8 ( 10.1186/1471-2156-8-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Estes S, Coleman-Hulbert AL, Hicks KA, de Haan G, Martha SR, Knapp JB, Smith SW, Stein KC, Denver DR. 2011. Natural variation in life history and aging phenotypes is associated with mitochondrial DNA deletion frequency in Caenorhabditis briggsae. BMC Evol. Biol. 11, 11 ( 10.1186/1471-2148-11-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayashi J, Ohta S, Kikuchi A, Takemitsu M, Goto Y, Nonaka I. 1991. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl Acad. Sci. USA 88, 10 614–10 618. ( 10.1073/pnas.88.23.10614) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porteous WK, et al. 1998. Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur. J. Biochem. 257, 192–201. ( 10.1046/j.1432-1327.1998.2570192.x) [DOI] [PubMed] [Google Scholar]
- 42.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. 2003. Mitochondrial threshold effects. Biochem. J. 370, 751–762. ( 10.1042/BJ20021594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. 1990. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 61, 931–937. ( 10.1016/0092-8674(90)90059-N) [DOI] [PubMed] [Google Scholar]
- 44.Konrad A, Thompson O, Waterston RH, Moerman DG, Keightley PD, Bergthorsson U, Katju V. 2017. Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol. Biol. Evol. 34, 1319–1334. ( 10.1093/molbev/msx051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katju V, Packard LB, Bu L, Keightley PD, Bergthorsson U. 2015. Fitness decline in spontaneous mutation accumulation lines of Caenorhabditis elegans with varying effective population sizes. Evolution 69, 104–116. ( 10.1111/evo.12554) [DOI] [PubMed] [Google Scholar]
- 46.Katju V, Packard LB, Keightley PD. 2018. Fitness decline under osmotic stress in Caenorhabditis elegans populations subjected to spontaneous mutation accumulation at varying population sizes. Evolution 72, 1000–1008. ( 10.1111/evo.13463) [DOI] [PubMed] [Google Scholar]
- 47.Lee Y, Hwang W, Jung J, Park S, Cabatbat JJ, Kim PJ, Lee SJ. 2016. Inverse correlation between longevity and developmental rate among wild C. elegans strains. Aging 8, 986–999. ( 10.18632/aging.100960) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR. 1992. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics 130, 471–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gitschlag BL, Kirby CS, Samuels DC, Gangula RD, Mallal SA, Patel MR. 2016. Homeostatic responses regulate selfish mitochondrial genome dynamics in C. elegans. Cell Metab. 24, 91–103. ( 10.1016/j.cmet.2016.06.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM. 2016. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. ( 10.1038/nature17989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ephrussi B, de Margerie-Hottinguer H, Roman H. 1955. Suppressiveness: a new factor in the genetic determinism of the synthesis of respiratory enzymes in yeast. Proc. Natl Acad. Sci. USA 41, 1065–1071. ( 10.1073/pnas.41.12.1065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernardi G. 1979. The petite mutation in yeast. Trends Biochem. Sci. 4, 197–201. ( 10.1016/0968-0004(79)90079-3) [DOI] [Google Scholar]
- 53.Beziat F, Morel F, Volz-Lingenhol A, Saint Paul N, Alziari S. 1993. Mitochondrial genome expression in a mutant strain of D. subobscura, an animal model for large scale mtDNA deletion. Nucleic Acids Res. 21, 387–392. ( 10.1093/nar/21.3.387) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips WS, Coleman-Hulbert AL, Weiss ES, Howe DK, Ping S, Wernick RI, Estes S, Denver DR. 2015. Selfish mitochondrial DNA proliferates and diversifies in small, but not large, experimental populations of Caenorhabditis briggsae. Genome Biol. Evol. 7, 2023–2037. ( 10.1093/gbe/evv116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ross JM, et al. 2013. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415. ( 10.1038/nature12474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF. 2014. Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 10, e1004369 ( 10.1371/journal.pgen.1004369) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinto M, Moraes CT. 2015. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 85, 250–258. ( 10.1016/j.freeradbiomed.2015.05.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallace DC. 1982. Structure and evolution of organelle genomes. Microbiol. Rev. 46, 208–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lang BF, Gray MW, Burger G. 1999. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. ( 10.1146/annurev.genet.33.1.351) [DOI] [PubMed] [Google Scholar]
- 60.Andersson SG, et al. 1998. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140. ( 10.1038/24094) [DOI] [PubMed] [Google Scholar]
- 61.Wallace DC. 1992. Mitochondrial genetics: a paradigm for aging and degenerative diseases. Science 256, 628–632. ( 10.1126/science.1533953) [DOI] [PubMed] [Google Scholar]
- 62.Tang Y, Manfredi G, Hirano M, Schon EA. 2000. Maintenance of human rearranged mitochondrial DNAs in long-term cultured transmitochondrial cell lines. Mol. Biol. Cell 11, 2349–2358. ( 10.1091/mbc.11.7.2349) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Diaz F, Bayona-Bafaluy MP, Rana M, Mora M, Hao H, Moraes CT. 2002. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 30, 4626–4633. ( 10.1093/nar/gkf602) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukui H, Moraes CT. 2009. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 18, 1028–1036. ( 10.1093/hmg/ddn437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Campbell G, Krishnan KJ, Deschauer M, Taylor RW, Turnbull DM. 2014. Dissecting the mechanisms underlying the accumulation of mitochondrial DNA deletions in human skeletal muscle. Hum. Mol. Genet. 23, 4612–4620. ( 10.1093/hmg/ddu176) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Fitness data values for four fitness-related traits are provided in the electronic supplementary material.