

SUMMARY
H7N9 avian influenza virus causes severe infections and might have the potential to trigger a major pandemic. Molecular determinants of human humoral immune response to N9 neuraminidase (NA) proteins, which exhibit unusual features compared with seasonal influenza virus NA proteins, are ill-defined. We isolated 35 human monoclonal antibodies (mAbs) from two H7N9 survivors and two vaccinees. These mAbs react to NA in a subtype-specific manner and recognize diverse antigenic sites on the surface of N9 NA, including epitopes overlapping with, or distinct from, the enzyme active site. Despite recognizing multiple antigenic sites, the mAbs use a common mechanism of action by blocking egress of nascent virions from infected cells, thereby providing an antiviral prophylactic and therapeutic protection in vivo in mice. Studies of breadth, potency, and diversity of antigenic recognition from four subjects suggest that vaccination with inactivated adjuvanted vaccine induce NA-reactive responses comparable to that of H7N9 natural infection.
Graphical Abstract
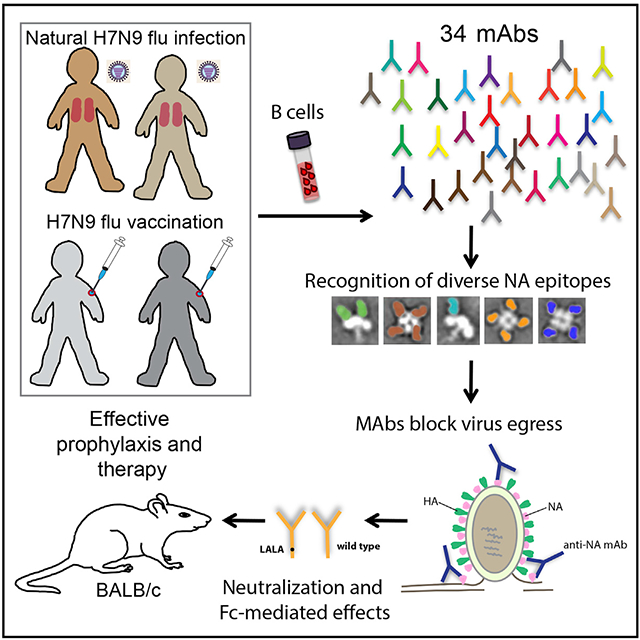
In Brief
Molecular determinants of the human B cell response to avian influenza N9 neuraminidase (NA) proteins, which differ from seasonal virus NAs, are ill-defined. Gilchuk et al. identify antibodies to multiple protective antigenic sites on N9 NA that block egress of nascent virions from infected cells and mediate protection in mice.
INTRODUCTION
Influenza viruses have two major surface glycoproteins, hemagglutinin (HA), and neuraminidase (NA). HA mediates attachment of virions to terminal sialic acids (SAs) on the cell surface and fusion of the virus with cellular membranes, whereas NA cleaves terminal SA residues present on host cell surfaces, thereby releasing progeny virions from infected cells. Influenza virus infection or vaccination in humans elicits a humoral immune response against both HA and NA. The level of hemagglutination inhibition (HAI) mediated by serum anti-HA antibodies has been used as a correlate of protective immunity and is the conventional regulatory measurement for evaluating potency of seasonal influenza vaccine preparations. The titer of naturally occurring influenza NA-inhibiting (NI) antibodies also is a predictor of immunity (Couch et al., 2013; Monto et al., 2015; Ng et al., 2019). In recent studies in a human challenge model with A/California/04/2009 H1N1 (2009 A(H1N1)pdm) influenza A virus (IAV), the baseline serum NI titer correlated better with disease severity metrics and exhibited a more robust independent effect on clinical outcomes than HAI titers (Memoli et al., 2016). Moreover, anti-influenza drugs targeting the active site of NA have been licensed for use in the U.S. since 1999 (Beard et al., 2018) and have shown clinical benefit.
Influenza A viruses are subtyped on the basis of HA and NA proteins. There are 16 HA subtypes and 9 NA subtypes known in IAVs (Fouchier et al., 2005); two bat-derived influenza-like viruses defined as H17N10 and H18N11 subtypes also have been identified (Tong et al., 2012; Tong et al., 2013). Only two IAV subtypes (H1N1 and H3N2) are currently in general circulation among people. Infection of humans by other IAV subtypes (such as avian H5N1, H7N2, H7N3, and H7N7 viruses) sporadically occur because of transmission from birds to humans (Mostafa et al., 2018). Since 2013, novel avian IAV H7N9 strains have caused six annual outbreaks in China (Wang et al., 2017; WHO, 2018). These outbreaks occur with high morbidity and mortality rates (39%). However, H7N9 infections occurred almost exclusively after exposure to poultry, and sustained human-to-human transmission has not been reported for this virus. Due to the health concern posed by avian H7N9 viruses and the emergence of two antigenically distinct lineages, numerous H7N9 candidate vaccines are being developed and evaluated (Jackson et al., 2015; WHO, 2017). However, currently, there is no licensed vaccine for prevention of H7N9 infection, and the treatment strategy consists of supportive medical care and the use of neuraminidase inhibitors. Like other influenza viruses, H7N9 is evolving in the field to become resistant to these inhibitors (Hai et al., 2013).
We and others have isolated and characterized human monoclonal antibodies (mAbs) to HA induced by H7 vaccination (Thornburg et al., 2016) or natural infection (Huang et al., 2019). Little is known, however, about human NA-specific antibodies elicited by H7N9 experimental vaccination or natural infection. The N9 NA, like some other avian NAs, contains a second SA binding site that is not present in the NAs of human seasonal influenza viruses (Sun et al., 2014; Uhlendorff et al., 2009; Varghese et al., 1997). N9 NA differs antigenically from N1 and N2 subtype NAs in human seasonal IAVs. Polyclonal serum NI antibodies are induced after experimental vaccination with H7N9 virus-like particles (VLPs) (Fries et al., 2013) or natural infection (Ma et al., 2018). However, the breadth, protective capacity, and epitopes targeted by human anti-N9 NA mAbs after vaccination or natural infection are mostly unknown.
Here, we examined the human B cell response to N9 NA after experimental H7N9 subunit vaccination or natural infection. We isolated and characterized in detail many N9-specific human antibodies targeting diverse antigenic sites on the surface of NA. The anti-N9 mAbs were mostly subtype-specific, suggesting that the N9 NA protein is antigenically distinct from other IAV NA subtypes. Mechanism of action studies showed that the mAbs neutralize virus infection in vitro by inhibiting egress of nascent virions. Many anti-N9 mAb exhibited robust NA inhibitory activity and provided prophylactic protection in a murine model of H7N9 infection.
RESULTS
H7N9 Vaccination or Infection Elicits B Cell Response against N9 NA
We obtained peripheral blood mononuclear cells (PBMCs) from two donors who had recovered from a naturally occurring H7N9 infection (Skowronski et al., 2016) or from 30 otherwise healthy subjects previously immunized with monovalent inactivated A/Shanghai/2/2013 (SH13) H7N9 vaccine (inactivated influenza vaccine [IIV]) by the NIH Vaccine Treatment and Evaluation Unit (DMID 13-0033) (Jackson et al., 2015) (Table S1). Blood samples were obtained approximately 11 months after recovery from natural infection or 42 days after immunization. PBMCs were transformed with Epstein-Barr virus and other stimuli and the number of cell clusters forming transformed B lymphoblastoid cell lines (LCLs) was determined. To identify H7N9-reactive B cell cultures, we screened the supernatants from the resulting LCLs via ELISA for binding to H7 HA or N9 NA antigens from the SH13 H7N9 virus strain. Interestingly, in the response of the donors that we tested, we did not observe a fixed relationship between the frequencies of N9-reactive and H7-reactive LCLs. Instead, we identified four distinct patterns of the response to H7N9 IIV or natural infection (Figure 1). Among the 30 vaccinees, only five responded to both H7 and N9 antigens, including 4 of 12 vaccinees who had been primed and boosted with IIV plus Adjuvant System 03 (AS03) (GlaxoSmithKline), and 1 of 4 vaccinees who had been primed with IIV plus AS03 and boosted with IIV. This finding suggests that an adjuvant like AS003 might be needed to optimize the induction of the responses to both major antigens with inactivated viral vaccine. Five donors responded only to H7 HA, and seven donors responded only to N9 NA (Figure 1; Table S1). Both of the H7N9 natural infection survivors responded to both H7 HA and N9 NA antigens. Of note is the fact that 12 of 12 donors with B cell responses to N9 NA received a vaccine containing AS03 (eight donors), AS03 and MF59 (three donors), or MF59 (one donor), whereas none of the donors receiving unadjuvanted vaccine exhibited a N9 NA B cell response over the threshold of detection.
Figure 1. H7N9 Influenza Virus Infection or Vaccination Induces NA- or HA-Reactive B Cells.

The frequency of N9-reactive LCLs was compared with the frequency of H7-reactive LCLs. The colors of the bars correspond to LCL specificity: black, N9 NA reactive and white, H7 HA. The gray dotted line indicates the threshold of positive response (0.25%). Arrows indicate donors from which anti-NA mAbs were isolated. Color shapes indicate antigenic exposure history of donors: brown, prime and boost vaccinations with IIV and AS03 adjuvant; red, prime with IIV and AS03 and boost with IIV only; pink, prime with IIV only and boost with IIV and AS03; violet, prime with IIV and AS03 and boost with IIV and MF59 adjuvant; lilac, prime with IIV and ME59 and boost with IIV and AS03; blue, prime and boost with IIV and MF59; yellow, prime and boost with IIV alone; green, H7N9 infection survivors.
Hybridomas secreting human antigen-specific mAbs were generated from B cell lines secreting virus-specific antibodies, as previously described (Crowe, 2009). To identify NA-reactive B cell cultures, the supernatants from the resulting LCLs from two natural disease survivors and two vaccinees were screened by ELISA for binding to NA antigen from the SH13 H7N9, A/Wisconsin/67/2005 H3N2, 2009 A(H1N1)pdm, or A/Pennsylvania/1/ 2007 H3N8 viruses. All four donors exhibited a narrow N9-specific response with negligible cross-reactivity to other NAs (Figure S1C). A total of 35 cloned hybridoma cell lines secreting anti-N9 NA human mAbs was isolated, including 16 lines from two vaccinees and 19 from the two survivors (Table S1). Most (33 of 35) mAbs were independent clones that displayed a high degree of antibody variable gene sequence diversity, including a unique HCDR3 sequence for each mAb (Table S2). Two mAbs, NA-22 and NA-69, were clonally related. We conclude from this analysis that either natural infection or vaccination with monovalent inactivated H7N9 virus that was formulated with adjuvant induces an anti-N9 NA B cell response.
Human Anti-N9 mAbs Elicited by H7N9 Infection or Vaccination Are N9 Subtype Specific
The discovery of heterosubtypic HA-specific antibodies has spurred interest in universal influenza vaccine immunity (Erbelding et al., 2018; Paules et al., 2017), but the extent of the ability of humans to make heterosubtypic NA antibodies is not clear. To determine reactivity of anti-NA human antibodies, we first assessed binding of serum samples from H7N9 survivors to diverse recombinant NA proteins from group 1 (N1, N4, and N8) or group 2 (N9, N6, N7, and N2) IAVs. Serum from survivors showed high reactivity to group 2 NA antigens (Figures S1A and S1B; Table S3), although it is not possible to discriminate activity of broadly reactive monoclonal from monospecific polyclonal responses to individual NAs.
To assess whether any of the N9-reactive mAbs that we isolated exhibited a heterosubtypic binding profile, we also tested them for binding to members of a diverse panel of recombinant NA proteins (Figures 2A and S2; Table S4). We found that most (32 of 35) mAbs in the panel that were identified by screening of LCLs with SH13 H7N9 antigen exhibited reactivity to the phylogenetically closest N9 antigen from the A/Hunan/02650/2016 strain. Fifteen mAbs exhibited some depth of reactivity for the N9 subtype as they cross-reacted with all three of the N9 antigens tested, including N9 from A/whale/Maine/1/1984 H13N9. The broadest mAb from the panel, NA-97, exhibited some heterosubtypic breadth as it reacted with all tested N9 antigens and two N6 NA antigens from recent H5N6 viruses. None of the anti-N9 mAbs cross-reacted with representative NA proteins from N2 or N7 of group 2 or N1, N4, or N8NA antigens of group 1. This finding demonstrates that the anti-N9 B cell response to infection or subunit vaccination is mostly N9 subtype specific and suggests that the observed broad reactivity in serum likely was mediated by a polyclonal collection of subtype-specific mAbs. There could also be a difference in the breadth of antibodies encoded by memory-B-cell-derived mAbs compared with the breadth of plasma-cell-derived serum antibodies.
Figure 2. Binding of N9-Reactive mAbs Is Subtype-Specific and Is not Affected by Mutations Causing Resistance to Neuraminidase Inhibitors.

(A) A panel of 35 human mAbs was isolated on the basis of reactivity to recombinant SH13 N9 NA antigen. Cross-reactivity of N9-reactive mAbs to recombinant NA proteins from group 1 was measured by ELISA. Representative EC50 values (ng/mL) from two independent experiments are plotted as a heatmap. NAs were clustered by amino acid sequence phylogeny. Three mAbs that bind N9 antigen with EC50 value higher than 10 μg/mL (highest tested concentration) excluded from representation.
(B) Binding of N9-reactive mAbs to recombinant wild-type N9 NA from A/Shanghai/2/2013 virus or NI-resistant mutants was measured by ELISA. Representative EC50 values (ng/mL) from two independent experiments are plotted as a heatmap. Four mAbs that bind N9 SH13 antigen with EC50 value higher than 10 μg/mL (highest tested concentration) excluded from representation.
Some circulating H7N9 viruses have developed resistance to small-molecule NA inhibitor drugs because of the emergence of virus variants with E119V, I222K, and R292K amino-acid substitutions in the enzyme active site (Marjuki et al., 2015; Zhu et al., 2017). Each member of the panel of human anti-N9 mAbs isolated here was assessed for binding to two N9 drug-resistant variant viruses, possessing E119V or R292K mutations; an NA protein with the third mutation (I222K) could not be expressed in insect cell culture. Enzyme-linked immunosorbent assay (ELISA) analysis revealed that all tested mAbs from the panel bound equally well to recombinant NA of wild-type SH13 or to N9 variant proteins carrying the E119K or R292K mutations (Table S4; Figure 2B). This finding demonstrates that binding of most human N9 mAbs is not affected by mutations causing resistance to neuraminidase inhibitors. Therefore, these mAbs might be useful for prophylaxis or treatment of NI-resistant H7N9 IAV strains.
In summary, N9-reactive human mAbs elicited by natural infection or vaccination bound to N9 and exhibited subtype-specific reactivity.
Anti-N9 mAbs Neutralize Influenza Virus In Vitro Mainly by Inhibiting Egress of Nascent Virions through Steric Hindrance of the NA Enzyme Active Site
N9 NA has two discrete functional sites: the sialidase enzyme site and a second SA binding site, known as the hemadsorption site that seems to be more characteristic of avian viruses. The hemadsorption site of N9 NA has been shown previously to possess cell receptor binding properties (Uhlendorff et al., 2009; Varghese et al., 1997). The IAV NA protein sialidase activity is thought to be the main contributor to efficiency of viral egress from infected cells. To determine the specific stage in the virus life-cycle and the mechanism of inhibitory action for the neutralizing anti-N9 mAbs that we isolated, we performed HAI and egress inhibition assays with SH13 IDCDC-RG32A H7N9 virus. None of anti-N9 mAbs had HAI activity (Table S5). In contrast, an anti-H7 mAb H7.167 that targets the receptor binding site on HA (Thornburg et al., 2016) was used as a positive control and exhibited potent HAI activity.
Inhibition of egress of virions from infected cells in vitro was tested using a previously described assay (Bangaru et al., 2018). Madin-Darby canine kidney (MDCK) cell monolayer cultures were inoculated with H7N9 virus at a multiplicity of infection (MOI) of one focus forming unit (FFU) per cell, and three h later the monolayer of cells was washed with virus growth medium (VGM) followed by application of anti-N9 antibodies, zanamivir, or an irrelevant mAb. Delaying the addition of antibodies ensured unhindered initial infection and allowed assessment of the effect on egress only. Twenty-one h after infection, the amount of newly produced viral particles released into the supernatant was analyzed by a hemagglutination assay, given that the presence of anti-N9 mAbs does not interfere with this test. Zanamivir, a licensed neuraminidase activity inhibitor, was used as an assay positive control for comparative purposes. The majority (27 of 31 tested) of mAbs completely abolished H7N9 virus release into infected cell supernatants (Figures 3A and 3D; Table S5). Thus, neutralizing N9-specific human mAbs inhibit influenza virus principally at the egress stage of the viral lifecycle. The antiviral activity of the most active anti-N9 mAbs from the panel, which had an inhibitory concentration of 0.03 nM, is substantially more potent than that of zanamivir (7.3 nM).
Figure 3. Anti-N9 Human mAbs Inhibit Egress of A/Shanghai/2/2013 IDCDC-RG32A H7N9 Virus, Mainly by Blocking the N9 Enzyme Active Site.

Individual mAbs were assessed for H7N9 virus neutralization by using hemagglutination inhibition (HAI), egress inhibition, enzyme-linked lectin (ELLA) neuraminidase inhibition (NI) and NA-Fluor NI assays.
(A) Egress inhibition of H7N9 virus by mAbs. IC100 values (nM) are shown as mean ± SD of three technical replicates. Four mAbs with low expression were excluded from the analysis.
(B) Inhibition of N9 enzymatic activity by mAbs measured in ELLA. The assay used fetuin as substrate and H7N9 virus as the enzyme source. Data represent one of two independent experiments, shown as mean ± SD of three technical replicates.
(C) Inhibition of N9 enzymatic activity by mAbs measured in NA-Fluor assay. The assay used H7N9 virus as the enzyme source. Data are shown as mean ± SD of three technical replicates.
(D) mAbs functional activity comparison. Representative IC100 values (nM) from HAI or egress inhibition assays and IC50 values (nM) from ELLA NI or NA-Fluor NI assays are plotted as a heatmap. mAb NA-152 with low expression were excluded from the analysis.
See also Table S5.
Anti-NA neutralizing antibodies might inhibit egress of virions by one of two ways: direct interaction with the NA active site or through steric hindrance of that site (Chen et al., 2018; Krammer et al., 2018). To address the mechanism of egress inhibition mediated by N9 antibodies, we evaluated the capacity of the mAbs to inhibit SA cleavage by using an enzyme-linked lectin assay (ELLA) or an NA-Fluor assay. In the ELLA assay, the SA substrate is presented on large glycans with variable sialylation on the fetuin glycoprotein surface. The ELLA assay allows detection of mAb NA inhibiting activity by both the direct binding and indirect steric interference mechanisms. The majority (26 of 34) of mAbs inhibited cleavage of the large substrate fetuin by the SH13 IDCDC-RG32A H7N9 virus. The most potent inhibitors from the panel, NA-80 and NA-108, exhibited complete inhibition of N9 enzymatic activity and had IC50 values of ~0.1 nM, which is a much lower concentration than that for zanamivir (7.6 nM) (Figures 3B and 3D). Of note, NA-80 and NA-108 mAbs were isolated from an H7N9 vaccinee and survivor, respectively. More than half of the mAbs (15 of 26) from the panel had partial activity and only inhibited viral N9 NA enzymatic activity ~70%–75%, leaving behind a residual uninhibited fraction.
The NA-Fluor assay uses a small substrate 4-MU-NANA that easily accesses the enzyme active site, unless a mAb binds directly to the site. Among all 26 mAbs that inhibited N9 activity in the ELLA assay, only three mAbs inhibited the small substrate cleavage by H7N9 virus: NA-108, NA-73, and NA-45 (Table S5; Figure 3C). This result suggested that, whereas these three antibodies likely bind directly to the enzyme active site, the most common mechanism of viral egress inhibition for anti-N9 mAbs is indirect steric hindrance of the NA active site. Of the three mAbs that inhibited the small substrate, NA-108 was the most potent with an IC50 value of 0.54 nM, which is substantially lower than that of the drug zanamivir (7.6 nM). NA-73 activity was comparable to that of zanamivir. Although the NA-45 mAb inhibited H7N9-virus-mediated small substrate cleavage, this mAb activity required a high (> 66.7 nM) concentration to achieve 50% maximum effective concentration. Although the proportion of mAbs that inhibit small substrate cleavage was low (3 of 34 mAbs), representative mAbs were isolated from both survivors and vaccinees.
In summary, these findings demonstrate that anti-N9 mAbs neutralize H7N9 virus principally by steric hindrance of the NA active site, resulting in inhibition of egress of progeny virions from the infected cell surface.
Human Anti-N9 Inhibiting mAbs Recognize Three Major Antigenic Sites on the N9 Surface
To evaluate whether the human N9 mAbs targeted diverse antigenic regions on NA, we performed a quantitative competition-binding assay based on surface plasmon resonance (SPR) by using a Wasatch Microfluidics device. We assessed competition binding to N9 NA from SH13 for 29 human N9-specific mAbs and one murine N9-specific mAb NC10, which binds to the top of NA near the rim of the enzyme active site (Figure 4A). The N9-specific mAbs that lacked NI activity, like NA-175 and NA-37, segregated into the competition-binding group I. MAbs that exhibited NA enzymatic-inhibition activity segregated into two competition binding groups: group II and III. The most potent NA inhibitors represent group II and compete for binding with the murine mAb NC10. The epitopes for NA-108, NA-73, and NA-45 appear to be within the enzyme active site, on the basis of their ability to inhibit enzymatic activity of the NA with the small molecular weight substrate. These mAbs also exhibited an asymmetrical competition-binding pattern with other potent N9 inhibiting mAbs like NA-80 and NA-95, which are not able to inhibit N9 cleavage of the small size substrate. Additional competition binding studies by SPR (using a different SPR instrument, a BiaCore K8) with four mAbs NA-73, NA-108, NA-80, and NA-95 confirmed that these mAbs bind in close proximity within one region, but each mAb has a unique epitope (Figure S3). The mAbs NA-73 and NA-95 likely recognized the most distal non-overlapping epitopes in this region (Figure S3). Therefore, the antigenic region II can be further divided into two areas, IIa (enzyme active site) and IIb (top of NA, near the active site rim). Notably, mAbs that exhibited partial N9 enzymatic activity inhibition segregated into the separate competition-binding group III.
Figure 4. NI Anti-N9 Human mAbs Targeted Two Main Regions on the N9 Surface.

(A) mAbs were assessed for competition binding by surface plasmon resonance by using a Wasatch Microfluidics device. mAbs were judged to compete for the same site if maximum binding of second antibody was reduced to ≤ 49% of its uncompeted binding (shown in black boxes). The mAbs were considered non-competing if maximum binding of second mAb was ≥ 57% of its uncompeted binding (shown in white boxes). Grey boxes indicate an intermediate phenotype (competition between 50% and 56% of uncompeted binding). Blue, green, and orange lines and Roman numerals indicate inferred competition-binding groups. Clones with in vitro inhibitory activity are indicated with bold font. mAb NA-22 did not bind to N9 NA when tested by surface plasmon resonance, so it was excluded from analysis.
(B) Binding affinities of two representative mAbs from Group II or Group III for N9 NA from A/Shanghai/2/2013, determined by surface plasmon resonance.
See also Figures S3 and S4.
We next determined the binding affinity of two selected mAbs to N9 NA by SPR (Figures 4B and S4) by using a Biacore K8 (GE Healthcare Life Sciences). We assessed the weakly neutralizing mAb NA-22 (clonally related to NA-69, group III) and the potently neutralizing mAb NA-80 (group II). These antibodies also were selected for structural and in vivo protection studies on the basis of distinct functional and structural properties for these mAbs (detailed below). Both, NA-22 and NA-80 possessed a high binding affinity with respective KD values 0.8 and 0.2 nM, which are comparable to the range of affinity reported for high-affinity human mAbs to 2009 A(H1N1)pdm virus NA (Yasuhara et al., 2019).
To localize functional epitopes on N9 NA surface, we performed two complementary analyses: hydrogen-deuterium exchange mass spectrometry (HDX-MS) and negative stain electron microscopy (nsEM) of N9/mAb complexes. In related work, these approaches were validated by co-crystallizing and solving the three-dimensional (3D) structure or single-particle electron cryo-microscopy (cryo-EM) 3D reconstruction of the N9/Fab complexes for NA-22, NA-45, NA-63, NA-73 and NA-80 mAbs (accompanying manuscript Zhu et al., 2019).
The HDX-MS studies revealed mAb-induced alterations of hydrogen-deuterium exchange in the N9 antigen in the presence of mAbs. Changes in deuteration level were calculated as the difference in deuterium exchange of SH13 N9 NA amide backbone in the presence or absence of NA-22, NA-45, NA-63, NA-73, or NA-80 mAbs and plotted within the amino-acid sequence of N9 NA for multiple incubation times (Figures S5 and S6). Identification of decreased deuterium exchange in the presence of the mAbs was determined for peptides within the epitope footprint determined by crystallography or cryo-EM and based on the combination of multiple criteria: (1) significant reduction in deuterium accumulation (> 10% of the maximum number of deuterons that can be exchanged onto a given peptide) and (2) qualitative agreement between data from multiple time points for the same peptide. HDX-MS studies identified distinct predicted epitope regions for NA-80 and NA-63 mAbs (amino acids 329–342). These epitope predictions agree well with crystallography and cryo-EM studies of NA/Fab complexes for NA-80 and NA-63 mAbs (Zhu et al., 2019). On the other hand, HDX-MS data alone were not sufficient to predict the epitopes for mAbs NA-45, NA-22, or NA-73, because HDX-MS suggested multiple peptides with decreased deuterium level that distributed separately within the NA sequence and in the N9 structure. However, some amino acid residues in the peptides identified by HDX-MS for these mAbs overlapped with the epitopes determined by crystallography or cryo-EM (Zhu et al., 2019). Altogether, the HDX-MS, crystallography and cryo-EM studies revealed three distinct major antigenic sites targeted by N9 inhibiting mAbs on the N9 surface: (1) the enzyme active site, (2) the lateral surface of the NA head (amino acids 342–372), and (3) the interface of adjacent NA monomers (Figure 5A).
Figure 5. Epitope Mapping for Anti-N9 mAbs.

For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.chom.2019.10.003.
(A) Epitope footprints and HDX-MS profiles of NA-73, NA-45, NA-80, NA-63, or NA-22 mAbs were mapped onto the surface of the N9 NA tetramer (PDB: 4MWL). Lines indicate the epitope footprint of N9-Fab complex structure for mAbs NA-45 (cyan), NA-73 (green), NA-63 (orange), NA-80 (blue), or NA-22 (brown) determined by crystallography or cryo-EM reconstruction. Amino-acid residues within the epitope with decreased deuteration level upon NA-45, NA-73, NA-63, NA-80, or NA-22 mAbs binding is indicated by cyan, green, orange, blue, or brown colors, respectively. Pale cyan color indicates the enzyme active site, and yellow color indicates the hemadsorption site.
(B) Two-dimensional class averages of N9 NA alone or in complex with anti-N9 Fabs. Secondary structure features are visible in a variety of different views of the complex. Differences in Fab angles of approach are shown.
See also Figures S5 and S6 and Zhu et al., 2019.
A cross-reactive memory B cell response to conserved epitopes of NA from seasonal and H7N9 stains might be predicted in H7N9 vaccinees and survivors if common antigenic sites are present in those NA proteins. However, none of the 32 tested mAbs cross-reacted with N2 antigen (Figure 2). Analysis of epitopes for five NA/Fab complexes that were identified by crystallography studies (mAbs NA-22, NA-48, NA-63, NA-73, and NA-80) (Zhu et al., 2019) revealed relatively low conservation between N9 and N2 NAs epitope sequences (Figure S7). mAb NA-45 recognizes the most conserved epitope, but even that epitope exhibits at least four amino acid differences between N9 and the N2 NAs tested. The lack of cross-reactivity to N2 NA suggests that N9 NA is highly distinct antigenically and thus might elicit a qualitatively different response than N2 immunogens.
Examination of 2D class averages from the nsEM images for complexes of N9 NA with Fab fragments of each of five mAbs, NA-22, NA-45, NA-63, NA-73, or NA-80, revealed diverse binding angles and stoichiometries of bound Fabs (Figure 5B). NA-45 and NA-73 Fabs appeared to bind in an almost vertical orientation to the enzyme active site, whereas NA-80 and NA-63 bind at a wide angle and situate below the NA active site toward the side of the protomer. NA-22 binds the interface of adjacent NA monomers and tilts toward the NA stalk. All combinations of the Fab/N9 NA complex stoichiometry were observed (from zero to four Fabs per N9 tetramer) for the tested mAbs.
Altogether, the results of competition binding, HDX-MS, nsEM, and functional assays agreed and defined three major groups of inhibiting N9 human mAbs. The first group represents the potent inhibiting mAbs that formed competition binding group IIa, inhibiting cleavage of small substrate and targets enzyme active site. The second group includes potent inhibitors from competition-binding group IIb and targets the lateral surface of the NA head domain. The third group consists of mAbs that partially inhibit N9 enzymatic activity, forming competition-binding group III, and that target the interface of adjacent NA monomers.
Anti-N9 mAbs with Diverse Epitope Specificities Mediate High Levels of Protection In Vivo in Mice
We next evaluated the protective capacity of anti-N9 human mAbs in vivo. For the protection study, we chose mAbs NA-22, NA-45, NA-73, and NA-80 that were elicited by H7N9 vaccination and represented distinct epitopes on the NA surface and various levels of NI activity (Figures 3 and 5). Single-dose 10 mg/kg treatment with mAbs NA-22, NA-45, NA-73, or NA-80 (all immunoglobulin G1 [IgG1]) one day before lethal intranasal (i.n.) challenge of BALB/c mice with SH13 IDCDC-RG32A H7N9 virus provided complete protection against weight loss, mortality, and morbidity (Figure 6A). Negative control mAb DENV-2D22-treated mice experienced severe weight loss and illness and succumbed by day 10 after viral challenge. Notably, the level of protection provided by anti-N9 mAbs was comparable to that provided by a recombinant form of the broadly neutralizing HA-specific stem region binding mAb rCR9114 (Dreyfus et al., 2012) given at the same dose. These data indicate a high prophylactic potency of isolated human anti-N9 mAbs for prevention of respiratory tract H7N9 influenza infection.
Figure 6. Anti-N9 mAbs Mediate a High Level of Protection In Vivo against Lethal H7N9 Challenge in Mice.

Groups of BALB/c mice were inoculated intraperitoneally (i.p.) one day before (A) or one day after (B) virus challenge with 10 mg/kg of anti-N9 mAb or with DENV 2D22 control mAb reactive to an irrelevant antigen (dengue virus) or with 10 mg/kg of a recombinant mAb on the basis of the sequence of the broadly neutralizing influenza stem-targeted mAb rCR9114. On day 0 (d0), mice were challenged intranasally (i.n.) with a lethal dose of influenza A/Shanghai/02/2013 IDCDC-RG32A virus and monitored for protection. The weights and clinical scores are represented as the group mean ± SEM. The lower dotted line indicates the no-recovery threshold (> 30% weight loss) and endpoint for euthanasia. Body weight change curves in (B) were compared by overall test using two-way ANOVA. Survival curves were estimated using the Kaplan-Meier method. Survival of each group that was treated with an anti-NA or anti-HA (rCR9114) mAb was compared with the control group as indicated using log rank (Mantel-Cox) test. A clinical score of 4 corresponds to a moribund state. Data represent one experiment, and “n” symbol in the plots indicates number of mice per group for each treatment condition (5–10 mice per group).
We next assessed the therapeutic efficacy of the most potent mAbs that were identified in the prophylaxis study, NA-80 and NA-73. We used rCR9114 and DENV 2D22 as positive and negative controls, respectively. Single-dose 10 mg/kg treatment with individual mAb NA-80 or NA-73 that was administered one day after lethal i.n. challenge of mice with H7N9 virus caused complete protection from weight loss and illness (Figure 6B). DENV-2D22-treated mice succumbed to the disease by day 10. Mice treated with the same dose of rCR9114 were protected from mortality but experienced severe weight loss and illness (Figure 6B), thus showing a lower therapeutic efficacy for this HA antibody for an H7N9 virus than the anti-NA mAbs tested. These data indicate a high therapeutic potency for representative human anti-N9 mAbs against H7N9 influenza infection.
Previous studies with human anti-NA antibodies against 2009 A(H1N1)pdm virus suggested a critical role for Fc-region-mediated effector functions in protection (DiLillo et al., 2016; Yasuhara et al., 2019). We expressed the NA-22, NA-45, NA-73, and NA-80 mAbs in recombinant form with the human IgG1 isotype (rIgG1) and also as LALA Fc mutants that bind only weakly to human Fcγ-receptors (FcγR) and have a diminished function (Hessell et al., 2007). The binding of LALA Fc mutants to high- or low-affinity mouse FcγRs also is reduced, indicating the utility of mouse models to study Fc-mediated function by human mAbs (Arduin et al., 2015; Dekkers et al., 2017). To evaluate whether Fc-mediated function contributed to protection in vivo mediated by the anti-N9 antibodies, we compared the efficacy of wild-type or LALA-variant IgG1s as treatment in mice lethally challenged with H7N9 (Figure 7). The potent neutralizing mAbs NA-73 and NA-80 each afforded complete protection by either IgG1 when delivered at 10 mg/kg dose one day before virus challenge (Figures 7A and 7B) suggesting that neutralizing activity alone is sufficient for protection with high-dose prophylactic treatment. rNA-45 LALA, similarly to rNA-45 IgG1, protected from mortality and severe weight loss, whereas greater protection was achieved by the wild-type IgG1. Notably, Fc function for the weakly neutralizing mAb NA-22 was required for protection. Mice treated with rNA-22 LALA showed signs of severe disease, with only 50% of animals surviving, whereas all animals in the wild-type IgG1 treatment group were protected. We next assessed the relevance of effector functions for protection with strongly neutralizing mAbs NA-73 and NA-80 by limiting their dose. At 2 mg/kg prophylaxis dose, both IgG1- and LALA-treated groups experienced low to moderate weight loss after viral challenge but were protected from mortality, as determined by comparing their survival curves (Figures 7C and 7D). Although greater protection was achieved by the wild-type IgG1 variant of each tested mAb, the effect of Fc-mediated activity was small (Figures 7C and 7D). These results demonstrate that a high level of protection against N7N9 infection can be achieved by direct virus neutralization with mAbs possessing high NA inhibitory activity but might require Fc-mediated effector functions for those mAbs that exhibit weak and/or partial NA inhibitory activity.
Figure 7. Contribution of Fc-Region-Mediated Effector Functions to Protection by Strongly or Weakly Neutralizing Anti-N9 mAbs.

Groups of BALB/c mice were inoculated by the i.p. route with 10 mg/kg (A and B) or with 2 mg/kg (C and D) of recombinant wild type IgG1 or IgG1 LALA Fc variants of indicated anti-NA mAbs or with DENV 2D22 control mAb or with 10 mg/kg of mAb rCR9114. The next day (d0) mice were challenged by the i.n. route with a lethal dose of influenza A/Shanghai/02/2013 IDCDC-RG32A virus and monitored for protection. The weights (A and C) are represented as the group mean ± SEM. The lower dotted line indicates the no-recovery threshold (> 30% weight loss) and endpoint for euthanasia. Body weight change curves were compared by overall test using two-way ANOVA. Survival curves were estimated using the Kaplan-Meier method, and compared as indicated using log rank (Mantel-Cox) test (B and D). Data represent one experiment, and “n” symbol in the plots indicates number of mice per group for each treatment condition (5–10 mice per group).
DISCUSSION
Here, we defined the human NA-specific B cell response to H7N9 infection or vaccination though isolation of human mAbs to N9 NA and detailed studies of their epitopes and mechanisms of action. These studies are of interest because NA-targeted antibody-mediated immunity is increasingly recognized as a correlate of protection against influenza disease, but very little was known prior to these studies about epitope specificity or mechanism of virus neutralization of human N9 NA-specific antibodies. The studies revealed several interesting features of the cellular and molecular basis of human B cell recognition of H7N9 antigens.
First, the B cell response of each individual to HA or NA proteins appears to be independent, as we observed NA-dominant, HA-dominant, or balanced responses in the subject group studied. Previously, serum antibody studies in the context of H7N9 vaccine clinical trials showed induction of substantial immune responses to H7 HA after two doses of an adjuvanted vaccine; seroconversion rates were ~85% among vaccines, and a peak titer observed on day 42 after first vaccination (day 21 after boost) had notable decline of seroconversion at day 385 (~18%) (Jackson et al., 2015; Madan et al., 2016). Comparable results for serum HAI-based seroconversion rates (90%) were shown for H7 after natural H7N9 infection on day 100 from symptom onset, and there was a decrease to 36.4% on day 300 (Ma et al., 2018). In contrast, there are limited data on anti-N9 antibody responses after H7N9 infection or vaccination. Overall patterns of the NI antibody titers and proportion of seroconversion in H7N9 survivors were reported to be similar to HAI antibody titers, and there was a decline to 64% on day 300. However, adjuvanted inactivated monovalent H7N9 vaccinees were not evaluated for the NA-targeted response. Furthermore, recent studies of seasonal influenza vaccines reported that inactivated split or subunit vaccines poorly display key NA epitopes and rarely induce NA-reactive B cells (Chen et al., 2018). Here, we report Epstein-Barr virus immortalization and detection of N9-reactive human peripheral blood memory B cells on day 42 after first vaccination or approximately eleven months after natural H7N9 infection. Our results indicate that vaccination with inactivated monovalent H7N9 vaccine can elicit a robust anti-N9 B cell immune response if formulated with adjuvant. Moreover, some vaccinees exhibited an immunodominant response focused on the N9 NA antigen. The difference in results of response to infection compared with the studies of seasonal vaccine-induced NA-reactive B cell response might be due to the difference in timing of response characterization after vaccination (7 days for seasonal versus 42 days after first vaccination for H7N9). In addition, we studied memory B cells whereas the previous seasonal vaccine studies relied on characterization of plasmablasts. Interestingly, vaccination with H5N1 monovalent inactivated adjuvanted vaccine also induced a substantial anti-NA response (Khurana et al., 2018). Our study did not show a relation in the frequency of N9- and H7-targeted B cells within donors, which is in agreement with the previous findings from comparison of N1- and H1- targeted individual B cell responses after natural human infection with 2009 A(H1N1)pdm virus (Karunarathna et al., 2019).
Second, the N9 NA-reactive response is overwhelmingly subtype-specific, with little evidence of heterosubtypic cross-reactivity. We did not identify any anti-N9 mAbs that cross-reacted with NA proteins from seasonal IAVs, despite the fact that serum samples of these donors contained polyclonal antibody mixes that cross-reacted with many NA antigens from group 2, including seasonal N2 NAs. These findings suggest the N9 NA is antigenically distinct from the other NA subtypes of IAVs. Relatively low amino acid sequence homology (44%) between the N9 and N2 NA antigens used in our study, and amino acid differences within epitopes targeted by NA-73, NA-80, NA-63, and NA-22 mAbs in N2 NAs when compared with that of N9 NAs, support this conclusion. It is possible that B cells encoding heterosubtypic mAbs are elicited at very low frequency and could not be isolated using the hybridoma method that we used here. Alternatively, there could be differences in breadth in mAbs derived from memory B cell compared with plasma-cell-derived serum antibodies. In contrast, many H7-reactive human mAbs cross-react with seasonal H3 HA antigens (Thornburg et al., 2016). Overall, previous studies of NA-targeted immune response in animal models (Walz et al., 2018; Wohlbold et al., 2015) or polyclonal antibody studies in humans (Chen et al., 2018) also suggested frequent induction of subtype-specific responses.
Third, the human response to N9 NA recognizes diverse antigenic sites on the surface of NA. About 10% of the mAbs isolated here bind directly to the NA active site, but most do not. Nevertheless, many of the mAbs that do not directly bind to residues in the enzyme active site inhibit SA cleavage when tested with a large-substrate assay, suggesting that indirect inhibition of NA activity by steric inhibition of binding is in fact the major mechanism of NA-specific mAb-mediated immunity. We also investigated whether these antibodies block virus attachment. N9 NA is particularly interesting because it possesses an additional SA binding site distinct from the enzyme active site (Laver et al., 1984). Although this site binds to SA, the SA specificity and the binding properties vary from those of the HA receptor-binding site. The N9 hemadsorption site was demonstrated recently to enhance viral binding to human-like SA receptors and, therefore, was implicated in the transmission of H7N9 virus (Benton etal., 2017). However, our data do not support a hypothesis of biologically relevant contribution of the NA hemadsorption site to H7N9 virus attachment, given that anti-N9 human mAbs failed to inhibit attachment, in contrast to the attachment blocking capacity of anti-H7 mAbs (Benton et al., 2017; Thornburg et al., 2016).
Fourth, although the N9 NA protein appears to be antigenically distinct from seasonal and other IAV NAs, the function of the antigenic regions in diverse NAs seems similar. Early studies with murine NI mAbs demonstrated the presence of an equivalent antigenic region on N2, N9, and N8 NA molecules that forms a nearly continuous surface across the top of the NA head protomer, encircling the enzyme active site (Colman et al., 1987; Gulati et al., 2002; Malby et al., 1994; Saito et al., 1994; Webster et al., 1984). Functional epitopes targeted by human anti-N9 NA antibodies have not been investigated previously. Here, we demonstrated the presence of three main regions targeted by anti-N9 human mAbs with NI activity. Two of these regions (the enzyme active site and the lateral surface of the NA head) overlap and are located on top of the NA head protomer, and they have been reported previously to contain epitopes for N9 NA murine mAbs. These two regions accumulate amino acid alterations under immune pressure and elicit protective antibodies after infection with 2009 A(H1N1)pdm in humans (Yasuhara et al., 2019). We report the recognition of a third N9 NA antigenic region, the interface of two adjacent NA protomers within the tetramer; however, previously, it was shown that N8 and N1 NAs contains epitopes in this region (Saito et al., 1994; Wan et al., 2015). It might be that the membrane-distal, carboxy-terminal part of the N9 NA is the site of recognition for non-inhibiting anti-N9 mAbs given that previously an anti-N1 mAb that lacked NA inhibiting activity was mapped to this region (Job et al., 2018).
The human mAbs isolated here with NI activity recognize both wild-type and neuraminidase inhibitor drug-resistant strain proteins, suggesting they might have utility for prevention of treatment of drug-resistant infections. There is no current alternative for treatment of NI-resistant strains.
Finally, representative human mAbs isolated here protected mice from lethal H7N9 challenge, suggesting they might be of high utility for use in prevention or treatment of human infections. NI antibodies are known to reduce the viral loads and symptoms in infected mice (Job et al., 2018), ferrets (Walz et al., 2018), and humans (Chen et al., 2018; Couch et al., 2013). Several murine anti-N9 mAbs have been tested in protection studies in mice (Wan et al., 2018; Wilson et al., 2016). Previously, murine anti-N9 NA mAbs that inhibited NA enzymatic activity with a small-molecule substrate in vitro provided superior in vivo protection compared with mAbs that inhibited NA activity only in the ELLA assay that utilize large substrate (Wan et al., 2018). Here, all four tested human mAbs to NA9 conferred protection when administered prophylactically and two tested mAbs offered therapeutic protection at a dose 10 mg/kg per animal. Our results from a comparison of wild-type and LALA variants of four anti-N9 mAbs demonstrated that Fc-mediated effector functions might be critical for in vivo protection by some anti-NA mAbs that possess weak and/or partial NI activity. Furthermore, the mAb studies suggest that infection or immunization with monovalent inactivated H7N9 virus vaccine formulated with adjuvant induces anti-N9 NA B cell responses encoding subtype-specific, potent NI, and protective mAbs.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to and be fulfilled by the corresponding author James E. Crowe, Jr. (james.crowe@vumc.org). Materials described in this paper are available for distribution under the Uniform Biological Material Transfer Agreement, a master agreement that was developed by the NIH to simplify transfers of biological research materials.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Donors
Blood samples from two survivors of natural H7N9 infection, as described (Skowronski et al., 2016), were collected after written informed consent approximately 11 months after recovery from infection. The study was approved by the Institutional Review Board of Vanderbilt University Medical Center. The vaccinated subject PBMC samples used in this study were obtained previously from subjects enrolled in a phase II randomized, doubled-blinded, controlled study in healthy adults to assess the safety, reactogenicity, and immunogenicity of a monovalent influenza A/H7N9 virus vaccine (inactivated influenza vaccine) at different dosages with or without AS03 or MF59 adjuvant (DMID 13-0033). The ClinicalTrials.gov number for that trial was . That previous vaccine study was approved by the Institutional Review Board of Vanderbilt University Medical Center.
Mice
BALB/c mice were purchased from Jackson Laboratories (Bar Harbor). Breeding, maintenance and experimentation complied with Vanderbilt Institutional Animal Care and Use Committee regulations. Details of mouse husbandry can be found in the “In vivo protection study” subsection.
Cell Lines and Viruses
A/Shanghai/2/2013 (H7N9)-PR8-IDCDC-RG32A (Influenza Reagent Resource) were propagated and titered in monolayer cultures of MDCK cells (ATCC). Cells were maintained in Dulbecco’s Modified Eagle Medium (GIBCO DMEM, Invitrogen) at 37°C in 5% CO2. H7N9 A/Shanghai/2/2013 (H7N9)-PR8-IDCDC-RG32A were manipulated under BSL-2 conditions with BSL-3 practices.
Antigens
Recombinant N9 NA protein based on the sequence from the influenza virus A/Shanghai/2/2013 (SH13) (H7N9) (GISAID accession number EPI_ISL_138738) and its mutated variants N9 SH13 E119V and N9 SH13 R292K, or N9 A/Hunan/02650/2016 (H7N9), N9 A/whale/Maine/1/1984 (H13N9), N6 A/Sichuan/26221/2014 (H5N6), N6 A/Hubei/29578/2016 (H5N6) or N7 A/Netherlands/219/2003 (H7N7) were expressed by a baculovirus expression system in insect cells. N2 A/mallard/Netherlands/3/ 1999, N2 A/Wisconsin/67/2005, N2 A/Brisbane/10/2007, N2 A/shorebird/Delaware/127/1997, N2 A/turkey/Wisconsin/1/1966, N1 A/Puerto Rico/8/1934, N1 A/New Caledonia/20/1999, N1 A/Brisbane/59/2007, N1 A/California/04/2009, N4 A/gray teal/Australia/ 2/1979, and N8 A/equine/Pennsylvania/1/2007 were purchased from BEI Resources.
METHOD DETAILS
Cloning, Expression and Purification of Recombinant Antigens
The cDNAs corresponding to the NA ectodomain plus stalk region (37 to 468 aa) of N9, N6 or N7 NAs were inserted into a baculovirus transfer vector, pFastbacHT-A (Thermo Fisher Scientific) with an N-terminal gp67 signal peptide, thrombin cleavage site, His6-tag, and an N-terminal tetramerization domain, essentially as previously described (Zhu et al., 2008) and (Zhu et al., 2012). For NA molecules from zanamivir-resistant N9 mutant viruses, the M-PIPE mutagenesis method (Klock and Lesley, 2009) was used to mutate the wild-type N9 plasmid. The constructed plasmids were used to transform DH10bac competent bacterial cells by site-specific transposition (Tn-7 mediated) to form a recombinant bacmid with β-galactosidase blue-white receptor selection. The purified recombinant bacmids were used to transfect Sf9 insect cells for overexpression. NA proteins were produced by inoculating suspension cultures of Sf9 cells with recombinant baculovirus at a MOI of 5 to 10 and incubating at 28°C while shaking at 110 RPM. After 72 h, Sf9 cells were removed by centrifugation and supernatants containing secreted, soluble NAs were concentrated and purified by metal affinity chromatography using Ni-nitrilotriacetic acid (NTA) resin (QIAGEN). The uncleaved NA ectodomain plus stalk region with tetramerization domain and His6-tag were buffered in 20 mM Tris pH 8.0, 150 mM NaCl.
A cDNA encoding the H7 HA gene from the A/Shanghai/02/2013 H7N9 virus was optimized for expression, and cDNAs were synthesized (Genscript) as soluble trimeric constructs by replacing the transmembrane and cytoplasmic domain sequences with cDNAs encoding the GCN4 trimerization domain and a His6-tag at the C terminus. Synthesized genes were subcloned into the pcDNA3.1(+) mammalian expression vector (Thermo Fisher Scientific). HA protein was expressed by transient transfection of 293F cells with polyethyleneimine transfection reagent and was grown in expression medium (Freestyle 293 Expression Medium; Thermo Fisher Scientific). The supernatants were harvested after 7 days, filter-sterilized with a 0.4-μm filter, and purified with HisTrap TALON FF crude columns (GE Healthcare Life Sciences).
Generation of Human Hybridomas and Antibody Isolation
Human hybridomas were generated as described previously (Smith and Crowe, 2015). Briefly, human B cells in the PBMC suspension were immortalized by transformation with Epstein-Barr virus in the presence of CpG10103, cyclosporin A and a Chk2 inhibitor. On day 8, the supernatants from transformed B cells were used to screen for the presence of antigen-reactive antibodies against antigens based on the HA and NA sequences from A/Shanghai/2/2013 (H7N9) and NA sequences from A/California/04/2009 (H1N1), A/Wisconsin/67/2005 (H3N2), A/Pennsylvania/1/2007 (H3N8) using a capture ELISA. Any well for which the binding of antibodies to antigen in supernatant was greater than two standard deviations above background was deemed positive, and the percentage of H7- or N9-reactive EBV-transformed B cells was calculated as the number of positive wells divided by the total number of LCLs in all wells, multiplied by 100. A threshold of 0.25% of antigen-reactive LCLs was used to determine a positive response. We identified one clone with confirmed reactivity to both recombinant NA and HA antigens, but further analysis revealed that the mAb obtained reacted with the His-tag that is present on both the HA and NA antigens used in the study. Cells from the wells containing B cells secreting NA-reactive antibodies were fused with HMMA2.5 myeloma cells using a BTX ECM 2001 electro cell manipulator, and human hybridomas were selected in medium with HAT solution containing ouabain. The hybridomas were cloned by flow cytometric sorting of single cells into 384-well plates and then expanded in culture. The selected cell line with highest level of IgG secretion for each clone was grown initially in hybridoma growth medium (ClonaCell-HY Medium E from STEMCELL Technologies) and then switched to serum-free medium (GIBCO Hybridoma-SFM, Thermo Fisher Scientific) for antibody expression and purification. IgG from the hybridoma cell line supernatants was purified by affinity chromatography using protein G columns (GE Healthcare Life Sciences, Protein G HP Columns). Purified IgG generated from hybridomas was used for all in vitro studies.
ELISA
For screening ELISA, plates were coated with antigen at 1 μg/mL for H7 HA or 2 μg/mL for NAs, in PBS. After blocking with 5% non-fat dry milk, 2% goat serum and 0.1% Tween-20 in PBS, plates were incubated with culture supernatants followed by incubation with 1:4,000 dilution of anti-human IgG conjugated with alkaline phosphatase (Meridian, Life Science Inc.). Plates were developed, and supernatants were counted as H7- or NA-reactive if their absorbance was 2.5-fold above the background from wells containing medium. For cross-reactivity assays, serum samples were assessed at three-fold dilutions starting from 1/20, in triplicate and purified mAbs were assessed at concentrations ranging from 10 μg/mL to 0.1 ng/mL, in triplicate. The serum antibody endpoint titer was calculated as the serum dilution that produced an optical density (OD) at 405 nm of 0.65 absorbance units in the ELISA assay using Prism 5.0 software (GraphPad). The 0.65 OD 405 nm reading fell in the linear part of the dilution curve. EC50 values for mAbs were determined using Prism 5.0 software (GraphPad) after log transformation of antibody concentration using sigmoidal dose-response nonlinear fit analysis with R2 values greater than 0.85, as described previously (Thornburg et al., 2013).
Characterization of Antibody Variable Genes
Antibody heavy- and light-chain variable region genes were sequenced from antigen-specific hybridoma lines that had been cloned biologically from flow cytometry. Briefly, total RNA was extracted using the RNeasy Mini kit (QIAGEN) and reverse-transcriptase PCR (RT-PCR) amplification of the antibody gene cDNAs was performed using the PrimeScript One Step RT-PCR kit (Clontech) according to the manufacturer’s protocols with gene-specific primers, as previously described (Thornburg et al., 2016). PCR products were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter) and sequenced directly using an ABI3700 automated DNA sequencer without cloning. The identities of gene segments and mutations from germlines were determined by alignment using the ImMunoGeneTics database (http://www.imgt.org) (Ruiz et al., 2000). Antibodies were considered as clonally related if they shared the same inferred IGHV and IGHJ genes and shared at least 75% nucleotide identity in the HCDR3 (Di Noia and Neuberger, 2007).
Cloning, Expression and Purification of Recombinant Antibodies
For the expression of recombinant forms of antibody clones, nucleotide sequences of antibody heavy- and light-chain antibody variable genes were codon-optimized for mammalian expression and synthesized at Twist Biosciences. Resulting gene fragments were directly cloned at Twist Biosciences into the pTwist CMV BetaGlobin WPRE NEO mammalian expression vector (Twist Biosciences) that had already been fused with the heavy chain Fc domain of human IgG1 or Fc LALA variant. Recombinant antibodies were expressed by transient transfection of 293F cells with polyethyleneimine transfection reagent and was grown in expression medium (Freestyle 293 Expression Medium; Thermo Fisher Scientific). The supernatants were harvested after 7 days, filter-sterilized with a 0.4-μm filter, and purified by affinity chromatography using HiTrap MabSelect Sure columns (GE Healthcare Life Sciences). Purified recombinant IgG were used for in vivo studies.
Affinity Measurements Using Surface Plasmon Resonance
Purified anti-N9 mAbs binding affinity to SH13 N9 NA was measured using a Biacore 8K instrument (GE Healthcare Life Sciences). Briefly, IgGs were captured on a Protein G sensor chip (GE Healthcare Life Sciences) with final surface densities of ~245 to 400 relative units (RU). Kinetic measurements were preformed using single-cycle kinetics experimental setup, by injecting a 0.156 μg/mL solution of IgG, and then bound with four-fold serial dilutions of N9 NA (starting from 100 nM). Dissociation data for IgGs were collected for 10 min. Binding data were globally fit to a bivalent analyte model (using Biacore 8K control software). This analysis determined the kinetic rate constants (Kon, Koff), from which the apparent KD was then calculated as Koff/Kon. Dissociation % indicates percent of IgG dissociated from antigen at time of dissociation step, according to relative unit (RU) values.
Hemagglutination Inhibition Assay
The hemagglutination inhibition (HAI) assay was performed with A/Shanghai/2/2013 (H7N9)-PR8-IDCDC-RG32A. For HAI, 25 μL of four hemagglutination units of virus were incubated for 1 h at room temperature with 25 μL two-fold serial dilutions of antibodies starting at 10 μg/mL in PBS. The 50 μL of antibody-virus mixture was incubated for 45 min at 4°C with 50 μL of turkey red blood cells (Rockland Immunochemicals,) diluted in PBS. The IC100 value was defined as the lowest antibody concentration that inhibited hemagglutination of red blood cells.
Egress Inhibition Assay
MDCK cells were seeded in plain Dulbecco Modified Eagle Medium (GIBCO DMEM, Thermo Fisher Scientific) containing 10% FBS in 96-well plates overnight. The cells were washed three times with Virus Growth Medium (VGM) (DMEM with 2% BSA and 2 μg/mL TPCK treated trypsin (Sigma-Aldrich)) and 100 μL of one multiplicity of infection of A/Shanghai/2/2013 (H7N9)-PR8-IDCDC-RG32A virus in VGM added to the cells and incubated for 3 h at 37°C in 5% CO2. The cells were then washed with VGM again and replenished with VGM containing three-fold serial dilutions of mAbs or zanamivir (GlaxoSmithKline), starting at the highest concentration of mAbs 10 μg/mL or equimolar. The plates were incubated for 21 h at 37°C in 5% CO2, and the supernatants were collected for performing the HA assay. For HA assay, we used turkey red blood cells (Rockland Immunochemicals) that were washed and diluted to 0.5% in PBS. A volume of 50 μL of the supernatants was incubated with 50 μL of the 0.5% turkey red blood cells in v-bottom plates for 1ch at 4°C. The IC100 values were defined as the lowest antibody concentration added to infected MDCK cells that correspondent to absence of virus in supernatant according to HA of red blood cells.
Enzyme-linked Lectin-based Assay (ELLA) and NA-Fluor Assay to Determine Neuraminidase Inhibition (NI)
Before performing assays, A/Shanghai/2/2013 (H7N9)-PR8-IDCDC-RG32A virus was titered to determine the optimum dilution. An NA inhibition assay with a small substrate was performed with the NA-Fluor Influenza Neuraminidase Assay kit (Thermo Fisher Scientific) according to protocol. For ELLA NI assays, 96-well ELISA plates were coated with 100 μL of 25 μg/mL of fetuin from fetal bovine serum (Sigma-Aldrich) diluted in 0.1 M PBS and incubated at overnight 4°C. 50 μL of three-fold serial dilutions of each antibody starting at a molar concentration of 66.7 nM (10 μg/mL) in PBS was added to 50 μL of pre-optimized virus dilution in PBS containing 0.9 mM CaCl2, 0.5 mM MgCl2, 1% BSA and 0.5% Tween. The fetuin-coated plates were washed, and the mAb-virus mixture was added to the plates and incubated for two h at 37°C. The plates were washed with PBST, and 100 μL of HRP-conjugated lectin from Arachis hypogaea (Sigma-Aldrich) at 5 μg/ mL was added to the plates and incubated for 1.5 h at RT. The plates were washed and 100 μL of TMB substrate was added to the plates, and the reaction was stopped with 1N HCl. The optical density values were measured at 450 nm wavelength on a BioTek plate reader. In both assays, each dilution was performed in triplicate, and the IC50 values were calculated in Prism software (GraphPad) using non-linear regression analysis.
Competition-Binding Groups
Competition-binding for the N9 mAb panel was performed using sequential binding on a surface plasmon resonance instrument (IBIS MX96, Wasatch Microfluidics). All experiments were conducted on a 96-ligand array with recombinant N9 NA from the A/Shanghai/2/2013 antigen. A heatmap was obtained by SPRi-premix experiments in which the first antibody and N9 NA were premixed before adding to the array printed with the second antibodies. Additional competition-binding experiments for mAbs NA-73, NA-80, NA-95 and NA-108 were performed using a Biacore 8K instrument (GE Healthcare Life Sciences, USA). Briefly, anti-His-tag monoclonal antibody was amine-coupled to a CM5 sensor chip (GE Healthcare Life Sciences) with final anti-His-tag capture surface densities of ~10,600 RU. Recombinant SH13 N9 NA was captured at concentration 1 μg/mL. Primary and secondary mAbs were loaded at concentration 10 μg/mL. The regeneration step was performed using 10 mM glycine buffer at pH 1.5.
Epitope Mapping Using Peptide Fragmentation and Hydrogen-Deuterium Exchange Mass Spectrometry
To maximize peptide probe coverage, the optimized quench condition was determined prior to deuteration studies (Hsu et al., 2009; Li et al., 2011). In short, NA antigen was diluted with H2O buffer 8.3 mM Tris, 150 mM NaCl, in H2O, pH 7.15 at 0°C and then quenched with 0.8% formic acid (v/v) containing various concentration of GuHCl (0.8 to 6.4 M) and Tris(2-carboxyethyl) phosphine (TCEP) (0.1 or 1.0 M). After incubating on ice for 5 min, the quenched samples were diluted 4-fold with 0.8% formic acid (v/v) containing 16.6% (v/v) glycerol and then were frozen at −80°C until they were transferred to the cryogenic autosampler. 6.4 M GuHCl, 1.0 M TCEP in 0.8% formic acid gave an optimal peptide coverage map.
The samples later were thawed automatically on ice and then immediately passed over an AL-20-pepsin column (16 μL bed volume, 30 mg/mL porcine pepsin (Sigma-Aldrich)). The resulting peptides were collected on a C18 trap and separated using a C18 reversed phase column (Vydac) running a linear gradient of 0.046% (v/v) trifluoroacetic acid, 6.4% (v/v) acetonitrile to 0.03% (v/v) trifluoroacetic acid, 38.4% (v/v) acetonitrile over 30 min with column effluent directed into an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Data were acquired in both data-dependent MS:MS mode and MS1 profile mode. Proteome Discoverer software (Thermo Finnigan Inc.) was used to identify the sequence of the peptide ions. DXMS Explorer (Sierra Analytics Inc., Modesto, CA) was used for the analysis of the mass spectra, as described previously (Hamuro et al., 2004). mAb-NA complexes were prepared by mixing N9 mAbs with tetrameric N9 NA at a 1:1.1 stoichiometric ratio. The mixtures were incubated at 25°C for 30 min. All functionally deuterated samples, with the exception of the equilibrium-deuterated control, and buffers were pre-chilled on ice and prepared in the cold room.
Functional deuterium-hydrogen exchange reactions were initiated by diluting free NA or antibody-bound NA stock solution with D2O buffer (8.3 mM Tris, 150 mM NaCl, in D2O, pDREAD 7.15) at a 1:2 vol/vol ratio. At 10, 100 or 1,000 s, quench was added to the respective samples, and then samples were frozen at −80°C. In addition, non-deuterated samples, equilibrium-deuterated back-exchange control samples were prepared, as previously described (Hsu et al., 2009; Li et al., 2011; Lu et al., 2012). The centroids of the isotopic envelopes of non-deuterated, functionally deuterated, and fully deuterated peptides were measured using DXMS Explorer, and then converted to corresponding deuteration levels with corrections for back-exchange (Zhang and Smith, 1993).
Negative Stain Electron Microscopy
Complexes were made by adding a 1:1 molar ratio of recombinant A/Shanghai/2/2013 N9 NA and Fab and incubated at room temperature for one h before keeping on ice. The complexes were added to 400 mesh copper grids coated with carbon and then stained with 2% uranyl formate. Raw micrographs were collected on a 120KeV Tecnai Spirit microscope equipped with a TemCam F426 detector. Data collection was used with Leginon (Potter et al., 1999) and the data were processed through Appion (Lander et al., 2009) with particles chosen with DoG Picker (Voss et al., 2009). Particle stacks were produced and 2D classes were made by MSA/MRA (Ogura et al., 2003). False coloring of Fabs was created using Photoshop software (Adobe), and Figure 5B was made with UCSF Chimera (Resource for Biocomputing, Visualization, and Informatics) (Pettersen et al., 2004).
In Vivo Protection Study
Mice were housed in individually ventilated cage racks with negative pressure ventilation and air filtering (Allentown, Inc., Allentown, NJ). To assess protective efficacy of mAbs, six- to eight-week old female BALB/c mice were inoculated i.p. with 200 μg (~10 mg/kg) or 40 μg (~2 mg/kg) of individual mAbs one day before or one day after virus challenge. Human anti-dengue virus mAb DENV 2D22 served as a mock control treatment. In ABSL-2 facilities, ketamine-xylazine anesthetized mice were inoculated i.n. with 104 FFU SH13 H7N9 (~4 x LD80) in 50 μL of sterile PBS. Mice were weighed and monitored daily for morbidity, and those losing over 30% of initial body weight were humanely euthanized as per IACUC requirements. The clinical symptoms were scored as follows: 0 - at healthy state, 1 - signs of ruffled fur and/or back arching, 2 - extensive fur ruffling and hunching posture, 3 – as above and little mobility or lethargy, 4 - moribund stage.
QUANTIFICATION AND STATISTICAL ANALYSIS
The descriptive statistics mean ± SEM or mean ± SD were determined for continuous variables as noted. Survival curves were estimated using the Kaplan-Meier method and curves compared using the two-sided log rank test (Mantel-Cox) with subjects right censored, if they survived until the end of the study. Body weight change curves were compared by overall test using two-way ANOVA. Statistical analyses were performed using Prism v5.0 (GraphPad). Statistical details of experiments can be found in the figure legends for Figures 3, 6, 7, S1, and S2. The “n” symbol in the plots indicates number of mice per group for each treatment condition.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study. This study did not generate code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NA-108 (hybridoma-produced Ig) | This study | N/A |
| NA-73 (hybridoma-produced Ig) | This study | N/A |
| NA-80 (hybridoma-produced Ig) | This study | N/A |
| NA-95 (hybridoma-produced Ig) | This study | N/A |
| NA-63 (hybridoma-produced Ig) | This study | N/A |
| NA-77 (hybridoma-produced Ig) | This study | N/A |
| NA-45 (hybridoma-produced Ig) | This study | N/A |
| NA-127 (hybridoma-produced Ig) | This study | N/A |
| NA-171 (hybridoma-produced Ig) | This study | N/A |
| NA-55 (hybridoma-produced Ig) | This study | N/A |
| NA-69 (hybridoma-produced Ig) | This study | N/A |
| NA-5 (hybridoma-produced Ig) | This study | N/A |
| NA-22 (hybridoma-produced Ig) | This study | N/A |
| NA-17 (hybridoma-produced Ig) | This study | N/A |
| NA-3 (hybridoma-produced Ig) | This study | N/A |
| NA-181 (hybridoma-produced Ig) | This study | N/A |
| NA-121 (hybridoma-produced Ig) | This study | N/A |
| NA-86 (hybridoma-produced Ig) | This study | N/A |
| NA-4 (hybridoma-produced Ig) | This study | N/A |
| NA-93 (hybridoma-produced Ig) | This study | N/A |
| NA-89 (hybridoma-produced Ig) | This study | N/A |
| NA-157 (hybridoma-produced Ig) | This study | N/A |
| NA-110 (hybridoma-produced Ig) | This study | N/A |
| NA-148 (hybridoma-produced Ig) | This study | N/A |
| NA-126 (hybridoma-produced Ig) | This study | N/A |
| NA-16 (hybridoma-produced Ig) | This study | N/A |
| NA-164 (hybridoma-produced Ig) | This study | N/A |
| NA-81 (hybridoma-produced Ig) | This study | N/A |
| NA-111 (hybridoma-produced Ig) | This study | N/A |
| NA-97 (hybridoma-produced Ig) | This study | N/A |
| NA-175 (hybridoma-produced Ig) | This study | N/A |
| NA-144 (hybridoma-produced Ig) | This study | N/A |
| NA-37 (hybridoma-produced Ig) | This study | N/A |
| NA-177 (hybridoma-produced Ig) | This study | N/A |
| NA-152 (hybridoma-produced Ig) | This study | N/A |
| rNA-80 IgG1 | This study | N/A |
| rNA-80 LALA | This study | N/A |
| rNA-73 IgG1 | This study | N/A |
| rNA-73 LALA | This study | N/A |
| rNA-22 IgG1 | This study | N/A |
| rNA-22 LALA | This study | N/A |
| rNA-45 IgG1 | This study | N/A |
| rNA-45 LALA | This study | N/A |
| H7.167 (hybridoma-produced Ig) | Thornburg et al., 2016 | N/A |
| DENV-2D22 (hybridoma-produced Ig) | Smith et al., 2015 | N/A |
| Goat anti-Human IgG (Fc)-AP | Meridian Life Science, Inc. | Cat# W99008A |
| Bacterial and Virus Strains | ||
| A/Shanghai/2/2013 (H7N9)-PR8-IDCDC-RG32A | Influenza Reagent Resource | Cat# FR-1389 |
| DH10Bac competent cells | Thermo Fisher Scientific | Cat# 10361012 |
| Biological Samples | ||
| PBMCs from H7N9 infection survivors | This paper | VVC Donor ID #957 and #958 |
| PBMCs from 30 otherwise healthy subjects previously immunized with monovalent inactivated A/Shanghai/02/2013 (Sh2) H7N9 vaccine by the NIH Vaccine Treatment and Evaluation Unit (DMID 13-0033). | Jackson et al., 2015 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Zanamivir (Relenza) | GlaxoSmithKline | NDC0713068101 |
| 1-Step Ultra TMB-ELISA | Thermo Fisher Scientific | Cat#34029 |
| Dulbecco’s Phosphate-Buffered Saline, 1X with calcium and magnesium | Corning Life Sciences | Cat#21-030-CM |
| Freestyle 293 expression medium | Thermo Fisher Scientific | Cat#12338002 |
| 50x HAT media supplement | Sigma-Aldrich | Cat#H0137 |
| Ouabain | Sigma-Aldrich | Cat#O3125 |
| HyClone insect cell culture medium | GE Healthcare Life Sciences | Cat#SH30280.03 |
| Fetal Bovine Serum, ultra-low IgG | Thermo Fisher Scientific | Cat#16250078 |
| TPCK treated trypsin | Sigma-Aldrich | Cat#T1426 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A3608 |
| 100x Penicillin Streptomycin Glutamine | Thermo Fisher Scientific | Cat#10378016 |
| ClonaCell-HY Medium E | Stem Cell Technologies | Cat#03805 |
| ClonaCell-HY Medium A | Stem Cell Technologies | Cat#03801 |
| DMEM, high glucose, GlutaMAX™ Supplement | Thermo Fisher Scientific | Cat#10566-024 |
| GIBCO Hybridoma-SFM | Thermo Fisher Scientific | Cat#12045076 |
| Recombinant N9 NA A/Shanghai/2/2013 (H7N9) | This study | N/A |
| Recombinant N9 NA A/Shanghai/2/2013 (H7N9) E119V | This study | N/A |
| Recombinant N9 NA A/Shanghai/2/2013 (H7N9) R292K | This study | N/A |
| Recombinant N9 NA A/Hunan/02650/2016 (H7N9) | This study | N/A |
| Recombinant N9 NA A/whale/Maine/1/1984 (H13N9) | This study | N/A |
| Recombinant N6 NA A/Sichuan/26221/2014 (H5N6) | This study | N/A |
| Recombinant N6 NA A/Hubei/29578/2016 (H5N6) | This study | N/A |
| Recombinant N7 NA A/Netherlands/219/2003 (H7N7) | This study | N/A |
| Recombinant N2 NA A/mallard/Netherlands/3/1999 | BEI Resources | Cat#NR-29011 |
| Recombinant N2 NA A/Wisconsin/67/2005 | BEI Resources | Cat#NR-19237 |
| Recombinant N2 NA A/Brisbane/10/2007 | BEI Resources | Cat#NR-43784 |
| Recombinant N2 NA A/shorebird/Delaware/127/1997 | BEI Resources | Cat#NR-657 |
| Recombinant N2 NA A/turkey/Wisconsin/1/1966 | BEI Resources | Cat#NR-43783 |
| Recombinant N1 NA A/Puerto Rico/8/1934 | BEI Resources | Cat#NR-19235 |
| Recombinant N1 NA A/New Caledonia/20/1999 | BEI Resources | Cat#NR-43779 |
| Recombinant N1 NA A/Brisbane/59/2007 | BEI Resources | Cat#NR-43785 |
| Recombinant N1 NA A/California/04/2009 | BEI Resources | Cat#NR-19234 |
| Recombinant N4 NA A/grey teal/Australia/2/1979 | BEI Resources | Cat#NR-656 |
| Recombinant N8 NA A/equine/Pennsylvania/1/2007 | BEI Resources | Cat#NR-13523 |
| Recombinant H7 HA A/Shanghai/2/2013 (H7N9) | Thornburg et al., 2016 | N/A |
| CpG10103 (TCGTCGTTTTTCGGTCGTTTT) | Synthesized by Invitrogen | N/A |
| Cyclosporin A | Sigma-Aldrich | Cat#C1832 |
| Chk2 inhibitor | Sigma-Aldrich | Cat#C3742 |
| Non-fat dry milk | Bio Rad | Cat#1706404 |
| Goat serum | Thermo Fisher Scientific | Cat#16210072 |
| Polyethylenimine (PEI) transfection reagent | Polysciences | Cat#23966 |
| Fetuin from fetal bovine serum | Sigma-Aldrich | Cat#F3004 |
| HRP-conjugated lectin from Arachis hypogaea | Sigma-Aldrich | Cat#L7759 |
| Critical Commercial Assays | ||
| RNeasy Mini kit | QIAGEN | Cat#74106 |
| PrimeScript One Step RT-PCR kit | Clontech | Cat#RR055A |
| NA-Fluor Influenza Neuraminidase Assay kit | Thermo Fisher Scientific | Cat#4457091 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Mouse-human HMAA 2.5 myeloma cell line | Dr. Marshall Posner | N/A |
| MDCK | ATCC | Cat#CCL-34 |
| Sf9 cells | ATCC | Cat#CRL-1711 |
| 293F cells | Thermo Fisher Scientific | Cat#11625019 |
| NA-108 hybridoma clone | This study | N/A |
| NA-73 hybridoma clone | This study | N/A |
| NA-80 hybridoma clone | This study | N/A |
| NA-95 hybridoma clone | This study | N/A |
| NA-63 hybridoma clone | This study | N/A |
| NA-77 hybridoma clone | This study | N/A |
| NA-45 hybridoma clone | This study | N/A |
| NA-127 hybridoma clone | This study | N/A |
| NA-171 hybridoma clone | This study | N/A |
| NA-55 hybridoma clone | This study | N/A |
| NA-69 hybridoma clone | This study | N/A |
| NA-5 hybridoma clone | This study | N/A |
| NA-22 hybridoma clone | This study | N/A |
| NA-17 hybridoma clone | This study | N/A |
| NA-3 hybridoma clone | This study | N/A |
| NA-181 hybridoma clone | This study | N/A |
| NA-121 hybridoma clone | This study | N/A |
| NA-86 hybridoma clone | This study | N/A |
| NA-4 hybridoma clone | This study | N/A |
| NA-93 hybridoma clone | This study | N/A |
| NA-89 hybridoma clone | This study | N/A |
| NA-157 hybridoma clone | This study | N/A |
| NA-110 hybridoma clone | This study | N/A |
| NA-148 hybridoma clone | This study | N/A |
| NA-126 hybridoma clone | This study | N/A |
| NA-16 hybridoma clone | This study | N/A |
| NA-164 hybridoma clone | This study | N/A |
| NA-81 hybridoma clone | This study | N/A |
| NA-111 hybridoma clone | This study | N/A |
| NA-97 hybridoma clone | This study | N/A |
| NA-175 hybridoma clone | This study | N/A |
| NA-144 hybridoma clone | This study | N/A |
| NA-37 hybridoma clone | This study | N/A |
| NA-177 hybridoma clone | This study | N/A |
| NA-152 hybridoma clone | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/cJ | The Jackson Laboratory | Cat#000651 |
| Recombinant DNA | ||
| pTwist_NA-80_hG1 | This paper | N/A |
| pTwist_NA-80_hK | This paper | N/A |
| pTwist_NA-80_hG1LALA | This paper | N/A |
| pTwist_NA-73_hG1 | This paper | N/A |
| pTwist_NA-73_hL | This paper | N/A |
| pTwist_NA-73_hG1LALA | This paper | N/A |
| pTwist_NA-22_hG1 | This paper | N/A |
| pTwist_NA-22_hL | This paper | N/A |
| pTwist_NA-22_hG1LALA | This paper | N/A |
| pTwist_NA-45_hG1 | This paper | N/A |
| pTwist_NA-45_hL | This paper | N/A |
| pTwist_NA-45_hG1LALA | This paper | N/A |
| pFastbacHT-A N9 NA A/Shanghai/2/2013 ectodomain | This paper | N/A |
| pFastbacHT-A N9 NA A/Shanghai/2/2013 E119V | This paper | N/A |
| pFastbacHT-A N9 NA A/Shanghai/2/2013) R292K | This paper | N/A |
| pFastbacHT-A N9 NA A/Hunan/02650/2016 ectodomain | This paper | N/A |
| pFastbacHT-A N9 NA A/whale/Maine/1/1984 ectodomain | This paper | N/A |
| pFastbacHT-A N6 NA A/Sichuan/26221/2014 ectodomain | This paper | N/A |
| pFastbacHT-A N6 NA A/Hubei/29578/2016 ectodomain | This paper | N/A |
| pFastbacHT-A N7 NA A/Netherlands/219/2003 ectodomain | This paper | N/A |
| pcDNA3.1(+) H7 HA A/Shanghai/2/2013 | Thornburg et al., 2016 | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.2 | GraphPad Software, Inc. | https://www.graphpad.com |
| ImMunoGeneTics database | Ruiz et al., 2000. | http://www.imgt.org/ |
| Appion | Lander et al., 2009 | http://emg.nysbc.org/redmine/projects/appion/wiki/Appion_Home |
| PyMOL | Schrödinger, LLC | https://www.pymol.org/ |
| Other | ||
| Turkey red blood cells | Rockland Immunochemicals | Cat#R313 |
| Agencourt AMPure XP magnetic beads | Beckman Coulter | Cat#A63880 |
| BD LSR2 (3-laser) flow cytometer | BD Biosciences | N/A |
| ECM 2001 Electro Cell Manipulator | BTX | N/A |
| ÄKTA pure chromatography system | GE Healthcare Life Sciences | N/A |
| Tecnai Spirit electron microscope with TemCam F416 4k x 4k CCD | Field Electron and Ion (FEI) Company | N/A |
| EL406 washer dispenser | BioTek | N/A |
| Biostack microplate stacker | BioTek | N/A |
| HiTrap Protein G High Performance | GE Healthcare Life Sciences | Cat#17-0404-01 |
| HiTrap MabSelect™ SuRe | GE Healthcare Life Sciences | Cat#11-0034-93 |
| HiTrap TALON crude | GE Healthcare Life Sciences | Cat#28-9537-66 |
| NTA resin | QIAGEN | Cat#30210 |
| Protein G sensor chip | GE Healthcare Life Sciences | Cat#29179316 |
| CM5 sensor chip | GE Healthcare Life Sciences | Cat#29104988 |
| CMD200M sensor chip | Xantec GmbH | N/A |
Highlights.
Human mAbs recognize several antigenic sites on influenza virus N9 NA
The mAbs act by blocking egress of nascent virions from infected cells
Human mAbs mediate prophylactic and therapeutic protection in vivo in mice
Protection is mediated by direct virus neutralization or Fc-region effector function
ACKNOWLEDGMENTS
We thank Merissa Mayo for assistance with acquisition of the survivor donor samples and Rachel Nargi and Rachel Sutton for protein expression and purification work. The original H7N9 vaccine studies (DMID 13-0033; Trials Registration: clinicaltrials.gov Identifier: ) conducted by the Vanderbilt Vaccine Research Program as part of the Vaccine Treatment and Evaluation Unit at Vanderbilt University Medical Center (HHSN272201300023I, PI: Creech). The network of VTEUs is supported by the National Institute of Allergy and Infectious Diseases, part of the National Institutes of Health. Additional support was provided by CTSA award No. UL1 TR002243 from the National Center for Advancing Translational Sciences. We thank Noah T. Ditto from Wasatch Microfluidics and Eric Roush from GE Healthcare Life Sciences for assistance, technical contribution, and advice. This project received support from the U.S. NIH HHSN272201400024C and U19 AI117905 to J.E.C. This work was supported by the National Center for Research Resources UL1 RR024975 and is now at the National Center for Advancing Translational Sciences UL1 TR000445. Flow-cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2019.10.003.
DECLARATION OF INTERESTS
J.E.C. has served as a consultant for Takeda Vaccines, Sanofi Pasteur, Pfizer, and Novavax; is on the Scientific Advisory Boards of CompuVax and Meissa Vaccines; and is Founder of IDBiologics, Inc. All other authors declare no conflict of interest. Vanderbilt University has applied for a patent related to the NA antibodies.
REFERENCES
- Arduin E, Arora S, Bamert PR, Kuiper T, Popp S, Geisse S, Grau R, Calzascia T, Zenke G, and Kovarik J (2015). Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol. Immunol 63, 456–463. [DOI] [PubMed] [Google Scholar]
- Bangaru S, Zhang H, Gilchuk I, Voss T, Irving R, Gilchuk P, Matta P, Zhu X, Lang S, Nieusma T, et al. (2018). A multifunctional human monoclonal neutralizing antibody that targets a unique conserved epitope on influenza HA. Nat. Commun 9, 2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard KR, Brendish NJ, and Clark TW (2018).Treatment of influenza with neuraminidase inhibitors. Curr. Opin. Infect. Dis 31, 514–519. [DOI] [PubMed] [Google Scholar]
- Benton DJ, Wharton SA, Martin SR, and McCauley JW (2017). Role of neuraminidase in influenza A(H7N9) virus receptor binding. J. Virol 91, e02293–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Wohlbold TJ, Zheng NY, Huang M, Huang Y, Neu KE, Lee J, Wan H, Rojas KT, Kirkpatrick E, et al. (2018). Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman PM, Laver WG, Varghese JN, Baker AT, Tulloch PA, Air GM, and Webster RG (1987). Three-dimensional structure of a complex of antibody with influenza virus neuraminidase. Nature 326, 358–363. [DOI] [PubMed] [Google Scholar]
- Couch RB, Atmar RL, Franco LM, Quarles JM, Wells J, Arden N, Niño D, and Belmont JW (2013). Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J. Infect. Dis 207, 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe JE Jr., (2009). Recent advances in the study of human antibody responses to influenza virus using optimized human hybridoma approaches. Vaccine 27 (Suppl 6), G47–G51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, Rispens T, and Vidarsson G (2017). Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 9, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, and Neuberger MS (2007). Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem 76, 1–22. [DOI] [PubMed] [Google Scholar]
- DiLillo DJ, Palese P, Wilson PC, and Ravetch JV (2016). Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest 126, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, et al. (2012). Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbelding EJ, Post DJ, Stemmy EJ, Roberts PC, Augustine AD, Ferguson S, Paules CI, Graham BS, and Fauci AS (2018). A universal influenza vaccine: the strategic plan for the national institute of allergy and infectious diseases. J. Infect. Dis 218, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, Munster V, Wallensten A, Bestebroer TM, Herfst S, Smith D, Rimmelzwaan GF, Olsen B, and Osterhaus AD (2005). Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol 79, 2814–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries LF, Smith GE, and Glenn GM (2013). A recombinant viruslike particle influenza A (H7N9) vaccine. N. Engl. J. Med 369, 2564–2566. [DOI] [PubMed] [Google Scholar]
- Gulati U, Hwang CC, Venkatramani L, Gulati S, Stray SJ, Lee JT, Laver WG, Bochkarev A, Zlotnick A, and Air GM (2002). Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98). J. Virol 76, 12274–12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai R, Schmolke M, Leyva-Grado VH, Thangavel RR, Margine I, Jaffe EL, Krammer F, Solórzano A, García-Sastre A, Palese P, and Bouvier NM (2013). Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nat. Commun 4, 2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y, Anand GS, Kim JS, Juliano C, Stranz DD, Taylor SS, and Woods VL Jr., (2004). Mapping intersubunit interactions of the regulatory subunit (RIalpha) in the type I holoenzyme of protein kinase A by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Mol. Biol 340, 1185–1196. [DOI] [PubMed] [Google Scholar]
- Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, et al. (2007). Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104. [DOI] [PubMed] [Google Scholar]
- Hsu S, Kim Y, Li S, Durrant ES, Pace RM, Woods VL Jr., and Gentry MS (2009). Structural insights into glucan phosphatase dynamics using amide hydrogen-deuterium exchange mass spectrometry. Biochemistry 48, 9891–9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KA, Rijal P, Jiang H, Wang B, Schimanski L, Dong T, Liu YM, Chang P, Iqbal M, Wang MC, et al. (2019). Structure-function analysis of neutralizing antibodies to H7N9 influenza from naturally infected humans. Nat. Microbiol 4, 306–315. [DOI] [PubMed] [Google Scholar]
- Jackson LA, Campbell JD, Frey SE, Edwards KM, Keitel WA, Kotloff KL, Berry AA, Graham I, Atmar RL, Creech CB, et al. (2015). Effect of varying doses of a monovalent H7N9 influenza vaccine with and without AS03 and MF59 adjuvants on immune response: a randomized clinical trial. JAMA 314, 237–246. [DOI] [PubMed] [Google Scholar]
- Job ER, Schotsaert M, Ibañez LI, Smet A, Ysenbaert T, Roose K, Dai M, de Haan CAM, Kleanthous H, Vogel TU, and Saelens X (2018). Antibodies directed toward neuraminidase N1 control disease in a mouse model of influenza. J. Virol 92, e01584–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunarathna HMTK, Perera RAPM, Fang VJ, Yen HL, Cowling BJ, and Peiris M (2019). Serum anti-neuraminidase antibody responses in human influenza A(H1N1)pdm09 virus infections. Emerg. Microbes Infect 8, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Coyle EM, Manischewitz J, King LR, Gao J, Germain RN, Schwartzberg PL, Tsang JS, Golding H, and The CHI Consortium. (2018). AS03-adjuvanted H5N1 vaccine promotes antibody diversity and affinity maturation, NAI titers, cross-clade H5N1 neutralization, but not H1N1 cross-subtype neutralization. NPJ Vaccines 3, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klock HE, and Lesley SA (2009). The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol 498, 91–103. [DOI] [PubMed] [Google Scholar]
- Krammer F, Fouchier RAM, Eichelberger MC, Webby RJ, Shaw-Saliba K, Wan H, Wilson PC, Compans RW, Skountzou I, and Monto AS (2018). NAction! How can neuraminidase-based immunity contribute to better influenza virus vaccines? MBio 9, e02332–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. (2009). Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol 166, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver WG, Colman PM, Webster RG, Hinshaw VS, and Air GM (1984). Influenza virus neuraminidase with hemagglutinin activity. Virology 137, 314–323. [DOI] [PubMed] [Google Scholar]
- Li S, Tsalkova T, White MA, Mei FC, Liu T, Wang D, Woods VL Jr., and Cheng X (2011). Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Biol. Chem 286, 17889–17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WD, Liu T, Li S, Woods VL Jr., and Hook V (2012). The prohormone proenkephalin possesses differential conformational features of subdomains revealed by rapid H-D exchange mass spectrometry. Protein Sci. 21, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MJ, Liu C, Wu MN, Zhao T,Wang GL, Yang Y, Gu HJ, Cui PW, Pang YY, Tan YY, et al. (2018). Influenza A(H7N9) virus antibody responses in survivors 1 year after infection, China, 2017. Emerg. Infect. Dis 24, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan A, Segall N, Ferguson M, Frenette L, Kroll R, Friel D, Soni J, Li P, Innis BL, and Schuind A (2016). Immunogenicity and safety of an AS03-adjuvanted H7N9 pandemic influenza vaccine in a randomized trial in healthy adults. J. Infect. Dis 214, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malby RL, Tulip WR, Harley VR, McKimm-Breschkin JL, Laver WG, Webster RG, and Colman PM (1994). The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure 2, 733–746. [DOI] [PubMed] [Google Scholar]
- Marjuki H, Mishin VP, Chesnokov AP, De La Cruz JA, Davis CT, Villanueva JM, Fry AM, and Gubareva LV (2015). Neuraminidase mutations conferring resistance to oseltamivir in influenza A(H7N9) viruses. J. Virol 89, 5419–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memoli MJ, Shaw PA, Han A, Czajkowski L, Reed S, Athota R, Bristol T, Fargis S, Risos K, Powers JH, et al. (2016). Evaluation of antihemagglutinin and antineuraminidase antibodies as correlates of protection in an influenza A/H1N1 virus healthy human challenge model. MBio 7, e00417–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto AS, Petrie JG, Cross RT, Johnson E, Liu M, Zhong W, Levine M, Katz JM, and Ohmit SE (2015). Antibody to influenza virus neuraminidase: an independent correlate of protection. J. Infect. Dis 212, 1191–1199. [DOI] [PubMed] [Google Scholar]
- Mostafa A, Abdelwhab EM, Mettenleiter TC, and Pleschka S (2018). Zoonotic potential of influenza A viruses: a comprehensive overview. Viruses 10, E497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S, Nachbagauer R, Balmaseda A, Stadlbauer D, Ojeda S, Patel M, Rajabhathor A, Lopez R, Guglia AF, Sanchez N, et al. (2019). Novel correlates of protection against pandemic H1N1 influenza A virus infection. Nat. Med 25, 962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, Iwasaki K, and Sato C (2003). Topology representing network enables highly accurate classification of protein images taken by cryo electron- microscope without masking. J. Struct. Biol 743, 185–200. [DOI] [PubMed] [Google Scholar]
- Paules CI, Marston HD, Eisinger RW, Baltimore D, and Fauci AS (2017). The pathway to a universal influenza vaccine. Immunity 47, 599–603. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Potter CS, Chu H, Frey B, Green C, Kisseberth N, Madden TJ, Miller KL, Nahrstedt K, Pulokas J, Reilein A, et al. (1999). Leginon: a system for fully automated acquisition of 1000 electron micrographs a day. Ultramicroscopy 77, 153–161. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Giudicelli V, Ginestoux C, Stoehr P, Robinson J, Bodmer J, Marsh SG, Bontrop R, Lemaitre M, Lefranc G, et al. (2000). IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 28, 219–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Taylor G, Laver WG, Kawaoka Y, and Webster RG (1994). Antigenicity of the N8 influenza A virus neuraminidase: existence of an epitope at the subunit interface of the neuraminidase. J. Virol 68, 1790–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronski DM, Chambers C, Gustafson R, Purych DB, Tang P, Bastien N, Krajden M, and Li Y (2016). Avian influenza A(H7N9) virus infection in 2 travelers returning from China to Canada, January 2015. Emerg. Infect. Dis 22, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, and Crowe JE Jr., (2015). Use of human hybridoma technology to isolate human monoclonal antibodies. Microbiol. Spectr 3, AID-0027–AID-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Zhou Y, Olivarez N, Broadwater A, de Silva A, and Crowe J Jr., (2012). Persistence of circulating memory B cell clones with potential for dengue virus disease enhancement for decades following infection. J. Virol 86, 2665–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Li Q, Wu Y, Wang M, Liu Y, Qi J, Vavricka CJ, and Gao GF (2014). Structure of influenza virus N7:the last piece of the neuraminidase “jigsaw” puzzle. J. Virol 88, 9197–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg NJ, Nannemann DP, Blum DL, Belser JA, Tumpey TM, Deshpande S, Fritz GA, Sapparapu G, Krause JC, Lee JH, et al. (2013). Human antibodies that neutralize respiratory droplet transmissible H5N1 influenza viruses. J. Clin. Invest 123, 4405–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg NJ, Zhang H, Bangaru S, Sapparapu G, Kose N, Lampley RM, Bombardi RG, Yu Y, Graham S, Branchizio A, et al. (2016). H7N9 influenza virus neutralizing antibodies that possess few somatic mutations. J. Clin. Invest 126, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, et al. (2012). A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 109, 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, et al. (2013). New world bats harbor diverse influenza A viruses. PLoS Pathog. 9, e1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlendorff J, Matrosovich T, Klenk HD, and Matrosovich M (2009). Functional significance of the hemadsorption activity of influenza virus neuraminidase and its alteration in pandemic viruses. Arch. Virol 154, 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese JN, Colman PM, van Donkelaar A, Blick TJ, Sahasrabudhe A, and McKimm-Breschkin JL (1997). Structural evidence for a second sialic acid binding site in avian influenza virus neuraminidases. Proc. Natl. Acad. Sci. USA 94, 11808–11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, and Carragher B (2009). DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol 166, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz L, Kays SK, Zimmer G, and von Messling V (2018). Neuraminidase-inhibiting antibody titers correlate with protection from heterologous influenza virus strains of the same neuraminidase subtype. J. Virol 92, e01006–e01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Yang H, Shore DA, Garten RJ, Couzens L, Gao J, Jiang L, Carney PJ, Villanueva J, Stevens J, and Eichelberger MC (2015). Structural characterization of a protective epitope spanning A(H1N1)pdm09 influenza virus neuraminidase monomers. Nat. Commun 6, 6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Qi L, Gao J, Couzens LK, Jiang L, Gao Y, Sheng ZM, Fong S, Hahn M, Khurana S, et al. (2018). Comparison of the efficacyof N9 neuraminidase-specific monoclonal antibodies against influenza A(H7N9) virus infection. J. Virol 92, e01588–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jiang H,Wu P, Uyeki TM, Feng L, Lai S, Wang L, Huo X,Xu K, Chen E, et al. (2017). Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013-17: an epidemiological study of laboratory-confirmed case series. Lancet Infect. Dis 17, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Brown LE, and Laver WG (1984). Antigenic and biological characterization of influenza virus neuraminidase (N2) with monoclonal antibodies. Virology 135, 30–42. [DOI] [PubMed] [Google Scholar]
- WHO (2017). Zoonotic influenza viruses: antigenic and genetic characteristics and development of candidate vaccine viruses for pandemic preparedness. Wkly. Epidemiol. Rec 92, 633–647.29052410 [Google Scholar]
- WHO (2018). Avian Influenza Weekly Update Number 663. https://iris.wpro.who.int/bitstream/handle/10665.1/14179/AI-20181116.pdf.
- Wilson JR, Guo Z, Reber A, Kamal RP, Music N, Gansebom S, Bai Y, Levine M, Carney P, Tzeng WP, et al. (2016). An influenza A virus (H7N9) anti-neuraminidase monoclonal antibody with prophylactic and therapeutic activity in vivo. Antiviral Res. 135, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold TJ, Nachbagauer R, Xu H, Tan GS, Hirsh A, Brokstad KA, Cox RJ, Palese P, and Krammer F (2015). Vaccination with adjuvanted recombinant neuraminidase induces broad heterologous, but not heterosubtypic, cross-protection against influenza virus infection in mice. MBio 6, e02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara A, Yamayoshi S, Kiso M, Sakai-Tagawa Y, Koga M, Adachi E, Kikuchi T, Wang IH, Yamada S, and Kawaoka Y (2019). Antigenic drift originating from changes to the lateral surface of the neuraminidase head of influenza A virus. Nat. Microbiol 4, 1024–1034. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Smith DL (1993). Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Xu X, and Wilson IA (2008). Structure determination of the 1918 H1N1 neuraminidase from a crystal with lattice-translocation defects. Acta Crystallogr. D Biol. Crystallogr D64, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, McBride R, Nycholat CM, Yu W, Paulson JC, and Wilson IA (2012). Influenza virus neuraminidases with reduced enzymatic activity that avidly bind sialic Acid receptors. J. Virol 86, 13371–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Zhou J, Li Z, Yang L, Li X, Huang W, Zou S, Chen W, Wei H, Tang J, et al. (2017). Biological characterisation of the emerged highly pathogenic avian influenza (HPAI) A(H7N9) viruses in humans, in mainland China, 2016 to 2017. Euro Surveill. 22, 30533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Turner HL, Lang S, McBride R, Bangaru S, Gilchuk IM, Yu W, Paulson JC, Crowe JEJ, Ward AB, et al. (2019). Structural basis of protection from H7N9 influenza virus by human anti-N9 neuraminidase antibodies. Cell Host Microbe 26, this issue, 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. This study did not generate code.