

Abstract
Consistent induction of donor‐specific unresponsiveness in the absence of continuous immunosuppressive therapy and toxic effects remains a difficult task in clinical organ transplantation. Transplant immunologists have developed numerous experimental treatments that target antigen‐presentation (signal 1), costimulation (signal 2), and cytokine production (signal 3) to establish transplantation tolerance. While promising results have been obtained using therapeutic approaches that predominantly target the adaptive immune response, the long‐term graft survival rates remain suboptimal. This suggests the existence of unrecognized allograft rejection mechanisms that contribute to organ failure. We postulate that trained immunity stimulatory pathways are critical to the immune response that mediates graft loss. Trained immunity is a recently discovered functional program of the innate immune system, which is characterized by nonpermanent epigenetic and metabolic reprogramming of macrophages. Since trained macrophages upregulate costimulatory molecules (signal 2) and produce pro‐inflammatory cytokines (signal 3), they contribute to potent graft reactive immune responses and organ transplant rejection. In this review, we summarize the detrimental effects of trained immunity in the context of organ transplantation and describe pathways that induce macrophage training associated with graft rejection.
Keywords: immunobiology, immunosuppression/immune modulation, infection and infectious agents, infectious disease, macrophage/monocyte biology: activation, rejection, tolerance: mechanisms, translational research/science
Short abstract
Ochando and colleagues describe the detrimental effects of trained immunity in organ transplantation and review mechanistic pathways that induce macrophage training associated with graft rejection.
Abbreviations
- BCG
Bacille Calmette Guérin
- CMV
Cytomegalovirus chronic infection model
- DAMP
Damage associated molecular pattern
- HMGB1
High mobility group box 1
- HMG‐CoA
Hydroxy‐methyl‐glutaryl coenzyme A
- IL6
Interleukin 6
- LDL
Low‐density lipoprotein
- mTOR
Mammalian target of rapamycin
- NLRP3
NOD like receptor pyrin domain containing 3
- NOD2
Nod‐like receptor 2
- OxLDL
Oxidized low density lipoprotein
- PRR
Pattern recognition receptor
- TLR4
Toll like receptor 4
- TNFα
Tumor necrosis factor α
- TRAF6
Tumor‐necrosis factor receptor–associated factor 6
1. INTRODUCTION
Transplantation is a therapeutic option that prolongs the life of thousands of patients with terminal organ failure. Patients who find a compatible donor and receive a transplant are normally treated daily with immunosuppressive drugs that inhibit the activated immune system. The immune response occurs in 2 different phases and is mediated by different cells. The first phase of the immune response is mediated by the innate immune system. This response has a very rapid time of onset and is mediated by myeloid cells, including macrophages, neutrophils, and dendritic cells. The second phase of the immune response is the adaptive response. This response occurs after the innate response, has a slower time of onset, and is mediated by lymphoid cells, including T and B lymphocytes. Conventional immunosuppressive therapies have only focused on the adaptive immune response. This is in part due to studies carried out by Miller in 1961, who demonstrated that T lymphocytes are necessary and sufficient to induce organ transplant rejection of skin allografts.1 Further confirmation of these results came from Pantelouris, who used recipient “nude” mice lacking T and B lymphocytes2 to confirm that the adaptive immune system mediates allograft rejection.3 Based on these studies and others, several laboratories aimed at preventing rejection by blocking T cell activation. This was accomplished by preventing signal 1 (antigen presentation), 2 (costimulation), and/or 3 (cytokine production), which are all required to effectively prime T cells.4 Although promising results were achieved through the use of either antagonistic monoclonal antibodies or agonistic immunoglobulins,5 the long‐term transplant survival rates remain suboptimal,6 underscoring the need for additional approaches to regulate the immune response.
Recent advances have highlighted the critical role of the innate immune system in initiating the immune response against the transplanted allograft7, 8 and mediating late, chronic rejection.9 These investigations are consistent with clinical data from more than 3 decades ago showing that macrophages represent the majority of cells in the transplanted organ during episodes of severe rejection.10 More recent data demonstrated that T cell–depleted human renal allograft recipients experience rejection characterized by infiltration of the allograft with monocytes and macrophages,11 confirming that human monocytes initiate the alloimmune response.12 Despite progress in understanding the pathways by which macrophages promote transplant rejection,13 the mechanisms by which these innate immune cells mediate the loss of the transplanted organ are not fully understood. Deciphering macrophage activation pathways that promote allograft immunity will promote the development of novel therapeutic paradigms to prevent allograft rejection.
Macrophages are heterogeneous cells, whose phenotype and function are regulated by the microenvironment. Macrophage polarization refers to the process by which macrophages adopt a particular phenotypic and functional program (M1/M2) in response to specific signals such as interleukins, interferons, colony‐stimulatory factors, and tumor necrosis factors.14 Polarized macrophages undergo epigenetic reprogramming, which leads to specific transcriptional changes associated with the secretion of effector molecules.15 Using an experimental mouse model of cardiac transplantation, we reported that macrophage polarization determines the outcome of the immune response. Graft‐infiltrating monocytes adopt an M1 pro‐inflammatory phenotype associated with organ rejection, while early macrophage polarization toward an M2 anti‐inflammatory state is characteristic of tolerance induction.16, 17 Li's laboratory further extended these findings to report that macrophage polarization into M1 and M2 subsets was dependent on tumor‐necrosis factor receptor–associated factor 6 (TRAF6) and mammalian target of rapamycin (mTOR), respectively.18 While mice deficient for TRAF6 in macrophages were defective in M1 polarization and developed severe transplant vasculopathy, deletion of mTOR from macrophages resulted in long‐term allograft survival without histological indications of chronic rejection, emphasizing the role of M2‐polarized macrophages for chronic allograft rejection.
More recently, we reported a previously unrecognized pathway of allograft rejection associated with macrophage activation.19 This novel mechanism involves long‐term functional reprogramming of myeloid cells and has been termed “trained immunity.” Trained immunity refers to the ability of innate immune cells to switch and maintain their functional, transcriptional, epigenetic, and metabolic programs after the engagement of specific pattern recognition receptors (PRR). This property explains classic epidemiological observations of vaccines, such as Bacille Calmette‐Guérin (BCG), to provide protection not only against the target disease (Mycobacterium tuberculosis), but also against other infections (Staphylococcus aureus and Candida albicans) and cancer.20 The term was originally defined as a form of “memory” of innate immunity in which myeloid cells, once exposed to certain ligands of infectious agents, undergo epigenetic and metabolic changes, enabling them to generate stronger and more effective subsequent protective immune responses to new infections. Therefore, trained immunity involves secondary stimulation of previously polarized macrophages that are activated and trained through specific signaling pathways (Figure 1). The long‐lasting effects of this protective immunity are explained by nonpermanent histone modifications. Histones are proteins in the cell nuclei that are highly conserved across species around which DNA winds and histone modifications determine the accessibility of genes for transcription during the immune response. While the precise histone modifications that occur in trained macrophages are currently under investigation, the addition of 3 methyl groups to the lysine 4 on the histone H3 protein (H3K4me3) and the acetylation of lysine 27 on histone 3 (H3K27Ac) correlate with BCG‐induced trained immunity. The training hypothesis extends the concept of macrophage polarization to include secondary spatiotemporal stimulation associated with a greater pro‐inflammatory response.
Figure 1.
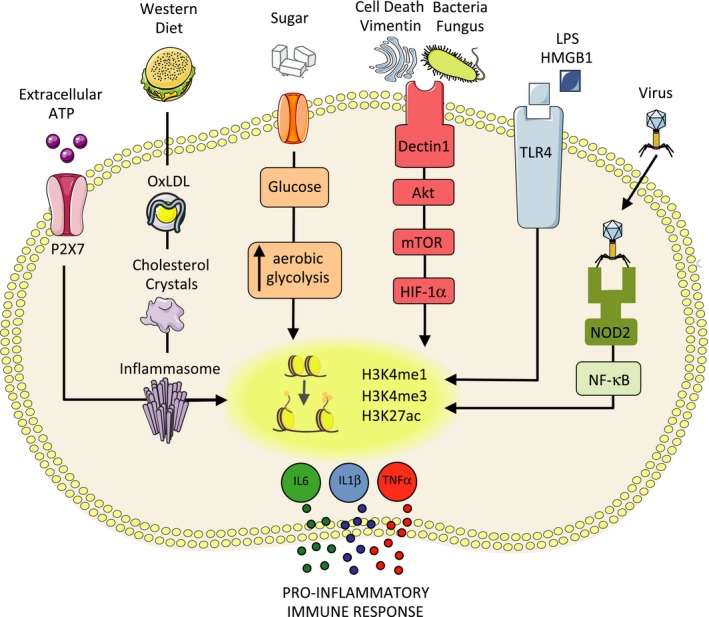
Trained immunity danger signals that compromise organ transplantation. Trained immunity–inducing agents, such as infection (viral, bacterial, and fungical), activation of the NLRP3 inflammasome, Western diet, sugar, OxLDL, and cell death are associated with increased morbidity and mortality in organ transplant patients. HMGB1, high mobility group box 1; IL, interleukin; mTOR, mammalian target of rapamycin; NOD2, Nod‐like receptor 2; NLRP, NOD‐like receptor pyrin domain‐containing‐3; OxLDL, oxidized low‐density lipoprotein; TLR4, Toll‐like receptor 4; TNFα, tumor necrosis factor α
In the context of organ transplantation, damage‐associated molecular patterns (DAMPs) are released in the donor organ during ischemia‐reperfusion injury and bind to trained immunity‐associated PRR. Consequently, these trained macrophages secrete higher quantities of pro‐inflammatory cytokines and more successfully activate the adaptive immune system than untrained macrophages, leading to allograft rejection.19 This may affect the long‐term function of the transplanted organ, as the immune protective function mediated by trained immunity is preserved for months in patients.21 Although trained immunity has not been demonstrated in transplant patients, macrophages with the potential to contribute to allograft rejection may be trained through signaling pathways that involve (1) vimentin/HMGB1, (2) infection (NOD2), (3) oxidized low‐density lipoprotein (OxLDL), and (4) the NLRP3‐inflammasome (Figure 1). Here, we describe the potential implications of these trained immunity pathways in the context of organ transplantation.
1.1. Vimentin and HMGB1
Macrophages adopt a long‐term pro‐inflammatory phenotype after stimulation through dectin‐1, which results in a nonspecific memory mediated by nonpermanent epigenetic reprogramming.22 Dectin‐1‐stimulated macrophages become hyperresponsive to stimulation (are “trained”), and they upregulate their glucose metabolism while increasing their lactate production. Following a second stimulation through Toll‐like receptor 4 (TLR4), trained macrophages produce high levels of pro‐inflammatory cytokines, such as interleukin 6 (IL‐6) and tumor necrosis factor α (TNFα),20 which are associated with organ transplant rejection.23
In the context of solid organ transplantation, dectin‐1‐expressing macrophages bind to vimentin, an endogenous protein involved in excisional wound healing.24 Vimentin is a dectin‐1 ligand 25 that is upregulated in the donor organ after transplantation and induces macrophage training early after transplantation and is associated with acute rejection.19 In line with our results, Azimzadeh and colleagues have previously demonstrated that vimentin is upregulated in nonhuman primate cardiac allografts during acute and chronic rejection.26 Since apoptosis has been detected in acute and chronic rejection 27 and vimentin is exposed on the surface of apoptotic cells,28 Azimzadeh and colleagues hypothesized that apoptotic cells in the donor allograft expose immunogenic vimentin to the immune surveillance system to activate various pro‐inflammatory effector mechanisms. We additionally hypothesize that dectin‐‐expressing macrophages, which are found in proximity to apoptotic tissue during rejection,29 become trained upon recognition of vimentin under sterile inflammatory conditions and secrete inflammatory cytokines that promote allograft rejection. Supporting this view, Rose and colleagues demonstrated accelerated rejection of cardiac allografts in vimentin‐immunized mice.30
Besides apoptosis, other danger signals released from dying cells in the donor allograft induce metabolic changes in innate immune cells when released into the extracellular compartment.31 Necrosis is induced during ischemia reperfusion injury (IRI)32 and necrotic cells release high mobility group box 1 (HMGB1) protein that mediates inflammation.33 Release of HMGB1 is induced in cardiac allografts during acute rejection, which is associated with an increase of the pro‐inflammatory cytokines IL6 and TNFα that are secreted by allograft‐infiltrated macrophages.34, 35 Therefore, HMGB1 released from necrotic cells during IRI promotes the secretion of the pro‐inflammatory cytokines by activated macrophages in the donor allograft. Upregulation of HMGB1 in the cardiac allografts may persist for at least 8 weeks posttransplantation, and is associated with accumulation of inflammatory CD11b+Ly6Chi myeloid cells that mediate chronic allograft rejection.36 These results suggest that HMGB1 represents a detrimental pro‐inflammatory signal after organ transplantation that is implicated in both acute and chronic rejection. To mediate its biological effects, HMGB1 binds to TLR4 in macrophages, which leads to the production of pro‐inflammatory cytokines.37 As discussed above, engagement of TLR4 induces epigenetic changes in trained macrophages that lead to pro‐inflammatory cytokine production.20 We therefore hypothesize that HMGB1‐mediated engagement of TLR4 induces training in graft‐infiltrating macrophages. Consistent with this view, Valdes‐Ferrer and colleagues observed that mouse splenocytes treated with HMGB1 produced higher TNFα when challenged with the TRL4 agonist lipopolysaccharide as a secondary inflammatory signal.38 On the contrary, we reported that the HMGB1‐TLR4 interaction favors the induction of transplantation tolerance under costimulatory blockade with anti‐CD40L mAb through the secretion of anti‐inflammatory IL‐10 by graft‐infiltrating macrophages.17 This suggests that the same endogenous DAMPs, such as HMGB1, may participate in both pro‐inflammatory and immune regulatory responses depending on the specific micro‐environmental stimuli. In addition, the same DAMP may bind through multiple receptors that trigger different immunological outcomes. In this respect, the danger signal HMGB1 secreted from necrotic cells was shown to associate with CD24 and Siglec‐G to reduce activation of nuclear factor κB and limit the inflammatory response.39 Thus, molecules that activate danger signaling pathways that trigger immune responses may also participate in the negative regulation of TLR responses as a control for excessive inflammation. Moreover, the same receptor that participates in training, such as dectin‐1, may exert opposing immunological functions according to the specific cell type that expresses this C‐type lectin‐like receptor.40 The above studies provide critical information for the design of future therapies that negatively regulate trained immunity. We conclude that graft‐infiltrating macrophages expressing dectin‐1 and TLR4 are trained by vimentin and HMGB1 in the donor allograft under sterile inflammatory conditions that occur during organ transplantation. These trained macrophages may represent an overlooked mechanism of allograft rejection that is not impacted by current clinical immunosuppressive regimens.
1.2. Infection and NOD2
Multiple pathways induce epigenetic and metabolic innate immune memory.41 Early studies demonstrated that the BCG induces Nod‐like receptor 2 (NOD2)‐dependent nonspecific protection to secondary infections, which occurs via epigenetic reprogramming of monocytes.42 Trained macrophages were shown to increase the trimethylation of histone H3 at lysine (H3K4me3) at the IL‐6 and TNFα promoters, which were associated with an increased secretion of these pro‐inflammatory cytokines. NOD2 is an intracellular PRR and represents a general sensor for the bacteria cell wall component muramyl dipeptide43 and viral RNA.44 This has critical impact in solid organ transplantation because immunosuppressed recipients face continuous risk of infections that are associated with graft rejection episodes.45 This is, in part, due to heterologous immunity, which describes the process by which T cell immunity to a previously encountered viral infection is directed against an organ allograft by the recognition of cross‐reactive allogeneic MHC complexes and which represents a significant barrier to tolerance induction.46
Using an experimental murine model of bacterial infection, elegant studies by Chong and colleagues demonstrated that infection with S. aureus at the time of transplantation prevents the induction of skin allograft acceptance induced by costimulatory blockade.47 Rejection was dependent on innate immune recognition and IL‐6 signaling, since MyD88‐, RAG‐, or IL‐6‐deficient recipients prolonged their skin allograft survival despite infection. Interestingly, T cell–directed therapy with either cyclosporine or sirolimus was unable to prevent graft loss, suggesting a critical role for the innate immune system in response to bacterial infection in transplant recipients. Previous work demonstrated that trained macrophages produce large amounts of pro‐inflammatory cytokines upon restimulation with S. aureus.48 While macrophages mediate potent cytokine responses upon NOD2 recognition of S. aureus,49 which confers nonspecific protection to secondary infections, this represents a potential risk in the context of solid organ transplantation, because an excessive inflammatory immune response may lead to graft loss. Similar results were obtained by Chong and colleagues using the intracellular enteric pathogenic bacteria Listeria monocytogenes. NOD2 is also involved in sensing intracellular L. monocytogenes,50 and infection with L. monocytogenes at the time of transplantation prevented both skin and heart allograft tolerance induced by costimulatory blockade.51
As mentioned above, heterologous immunity refers to the immunity that can develop to one antigen after the host has had exposure to a different antigen through cross‐reactivity. Heterologous immunity has been well described as a potent barrier to transplantation tolerance regimens in animal models, since viral‐specific T cells are cross‐reactive with alloantigen.46 There are few murine models of human chronic viral infections. Of the few existing models, the murine cytomegalovirus chronic infection model (mCMV) provides an excellent platform for trying to understand the pathology and graft loss associated with viral infections, which are manifested mainly in immunosuppressed hosts.52 Given that CMV activates NOD253 and that mCMV DNA is present only in cells of the myeloid lineage of latently infected mice,54 we hypothesize that mCMV infection may induce cytokine production by trained macrophages, such as IL‐6, which participate in the stimulation of viral‐specific T cells and/or antibody‐mediated responses that may cross‐react with the allograft in transplant recipients.55, 56, 57 Supporting this hypothesis, latent infection with mCMV prior to transplantation prevented the induction of prolonged allograft survival in heart transplant recipients.58 We argue that infectious agents represent a risk factor in organ transplant rejection due to the mechanisms associated with trained immunity‐mediated immune responses.
1.3. Oxidized low‐density lipoprotein and the NLRP3‐inflammasome
Monocytes primed with OxLDL switch to glycolysis and exhibit increased pro‐inflammatory cytokine production upon restimulation.59 OxLDL is DAMP that binds to the receptor CD36 expressed in myeloid cells and induces trained immunity.60 This represents a risk in organ transplantation because OxLDL present in transplant recipients is associated with an increased probability of graft rejection.61
A vast majority of transplant patients (40%‐80%) are reported to have hyperlipidemia.62 Circulating low‐density lipoprotein (LDL) is enhanced in transplant patients and high LDL content is associated with increased susceptibility to LDL oxidation.63, 64 Consequently, kidney transplant recipients exhibit increased levels of OxLDL after transplantation.65 OxLDL has been associated with development of chronic rejection in transplant recipients66 and represents a prognostic marker of transplant‐associated chronic allograft nephropathy and coronary artery disease.67, 68 Mechanistically, OxLDL promotes transplant interstitial fibrosis and arteriosclerosis through the stimulation of collagen production69, 70 and the development of autoantibodies.71, 72 This has a critical impact on the management of transplant patients because cyclosporin A is one of the most widely used immunosuppressive agents in organ transplantation, but at blood levels >100 ng/mL it is associated with increased OxLDL levels in kidney transplant recipients.73 This argues for the use of alternative immunosuppressive agents, such as tacrolimus or azathioprine, to reduce the OxLDL levels in these patients.74, 75 Additionally, inhibitors of the hydroxy‐methyl‐glutaryl coenzyme A (HMG‐CoA) reductase, the rate‐limiting step in cholesterol biosynthesis, are used to lower the serum levels of LDL and reduce the damage caused by OxLDL, such as transplant atherosclerosis.76, 77 Interestingly, inhibitors of HMG‐CoA have been shown to prevent trained immunity by epigenetic reprograming of macrophages through downregulation of H3K4me378 and to reduce vessel wall inflammation in atherosclerotic mice.79 However, in a therapeutic setting, statins are not able to reverse induction of trained immunity.80 This suggests that OxLDL induces trained macrophages that secrete pro‐inflammatory cytokines60 that may be involved in the development of atherosclerosis during chronic rejection.81 The dual effect of OxLDL may be explained by the NOD‐like receptor pyrin domain‐containing‐3 (NLRP3), which plays a pivotal function in distinct immunological scenarios. On the one hand, a Western‐type diet increases the OxLDL levels that favor the formation of cholesterol crystals and promote trained immunity through NLRP3 activation. This results in a long‐lasting inflammatory response characterized by release of IL‐1β and other pro‐inflammatory cytokines.82, 83 On the other hand, internalization of OxLDL following CD36 engagement leads to the formation of cholesterol crystals that activate the NLRP3 inflammasome in macrophages that promote atherogenesis.84 Therefore, it is plausible to hypothesize that trained macrophages contribute to allograft fibrosis and arteriosclerosis, which promote chronic rejection.
NLRP3 inflammasome is also triggered by necrotic cells.85 Different forms of cell death, including necrosis, are upregulated in the donor allograft due to ischemia‐reperfusion injury during the organ transplant procedure.86 This suggests that trained immunity may be triggered by graft‐infiltrating macrophages that encounter NLRP3 derived from necrotic cells under sterile inflammatory conditions. Additionally, recent data have documented that extracellular release of ATP from apoptotic cells attracts phagocytes.87 Using an experimental model of skin transplantation, Pelegrin and colleagues demonstrated that extracellular ATP represents a danger signal that contributes to allograft rejection. More specifically, the study revealed that under allogeneic transplant conditions, large amounts of ATP bound to the macrophage cell surface purinergic receptor P2X7. Activation of P2X7 led to the production of the pro‐inflammatory cytokines through NLRP3‐dependent pathways that were associated with graft rejection. Interestingly, the study demonstrated that P2X7 blockade decreased extracellular ATP concentrations and increased graft survival.88 We postulate that activation of the NLRP3 inflammasome through Western diet and cell death (discussed above) represents a potential risk for organ rejection in transplant patients because all these pathways have been demonstrated to induce macrophage training and promote potent immune responses.41
2. DISCUSSION
While lifelong immunosuppressive therapy has dramatically improved the short‐term results of organ transplantation, their side effects and toxicity compromise long‐term outcomes.89 As a result, the median survival time of a transplanted kidney from a deceased donor is 10 years.90 The pressing need for less toxic and more specific transplantation therapeutic treatment represents an elusive goal in organ transplantation. To prevent metabolic toxicity and other undesirable side effects of continuous immunosuppression,91, 92 novel therapeutic therapies that target the adaptive immune response are currently being evaluated in the clinic. These approaches include costimulatory blockade,93 lymphodepletion,94 in vivo induction of regulatory T cells,95 and bone marrow chimerism.96, 97 While promising results have been obtained using these methodologies, the induction of consistent, toxicity‐free, donor‐specific unresponsiveness remains elusive98 and underscores the need for novel approaches in the development of tolerance‐inducing protocols.
While most therapeutic approaches for the induction of indefinite allograft survival aim at targeting the adaptive immune response, recent data from the Fadi Lakkis laboratory demonstrated that the innate immune system initiates allogeneic nonself recognition and graft rejection. The study demonstrated that monocytes recall cell‐associated primary immunization and are able to mount an immune response up to 4 weeks after the initial stimuli, demonstrating that monocytes exhibit features of immune memory.8 Similarly, trained immunity is characterized by nonpermanent epigenetic reprogramming of macrophages that persists for weeks‐to‐months after the elimination of the initial stimulus.20, 22 This form of inflammation was originally described as a defense mechanism against nonself‐derived exogenous bacterial‐ and fungal‐derived components that represent pathogen‐associated molecular patterns (PAMPs). This ancient mechanism of immunological defense is also triggered by DAMPs that are released during tissue injury. Both PAMPs and DAMPs are recognized through PRRs expressed by macrophages, which activate the immune response leading to increased inflammatory cytokine production.
Inflammation compromises the outcome of the transplanted organ, and inflammatory responses associated with the innate immune system may have been historically underappreciated. This may be due to the accepted dogma that suggests that the innate immune system does not recall an immune response and does not have antigen specificity. However, early studies from William Hildermann and colleagues demonstrated transplantation immunity with a specific memory component in lower invertebrates that lack adaptive immunity. Second set of transplants is rejected significantly faster than first‐set transplants in corals (MST 22.0 first vs 11.6 second transplant) and in sponges (MST 9.0 first vs 3.8 second transplant), demonstrating that specific memory was induced upon secondary contact.99, 100 Interestingly, when duration of the alloimmune memory was evaluated by second sets of grafting pairs at different time points, reactivity was shown to fade considerably after 4 weeks of the first transplant. This suggests that the innate immune memory, which mediates organ transplant rejection, is not permanent. These studies are in line with the concept of trained immunity, which argues in favor of nonpermanent epigenetic remodeling of the innate immune system as a critical component of alloimmune memory events. Consistent with this view, Thomson and colleagues demonstrated that second‐set murine liver transplants were rejected by nonparenchymal MHC class II+ cells of hematopoietic origin (presumptive dendritic cells),101 and recent data from Li’s laboratory demonstrated that macrophages reject allogeneic cells in presensitized hosts.102 Collectively, the data confirm the traditional role of macrophages as inflammatory cells and further expand the mechanistic view by which macrophages contribute to the immunological memory. While trained immunity is associated with a negative outcome (rejection), it is important to highlight that epigenetic modifications also promote immune regulatory function of macrophages that may favor a positive outcome (graft survival).103 Moreover, immunological memory may also be associated with tolerance, as data from Chong’s laboratory demonstrated that transplantation tolerance is restored following infection‐mediated acute rejection, suggesting that memory of allograft tolerance dominates over the memory of transplant rejection.104
Immune metabolic pathways are associated with epigenetic rewiring of the innate immune system and trained immunity.78 At the center of different metabolic pathways associated with trained immunity, the mTOR plays a critical role. Prevention of transplant rejection can be achieved by daily rapamycin treatment, but its systemic administration affects various different cell types including podocytes, epithelial cells, endothelial cells, myeloid cells, B cells, T cells, NK, and NKT cells.105 In addition, poor water solubility and low bioavailability of rapamycin makes its systemic administration difficult106 and leads to life‐threatening toxic effects when used chronically in transplant recipient patients. To inhibit trained immunity and at the same time prevent the detrimental effects of lifelong rapamycin therapy, our laboratories developed a novel immune therapy based on nanobiologics. These nanobiologics are composed of apolipoprotein A‐1 obtained from the blood that together with phospholipids encapsulate the mTOR inhibitor rapamycin (mTORi‐nanoimmunotherapeutic). Our nanoimmunotherapeutic was able to prevent the induction of trained immunity in macrophages and promoted the acceptance of heart allografts in mice.19 This novel approach prevents the innate immune memory response and represents a new strategy for the development of future clinical treatments that aim at eliminating the need for continuous immunosuppression in transplant patients. Future experiments will determine whether the mTORi‐nanoimmunotherapeutic also prevents other trained immunity–related scenarios, including infection (viral, bacterial, and fungal),41 activation of the NLRP3 inflammasome in type 2 diabetic patients,97 or other chronic inflammatory conditions107 that are associated with increased morbidity and mortality in organ transplant patients.
Trained immunity is a recent term that describes the ability of innate immune cells, including monocytes and macrophages, to mount intensified immune responses that protect against pathogenic secondary stimuli. Murine studies demonstrated that organ transplantation induces macrophage training and represents a previously unrecognized mechanism that contributes to allograft rejection. Investigations to determine the precise locations (allograft, secondary lymphoid organs, bone marrow…), duration (days, weeks, months…), and participating innate immune cells (macrophages, NK cells, endothelial cells…) in trained immunity will provide valuable information to design more effective treatments in transplantation. In this respect, immunologic, metabolic, and epigenetic approaches that prevent trained immunity may be considered as synergistic therapeutic options to promote optimal long‐term survival rates in nonhuman primates and humans.
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. JO, WJMM, ZAF, and MGN declare that they are scientific founders of Trained Therapeutics Discovery.
AUTHOR CONTRIBUTION
All authors contributed to the writing, editing, and review of the manuscript.
ACKNOWLEDGMENTS
The authors’ work is supported by National Institutes of Health grants R01 AI139623AI (JO); R01 CA220234, R01 HL144072, P01 HL131478, and Netherlands Organization for Scientific Research (NWO) grant ZonMW Vici 91818622 (WJMM); R01 HL143814 and P01HL131478 (ZAF); European Research Council (ERC) Consolidator Grant (310372) and Spinoza Grant of the Netherlands Organization for Scientific Research (MGN); and UO1 AI131470 (JCM).
Ochando J, Fayad ZA, Madsen JC, Netea MG, Mulder WJM. Trained immunity in organ transplantation. Am J Transplant. 2020;20:10–18. 10.1111/ajt.15620
DATA AVAILABILITY STATEMENT
No data are presented as this is a review article.
REFERENCES
- 1. Miller JF. Immunological function of the thymus. Lancet. 1961;2(7205):748‐749. [DOI] [PubMed] [Google Scholar]
- 2. Flanagan SP. 'Nude', a new hairless gene with pleiotropic effects in the mouse. Genet Res. 1966;8(3):295‐309. [DOI] [PubMed] [Google Scholar]
- 3. Pantelouris EM. Observations on the immunobiology of 'nude' mice. Immunology. 1971;20(2):247‐252. [PMC free article] [PubMed] [Google Scholar]
- 4. Corthay A. A three‐cell model for activation of naive T helper cells. Scand J Immunol. 2006;64(2):93‐96. [DOI] [PubMed] [Google Scholar]
- 5. Page EK, Dar WA, Knechtle SJ. Tolerogenic therapies in transplantation. Front Immunol. 2012;3:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lamb KE, Lodhi S, Meier‐Kriesche HU. Long‐term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011;11(3):450‐462. [DOI] [PubMed] [Google Scholar]
- 7. Oberbarnscheidt MH, Zeng Q, Li QI, et al. Non‐self recognition by monocytes initiates allograft rejection. J Clin Invest. 2014;124(8):3579‐3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zecher D, van Rooijen N, Rothstein DM, Shlomchik WD, Lakkis FG. An innate response to allogeneic nonself mediated by monocytes. J Immunol. 2009;183(12):7810‐7816. [DOI] [PubMed] [Google Scholar]
- 9. Kitchens WH, Chase CM, Uehara S, et al. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am J Transplant. 2007;7(12):2675‐2682. [DOI] [PubMed] [Google Scholar]
- 10. Hancock WW, Buelow R, Sayegh MH, Turka LA. Antibody‐induced transplant arteriosclerosis is prevented by graft expression of anti‐oxidant and anti‐apoptotic genes. Nat Med. 1998;4(12):1392‐1396. [DOI] [PubMed] [Google Scholar]
- 11. Kirk AD, Hale DA, Mannon RB, et al. Results from a human renal allograft tolerance trial evaluating the humanized CD52‐specific monoclonal antibody alemtuzumab (CAMPATH‐1H). Transplantation. 2003;76(1):120‐129. [DOI] [PubMed] [Google Scholar]
- 12. Xu H, Dhanireddy KK, Kirk AD. Human monocytes as intermediaries between allogeneic endothelial cells and allospecific T cells: a role for direct scavenger receptor‐mediated endothelial membrane uptake in the initiation of alloimmunity. J Immunol. 2006;176(2):750‐761. [DOI] [PubMed] [Google Scholar]
- 13. Dai H, Friday AJ, Abou‐Daya KI, et al. Donor SIRPalpha polymorphism modulates the innate immune response to allogeneic grafts. Sci Immunol. 2017;2(12):pii eaam6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453‐461. [DOI] [PubMed] [Google Scholar]
- 15. Takeuch O, Akira S. Epigenetic control of macrophage polarization. Eur J Immunol. 2011;41(9):2490‐2493. [DOI] [PubMed] [Google Scholar]
- 16. Braza MS, Conde P, Garcia M, et al. Neutrophil derived CSF1 induces macrophage polarization and promotes transplantation tolerance. Am J Transplant. 2018;18(5):1247‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conde P, Rodriguez M, van der Touw W, et al. DC‐SIGN(+) macrophages control the induction of transplantation tolerance. Immunity. 2015;42(6):1143‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Y, Chen S, Lan P, et al. Macrophage subpopulations and their impact on chronic allograft rejection versus graft acceptance in a mouse heart transplant model. Am J Transplant. 2018;18(3):604‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braza MS, van Leent M, Lameijer M, et al. Inhibiting inflammation with myeloid cell‐specific nanobiologics promotes organ transplant acceptance. Immunity. 2018;49(5):819‐828 e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Netea MG, Joosten L, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kleinnijenhuis J, Quintin J, Preijers F, et al. Long‐lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun. 2014;6(2):152‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saeed S, Quintin J, Kerstens H, et al. Epigenetic programming of monocyte‐to‐macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen H, Goldstein DR. IL‐6 and TNF‐alpha synergistically inhibit allograft acceptance. J Am Soc Nephrol. 2009;20(5):1032‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eckes B, Colucci‐Guyon E, Smola H, et al. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci. 2000;113(Pt 13):2455‐2462. [DOI] [PubMed] [Google Scholar]
- 25. Thiagarajan PS, Yakubenko VP, Elsori DH, et al. Vimentin is an endogenous ligand for the pattern recognition receptor Dectin‐1. Cardiovasc Res. 2013;99(3):494‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Azimzadeh AM, Pfeiffer S, Wu GS, et al. Humoral immunity to vimentin is associated with cardiac allograft injury in nonhuman primates. Am J Transplant. 2005;5(10):2349‐2359. [DOI] [PubMed] [Google Scholar]
- 27. Miller LW, Granville DJ, Narula J, McManus BM. Apoptosis in cardiac transplant rejection. Cardiol Clin. 2001;19(1):141‐154. [DOI] [PubMed] [Google Scholar]
- 28. Rose ML. De novo production of antibodies after heart or lung transplantation should be regarded as an early warning system. J Heart Lung Transplant. 2004;23(4):385‐395. [DOI] [PubMed] [Google Scholar]
- 29. Szabolcs MJ, Ravalli S, Minanov O, Sciacca RR, Michler RE, Cannon PJ. Apoptosis and increased expression of inducible nitric oxide synthase in human allograft rejection. Transplantation. 1998;65(6):804‐812. [DOI] [PubMed] [Google Scholar]
- 30. Mahesh B, Leong HS, McCormack A, Sarathchandra P, Holder A, Rose ML. Autoantibodies to vimentin cause accelerated rejection of cardiac allografts. Am J Pathol. 2007;170(4):1415‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ochando J, Ordikhani F, Boros P, Jordan S. The innate immune response to allotransplants: mechanisms and therapeutic potentials. Cell Mol Immunol. 2019;16(4):350‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Linkermann A, Brasen JH, Darding M, et al. Two independent pathways of regulated necrosis mediate ischemia‐reperfusion injury. Proc Natl Acad Sci USA. 2013;110(29):12024‐12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191‐195. [DOI] [PubMed] [Google Scholar]
- 34. Huang Y, Yin H, Han J, et al. Extracellular hmgb1 functions as an innate immune‐mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7(4):799‐808. [DOI] [PubMed] [Google Scholar]
- 35. Wu H, Ma J, Wang P, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21(11):1878‐1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zou H, Yang Y, Gao M, et al. HMGB1 is involved in chronic rejection of cardiac allograft via promoting inflammatory‐like mDCs. Am J Transplant. 2014;14(8):1765‐1777. [DOI] [PubMed] [Google Scholar]
- 37. Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll‐like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107(26):11942‐11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valdés‐Ferrer SI, Rosas‐Ballina M, Olofsson PS, et al. High‐mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock. 2013;40(6):492‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec‐10 selectively repress tissue damage‐induced immune responses. Science. 2009;323(5922):1722‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Joo HyeMee, Upchurch K, Zhang W, et al. Opposing roles of Dectin‐1 expressed on human plasmacytoid dendritic cells and myeloid dendritic cells in Th2 polarization. J Immunol. 2015;195(4):1723‐1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mulder W, Ochando J, Joosten L, Fayad ZA, Netea MG. Therapeutic targeting of trained immunity. Nat Rev Drug Discov. 2019;18(7):553‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette‐Guerin induces NOD2‐dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. 2012;109(43):17537‐17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869‐8872. [DOI] [PubMed] [Google Scholar]
- 44. Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chong AS, Alegre ML. The impact of infection and tissue damage in solid‐organ transplantation. Nat Rev Immunol. 2012;12(6):459‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams AB, Williams MA, Jones TR, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111(12):1887‐1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahmed EB, Wang T, Daniels M, Alegre ML, Chong AS. IL‐6 induced by Staphylococcus aureus infection prevents the induction of skin allograft acceptance in mice. Am J Transplant. 2011;11(5):936‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng SC, Quintin J, Cramer RA, et al. mTOR‐ and HIF‐1alpha‐mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kapetanovic R, Nahori M‐A, Balloy V, et al. Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus‐activated macrophages. Infect Immun. 2007;75(2):830‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2‐dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731‐734. [DOI] [PubMed] [Google Scholar]
- 51. Wang T, Chen L, Ahmed E, et al. Prevention of allograft tolerance by bacterial infection with Listeria monocytogenes. J Immunol. 2008;180(9):5991‐5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thornley TB, Brehm MA, Markees TG, et al. TLR agonists abrogate costimulation blockade‐induced prolongation of skin allografts. J Immunol. 2006;176(3):1561‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kapoor A, Forman M, Arav‐Boger R. Activation of nucleotide oligomerization domain 2 (NOD2) by human cytomegalovirus initiates innate immune responses and restricts virus replication. PLoS ONE. 2014;9(3):e92704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mitchell BM, Leung A, Stevens JG. Murine cytomegalovirus DNA in peripheral blood of latently infected mice is detectable only in monocytes and polymorphonuclear leukocytes. Virology. 1996;223(1):198‐207. [DOI] [PubMed] [Google Scholar]
- 55. Castellino F, Germain RN. Chemokine‐guided CD4+ T cell help enhances generation of IL‐6RalphahighIL‐7Ralpha high prememory CD8+ T cells. J Immunol. 2007;178(2):778‐787. [DOI] [PubMed] [Google Scholar]
- 56. Gagnon J, Ramanathan S, Leblanc C, Cloutier A, McDonald PP, Ilangumaran S. IL‐6, in synergy with IL‐7 or IL‐15, stimulates TCR‐independent proliferation and functional differentiation of CD8+ T lymphocytes. J Immunol. 2008;180(12):7958‐7968. [DOI] [PubMed] [Google Scholar]
- 57. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin‐6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. 2011;334(6057):825‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cook CH, Bickerstaff AA, Wang J‐J, et al. Disruption of murine cardiac allograft acceptance by latent cytomegalovirus. Am J Transplant. 2009;9(1):42‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bekkering S, Blok BA, Joosten LA, Riksen NP, van Crevel R, Netea MG. In vitro experimental model of trained innate immunity in human primary monocytes. Clin Vaccine Immunol. 2016;23(12):926‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low‐density lipoprotein induces long‐term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. 2014;34(8):1731‐1738. [DOI] [PubMed] [Google Scholar]
- 61. Bosmans JL, Holvoet P, Dauwe SE, et al. Oxidative modification of low‐density lipoproteins and the outcome of renal allografts at 1 1/2 years. Kidney Int. 2001;59(6):2346‐2356. [DOI] [PubMed] [Google Scholar]
- 62. Agarwal A, Prasad GV. Post‐transplant dyslipidemia: mechanisms, diagnosis and management. World J Transplant. 2016;6(1):125‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Regnstrom J, Nilsson J, Tornvall P, Landou C, Hamsten A. Susceptibility to low‐density lipoprotein oxidation and coronary atherosclerosis in man. Lancet. 1992;339(8803):1183‐1186. [DOI] [PubMed] [Google Scholar]
- 64. Riella LV, Gabardi S, Chandraker A. Dyslipidemia and its therapeutic challenges in renal transplantation. Am J Transplant. 2012;12(8):1975‐1982. [DOI] [PubMed] [Google Scholar]
- 65. Ghanem H, van den Dorpel MA, Weimar W, Man in'T Veld AJ, El‐Kannishy MH, Jansen H. Increased low density lipoprotein oxidation in stable kidney transplant recipients. Kidney Int. 1996;49(2):488‐493. [DOI] [PubMed] [Google Scholar]
- 66. Isoniemi H, Nurminen M, Tikkanen MJ, et al. Risk factors predicting chronic rejection of renal allografts. Transplantation. 1994;57(1):68‐72. [DOI] [PubMed] [Google Scholar]
- 67. Holvoet P, Van Cleemput J, Collen D, Vanhaecke J. Oxidized low density lipoprotein is a prognostic marker of transplant‐associated coronary artery disease. Arterioscler Thromb Vasc Biol. 2000;20(3):698‐702. [DOI] [PubMed] [Google Scholar]
- 68. Swan SK. Role of lipids in chronic renal allograft rejection. Contrib Nephrol. 1997;120:62‐67. [DOI] [PubMed] [Google Scholar]
- 69. Jimi S, Saku K, Uesugi N, Sakata N, Takebayashi S. Oxidized low density lipoprotein stimulates collagen production in cultured arterial smooth muscle cells. Atherosclerosis. 1995;116(1):15‐26. [DOI] [PubMed] [Google Scholar]
- 70. Stribos E, Nielsen SH, Brix S, et al. Non‐invasive quantification of collagen turnover in renal transplant recipients. PLoS ONE. 2017;12(4):e0175898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fang JC, Kinlay S, Behrendt D, et al. Circulating autoantibodies to oxidized LDL correlate with impaired coronary endothelial function after cardiac transplantation. Arterioscler Thromb Vasc Biol. 2002;22(12):2044‐2048. [DOI] [PubMed] [Google Scholar]
- 72. Salonen JT, Korpela H, Salonen R, et al. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet. 1992;339(8798):883‐887. [DOI] [PubMed] [Google Scholar]
- 73. Apanay DC, Neylan JF, Ragab MS, Sgoutas DS. Cyclosporine increases the oxidizability of low‐density lipoproteins in renal transplant recipients. Transplantation. 1994;58(6):663‐669. [PubMed] [Google Scholar]
- 74. Cofan F, Cofan M, Campos B, Guerra R, Campistol JM, Oppenheimer F. Effect of calcineurin inhibitors on low‐density lipoprotein oxidation. Transplant Proc. 2005;37(9):3791‐3793. [DOI] [PubMed] [Google Scholar]
- 75. van den Dorpel MA, Ghanem H, Rischen‐Vos J, Man in't Veld AJ, Jansen H, Weimar W. Conversion from cyclosporine A to azathioprine treatment improves LDL oxidation in kidney transplant recipients. Kidney Int. 1997;51(5):1608‐1612. [DOI] [PubMed] [Google Scholar]
- 76. Jones T. The effect of HMG‐CoA reductase inhibitors on chronic allograft rejection. Expert Opin Emerg Drugs. 2001;6(1):95‐109. [DOI] [PubMed] [Google Scholar]
- 77. Motomura N, Saito S, Foegh ML. HMG‐CoA reductase inhibitors in organ transplantation. J Nephrol. 1997;10(2):68‐76. [PubMed] [Google Scholar]
- 78. Bekkering S, Arts R, Novakovic B, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. 2018;172(1–2):135‐146 e139. [DOI] [PubMed] [Google Scholar]
- 79. Duivenvoorden R, Tang J, Cormode DP, et al. A statin‐loaded reconstituted high‐density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bekkering S, Stiekema L, Bernelot Moens S, et al. Treatment with statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab. 2019;30(1):1–2. [DOI] [PubMed] [Google Scholar]
- 81. Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci USA. 1987;84(9):2995‐2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Christ A, Günther P, Lauterbach M, et al. Western diet triggers NLRP3‐dependent innate immune reprogramming. Cell. 2018;172(1–2):162‐175 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sheedy FJ, Grebe A, Rayner KJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14(8):812‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iyer SS, Pulskens WP, Sadler JJ, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106(48):20388‐20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Salvadori M, Rosso G, Bertoni E. Update on ischemia‐reperfusion injury in kidney transplantation: pathogenesis and treatment. World J Transplant. 2015;5(2):52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find‐me signal to promote phagocytic clearance. Nature. 2009;461(7261):282‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Amores‐Iniesta J, Barberà‐Cremades M, Martínez CM, et al. Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep. 2017;21(12):3414‐3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Israni AK, Snyder JJ, Skeans MA, Kasiske BL, Investigators P. Clinical diagnosis of metabolic syndrome: predicting new‐onset diabetes, coronary heart disease, and allograft failure late after kidney transplant. Transpl Int. 2012;25(7):748‐757. [DOI] [PubMed] [Google Scholar]
- 90. Rana A, Gruessner A, Agopian VG, et al. Survival benefit of solid‐organ transplant in the United States. JAMA Surg. 2015;150(3):252‐259. [DOI] [PubMed] [Google Scholar]
- 91. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4(2):481‐508. [DOI] [PubMed] [Google Scholar]
- 92. Engels EA, Pfeiffer RM, Fraumeni JF, et al. Spectrum of cancer risk among US solid organ transplant recipients. JAMA. 2011;306(17):1891‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vincenti F, Rostaing L, Grinyo J, et al. Belatacept and long‐term outcomes in kidney transplantation. N Engl J Med. 2016;374(4):333‐343. [DOI] [PubMed] [Google Scholar]
- 94. Xu H, Samy KP, Guasch A, et al. Postdepletion lymphocyte reconstitution during belatacept and rapamycin treatment in kidney transplant recipients. Am J Transplant. 2016;16(2):550‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koreth J, Matsuoka K‐I, Kim HT, et al. Interleukin‐2 and regulatory T cells in graft‐versus‐host disease. N Engl J Med. 2011;365(22):2055‐2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kawai T, Cosimi AB, Spitzer TR, et al. HLA‐mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. 2008;358(4):353‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Espinosa JR, Samy KP, Kirk AD. Memory T cells in organ transplantation: progress and challenges. Nat Rev Nephrol. 2016;12(6):339‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hildemann WH, Johnson IS, Jokiel PL. Immunocompetence in the lowest metazoan phylum: transplantation immunity in sponges. Science. 1979;204(4391):420‐422. [DOI] [PubMed] [Google Scholar]
- 100. Hildemann WH, Raison RL, Cheung G, Hull CJ, Akaka L, Okamoto J. Immunological specificity and memory in a scleractinian coral. Nature. 1977;270(5634):219‐223. [DOI] [PubMed] [Google Scholar]
- 101. Fu F, Thai NL, Li Y, et al. Second‐set rejection of mouse liver allografts is dependent on radiation‐sensitive nonparenchymal cells of graft bone marrow origin. Transplantation. 1996;61(8):1228‐1233. [DOI] [PubMed] [Google Scholar]
- 102. Liu W, Xiao X, Demirci G, Madsen J, Li XC. Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol. 2012;188(6):2703‐2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Seeley JJ, Baker RG, Mohamed G, et al. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature. 2018;559(7712):114‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Miller ML, Daniels MD, Wang T, et al. Spontaneous restoration of transplantation tolerance after acute rejection. Nat Commun. 2015;6:7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fantus D, Rogers NM, Grahammer F, Huber TB, Thomson AW. Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat Rev Nephrol. 2016;12(10):587‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Napoli KL, Wang ME, Stepkowski SM, Kahan BD. Distribution of sirolimus in rat tissue. Clin Biochem. 1997;30(2):135‐142. [DOI] [PubMed] [Google Scholar]
- 107. Arts R, Joosten L, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol. 2018;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data are presented as this is a review article.