

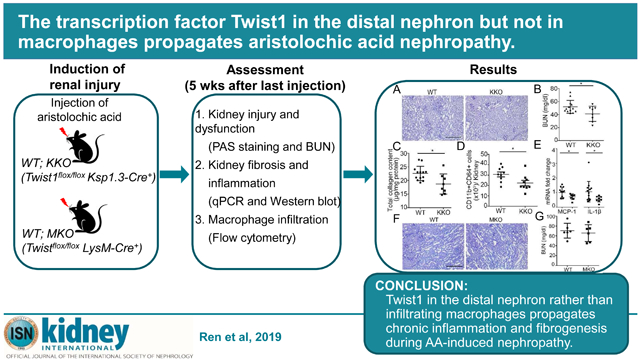
Abstract
Tubulointerstitial disease in the kidney culminates in renal fibrosis that portents organ failure. Twist1, a basic helix-loop-helix protein 38 transcription factor, regulates several essential biological functions, but inappropriate Twist1 activity in the kidney epithelium can trigger kidney fibrogenesis and chronic kidney disease. By contrast, Twist1 in circulating myeloid cells may constrain inflammatory injury by attenuating cytokine generation. To dissect the effects of Twist1 in kidney tubular versus immune cells on renal inflammation following toxin-induced renal injury, we subjected mice with selective deletion of Twist1 in renal epithelial cells or macrophages to aristolochic acid-induced chronic kidney disease. Ablation of Twist1 in the distal nephron attenuated kidney damage, interstitital fibrosis, and renal inflammation after aristolochic acid exposure. However, macrophage-specific deletion of Twist1 did not impact the development of aristolochic acid-induced nephropathy. In vitro studies confirmed that Twist1 in renal tubular cells underpins their susceptibility to apoptosis and propensity to generate pro-fibrotic mediators in response to aristolochic acid. Moreover, co-culture studies revealed that Twist1 in renal epithelia augmented the recruitment and activation of pro-inflammatory CD64+ macrophages. Thus, Twist1 in the distal nephron rather than in infiltrating macrophages propagates chronic inflammation and fibrogenesis during aristolochic acid-induced nephropathy
Keywords: chronic kidney disease, distal tubule, macrophages
Graphical Abstract
Introduction
Renal fibrosis complicates persistent tubulointerstitial injury in the kidney, and predicts progression of virtually all forms of chronic kidney disease (CKD). After injury, a complex cascade of cellular events including macrophage and T cell infiltration, tubular cell atrophy/death with microvascular rarefaction, and pericyte/fibroblast activation coordinate to generate and deposit extra-cellular matrix (ECM) in multiple kidney compartments1-3. A variety of insults such as ischemia, toxin exposure, and infection can provoke tubulointerstitial injury. Aristolochic acid (AA) as an important cause of drug-associated renal injury, was first reported in Belgium in patients with prolonged intake of a slimming regimen containing AA, and is implicated in the pathogenesis of Balkan and Chinese herbal nephropathies.4-7 AA mainly targets tubular epithelial cells, and provokes death of tubules via formation of DNA-adducts.8 Whereas acute AA exposure in human patients damages the proximal tubule, distal tubular injury and dysfunction is more prominent with chronic AA exposure.9 Damaged tubular cells release chemokines and inflammatory cytokines to incite a secondary immune response with accumulation of activated myeloid cells in the kidney. Thus, macrophage/monocyte infiltration and activation is a hallmark of both human and experimental CKD induced by AA.5 However, the molecular mechanisms governing interactions between tubular and myeloid cells during AA-related renal inflammation require elucidation.
Twist1, as a basic helix-loop-helix (bHLH) transcription factor is required for mesoderm differentiation in embryonic development. Accordingly, Twist1 mutations in humans cause Saethre-Chotzen syndrome,10 and Twist mutant embryos in rodents 11 fail to form the cranial neural tube. On the other hand, Twist1 expression may be inappropriately upregulated in disease states leading to maladaptive healing.12 For example, following renal injury, Twist1 in proximal tubular cells induces pro-fibrotic gene expression programs that promote extracellular matrix deposition.13 Moreover, Twist1 upregulates the chemokine CCL2 that attracts myeloid cells into injured tissues.14 Nevertheless, in myeloid cells and T lymphocytes, Twist1 limits the generation of pro-inflammatory cytokines, including IFN-γ, TNFα, IL-17, and IL-2 by suppressing the NF-κB-dependent signaling cascade.15-18 Given that Twist1 impacts crosstalk between multiple cell lineages,14, 19, 20 the relative effects of Twist1 in kidney tubular cells versus infiltrating myeloid cells on renal inflammation following toxin-induced injury remain unclear.
To discriminate these opposing, cell-specific actions of Twist1 in toxin-induced CKD, we subjected mice with renal tubular cell (RTC)- or macrophage-specific Twist1 deficiency to chronic AA nephropathy (AAN).
Results
Deletion of Twist1 in tubular cells but not in macrophages attenuates AA-induced CKD
As injury and fibrosis caused by AA impact the deep cortex and medulla,21-23 we bred mice lacking Twist1 selectively in the distal nephron (Twist1flox/flox Ksp1.3-Cre+= “KKO”) and controls with normal Twist1 expression in all tissues (Twist1flox/flox Ksp1.3-Cre− = “WT”), and injected these animals with AA. At baseline, despite the KKO kidneys having decreased mRNA levels for cadherin 16 (Cdh16) (Supplementary Figure S1C), WT and KKO kidneys showed similar architecture with similar protein levels and staining for Cdh16 and aquaporin 1 (Aqp1) (Supplementary Figure S1A-B, and S1D-E). WT and KKO animals also had similar kidney function as measured by BUN levels (Supplementary Figure S1F). Five weeks following AA injection, all mice had kidney injury with interstitial matrix expansion and inflammatory cell infiltration. Consistent with AA’s capacity to drive tubulointerstitial disease, dilation of tubular lumen, loss of brush border, and tubular epithelial flattening and detachment were evident in sections from WT mice (Figure 1A). However, under the same conditions, the KKO kidneys showed relative preservation of kidney morphology with less tubular injury and inflammatory cell infiltration (Figure 1A). These differences were confirmed by blinded injury scores (2.7 ± 0.16 vs 2.0 ± 0.26; p<0.05) (Figure 1B). At a molecular level, mRNA levels for neutrophil gelatinase-associated lipocalin (NGAL) and Kim-1, markers of kidney tubular injury, were significantly lower in AA-treated KKO kidneys compared with WT controls (Figure 1C). Twist1 KKO mice had lower blood urea nitrogen (BUN) levels (41 ± 4 vs 52 ± 3 mg/dl; p<0.05) but similar serum creatinine levels compared to WTs (Figure 1D-E). Body weights were also similar between the groups (Supplementary Figure S2). These data suggest that tubular Twist1 instigated tubular damage but did not clearly influence kidney function. On balance, these results indicate that Twist1 in tubular cells promotes AA-induced CKD.
Figure 1. Twist1 in the distal nephron aggravates aristolochic acid (AA)-induced CKD.

(A) Representative images of kidney sections from WT and KKO mice at 5wks after AA injection. Scale bar =100 μm (B) WT and KKO kidney pathology scores (n≥8). (C) Renal mRNA expression of NGAL and Kim-1 (n≥8). (D) Blood urea nitrogen (BUN) (n≥8) (E) Serum creatinine (Scr) (n≥8). Data represent the mean± SD (*p<0.05).
Macrophages accumulate and undergo activation in the kidney interstitium during the pathogenesis of AA-induced nephropathy, and Twist1 regulates the generation of pro-inflammatory cytokines in myeloid cells.5, 16 We have seen in other models that expression of lysozyme M (LysM) marks macrophages that infiltrate the kidney from the circulation.24 Using a mouse line bearing green fluorescent LysM-expressing macrophages, we confirmed robust accumulation of these myeloid cells in the kidney after AA injection (Figure 2A). Thus, to determine the effects of Twist1 in myeloid cells on severity of AA-induced CKD, we generated mice with macrophage-specific Twist1 deletion (Twistflox/flox LysM-Cre+ = “MKO”) and wild-type littermate controls (Twistflox/flox LysM-Cre− = “WT”), and subjected these animals to our AA model as above. We noted robust tubulointerstitial disease in both the WT and MKO kidneys at 5 weeks post-AA (Figure 2B). However, there were no differences between the WT and MKO mice in kidney injury scores, renal NGAL mRNA levels, or BUN (Figure 2C-E). These results demonstrate that Twist1 in macrophages does not influence renal injury after AA treatment. Thus, Twist1 in tubular cells but not in macrophages drives renal damage during AA-induced chronic nephropathy.
Figure 2. Twist1 in macrophages does not influence AA-induced CKD.

(A) Representative kidney sections from LysM-Cre+ mT/mG mice at 5 weeks after vehicle (left) and AA injection (right). Scale bar = 50 μm. (B) Representative images of kidney sections from WT and MKO mice at 5wks after AA injection. Scale bar =100 μm. (C) WT and MKO kidney pathology scores (n=6). (D) Renal mRNA expression of NGAL (n=6). (E) BUN (n=6). Data represent the mean± SD.
Twist1 in the distal nephron propagates AA-induced renal fibrosis
As unresolved renal tubular injury culminates in deposition of extracellular matrix in the kidney interstitium after AA injection, we quantitated markers and mediators of renal fibrosis in the WT and KKO kidneys. As shown in Figure 3A, tubular-specific loss of Twist1 in the KKO mice significantly blunted mRNA expression of type I collagen and pro-fibrotic mediators, PAI-1 and TGFβ1 but preserved mRNA expression for the epithelial cell marker E-cadherin after AA treatment compared to controls (Figure 3A). Similarly, quantitation of collagen protein by Sirius red / fast green analysis and blinded scoring of collagen-1 stains revealed diminished collagen protein deposition in the KKO kidneys compared to controls (Figure 3B-D). Western analysis confirmed that the injured KKO kidneys expressed lower levels of collagen-1, fibronectin (FN), and α-smooth muscle actin (a-SMA), a marker for the activated myofibroblasts that secrete collagen (Figure 3E-F). Collectively, these data indicate that Twist1 in the distal nephron can propagate scar formation in the kidney following AA exposure.
Figure 3. Twist1 in the distal nephron exacerbates kidney fibrosis induced by AA.

(A) mRNA expression of Col-I, Pai-1, TGFβ1, and E-cadherin in the kidneys from WT and KKO mice at 5wks after AA injection (n≥8). (B) Total collagen content measured by Sirius red / fast green analysis of tissue sections from WT and KKO kidneys after AA exposure. (C) Representative sections from WT and KKO kidneys stained with Col-I at 5wks after AA injection. Scale bar =100 μm. (D) Blinded morphometric quantitation of Col-I (n≥8). (E) Western blot for FN, Col-I and α-SMA in whole kidney at 5wks after AA injection. (F) Semi-quantification of FN, Col-I and α-SMA from (E) (n≥8). Data represent the mean± SD (*p<0.05, **p<0.01, ***p<0.001).
To assess the effects of Twist1 in macrophages on kidney fibrosis after AA-induced injury, we quantitated mRNA expression for type I collagen, PAI-1, and TGFβ1 in WT and MKO kidneys but detected no differences between groups (Figure 4A). Moreover, at the protein level type I collagen, FN, and α-SMA levels were similar in the WT and MKO kidneys (Figure 4B-C), indicating that Twist1 in macrophages does not impact extracellular matrix deposition in the kidney during AA nephropathy.
Figure 4. Myeloid Twist1 does not impact kidney interstitial fibrosis following AA exposure.

(A) mRNA expression of Col-I, Pai-1 and TGFβ1 in kidneys from WT and MKO mice at 5wks after AA injection (n=6). (B) Western blot for Col-I, FN and α-SMA in whole kidney at 5wks after AA injection (n=6). (C) Semi-quantification of Col-I, FN and α-SMA from (B) (n=6). Data represent the mean± SD.
Twist1 in the tubule but not in myeloid cells regulates macrophage accumulation in the kidney after AA-mediated injury
Macrophages orchestrate both injury and repair of the kidney following a toxic insult. To assess the role of Twist1 in the distal nephron on macrophage accumulation in the kidney after AA treatment, we isolated infiltrating CD45+ cells from the injured kidney via flow cytometry (Supplementary Figure S3) and quantified the number of CD11b+CD64+ macrophages via flow cytometry. As shown in Figure 5A and B, the number of CD11b+CD64+ macrophages from KKO kidneys after AA injection is higher than that from WTs (30725±2352 vs. 22393±2575; p<0.05). Immunohistochemistry for F4/80 showed the macrophages distributed across the renal interstitium (Figure 5C). Macrophages elaborate chemokines/proinflammatory cytokines that mediate tissue injury and fibrosis. In this regard, mRNA levels for MCP-1 and IL1β in the KKO kidney were 70% and 50%, respectively, of WT values, whereas IFN-γ was similarly expressed in the two groups (Figure 5D). By contrast, robust deletion of Twist1 directly from myeloid cells in the Twist1 MKO cohort (>80%, p < 0.0001, data not shown) had no impact at 5 weeks on renal macrophage accumulation (Figure 5E-F) or cytokine expression (Figure 5G).
Figure 5. Twist1 in kidney epithelial cells but not macrophages enhances MCP-1 and IL-1β mRNA expression, and promotes macrophage accumulation in the AA induced kidneys.

(A) Representative flow plots of CD11b+CD64+ macrophages from kidneys of AA-treated WT and KKO animals. (B) Absolute numbers of total CD11b+CD64+ macrophages from the WT and KKO kidneys (n=9). (C) Representative F4/80 stains of kidney sections from AA-treated WT and KKO mice. Scale bar =100 μm. (D) mRNA expressions for MCP-1, IL-1β and IFN-γ in the kidneys from AA-treated WT and KKO mice (n≥8). (E) Representative flow plots of CD11b+CD64+ macrophages from kidneys of AA-treated WT and MKO animals. (F) Absolute numbers of total CD11b+CD64+ macrophages from the WT and MKO kidneys (n≥7). (G) mRNA expression of MCP-1, IL-1β and IFN-γ in the kidneys from AA-treated WT and MKO mice (n=6). Data represent the mean± SD (*p<0.05).
Twist1 ablation sensitizes AA-induced tubular epithelial cell apoptosis in vitro
To determine the effect of Twist1 on epithelial cell survival in culture, we isolated primary tubular cells from WT and KKO kidneys and exposed them to AA. Tubular cell cultures expressed mRNA for both Aqp1 and Cdh16, and the KKO cells showed markedly lower levels of Twist1 mRNA expression versus WT controls (Supplementary Figure S4). In WT tubular cells, AA dramatically induced caspase 3 cleavage in a dose-dependent manner (Figure 6A-B), an effect that was blunted in Twist1 KO tubular cells. At 24 hrs after Vehicle or AA treatment, WT and Twist1 KO tubular cells were harvested, co-stained with Annexin V and Propidium Iodide (PI), and analyzed by FACS analysis (Supplementary Figure S5) to identify apoptotic cells. Flow cytometric analysis showed that the percentage of dead cells including early cell apoptosis (Annexin V+/PI−) and late cell apoptosis (Annexin V+/PI+) was markedly elevated after AA treatment (Figure 6C-D), whereas Twist1 KO tubular cells exhibited less apoptosis after AA treatment compared with WTs. Thus, Twist1 contributes to AA-induced tubular cell apoptosis.
Figure 6. Twist1 sensitizes kidney epithelial cells to AA-induced apoptosis in vitro.

(A) Western blot for Cleaved-caspase 3 from WT and Twist1 KKO (“KO”) epithelial cells induced by AA as indicated. (B) Semi-quantitative analysis for Cleaved-caspase 3 from (A) (n=3). (C) Representative plots of FACS analysis wherein WT and Twist1 KO epithelial cells treated with vehicle or AA were stained with annexin-V-FITC and PI to identify cellular apoptosis. (D) Summary data quantifying apoptosis among different groups (n=3-5). Data represent the mean± SD (*p<0.05, #p<0.05).
Twist1 in tubular cells promotes matrix protein production
Twist1 is crucial for cellular transdifferentiation in physiological and pathological settings12, 13, 25, 26, but the effects of Twist1 in tubular cells on the phenotype of infiltrating macrophages that could impact renal scar formation requires elucidation. To explore these paracrine effects, we first treated WT and Twist1 KO tubular cells with TGFβ1 (5ng/ml). As shown in Figure 7A-B, under TGFβ1 stimulation, the protein levels of FN were increased in WT tubular cells compared with vehicle treatment. However, Twist1 deletion in tubular cells blunted the production of FN in response to TGFβ1. Prior studies have shown FN is an endogenous TLR agonist released by the injured to activate macrophages, enhancing their production of inflammatory cytokines.27 To assess effects of FN on macrophage function, we stimulated peritoneal macrophages with FN fragment III and detected IL1β and TNFα mRNA expression. FN-fragment III could significantly upregulate IL1β and TNFα mRNA expression in macrophages compared with vehicle treatment (Figure 7C). Together, these results suggest that Twist1 in tubular cells promotes matrix production directly by upregulating FN, but also indirectly by stimulating FN-dependent generation of pro-fibrotic cytokines in neighboring macrophages.
Figure 7. Twist1 in kidney epithelial cells promotes TGF-β1-induced fibronectin (FN) expression.

(A) Western blot for FN in each group as indicated. (B) Semi-quantification of FN from (A) (n=3). (C) mRNA expression of IL1β and TNFα from macrophages treated with vehicle or recombinant FN fragment 3 (n=3). Data represent the mean± SD (*p<0.05, **p<0.01, #p<0.05).
Twist1 in kidney epithelial cells exaggerates IL-1β expression in macrophages
To further explore the effects of Twist1 in tubular cells on macrophage phenotype, we employed a co-culture system as shown schematically in Figure 8A. Primary WT and Twist1 KKO tubular cells pre-treated with vehicle or AA in the lower chamber of a transwell plate were co-cultured with WT macrophages in the upper chamber for 24 hours, after which the tubular cells and macrophages were harvested for mRNA expression analysis. Compared with WT tubular cells, Twist1-deficient tubular cells treated with AA induced significantly less IL1β and MCP-1 expression in the co-cultured macrophages (Figure 8B-C). Similarly, knockdown of the Twist1 target fibronectin in WT tubular cells using siRNA abrogated the induction of mRNA for IL-1β and MCP-1 in the co-cultured macrophages, suggesting that tubular Twist1 drives myeloid cell activation via a fibronectin-dependent pathway (Supplementary Figure S6). By contrast, TNFα mRNA expression in the macrophages was similar between the groups under different treatment (Figure 8D). Within the tubular cells, Twist1-deficiency abrogated the AA-induced pregulation of the kidney injury markers NGAL and Kim-1. Indeed, co-culture with macrophages, even in the absence of AA, was sufficient to elicit the difference in tubular NGAL expression accruing from Twist1 expression (Figure 8E-F). Thus, Twist1 in tubular cells directly augments AA-induced tubular injury but also drives macrophage expression of IL-1β and MCP-1, both of which are known to provoke renal scar formation.
Figure 8. Twist1 in kidney epithelial cells enhances IL-1β and MCP-1 expression in macrophages.

(A) Schematic diagram showing the experimental setup of testing gene levels from macrophages and epithelial cells. (B-D) mRNA expression of IL1β (B), MCP-1(C) and TNFα (D) from co-cultured macrophages among group as indicated (n=3). (E-H) mRNA expression of NGAL (E), Kim-1 (F), Pai-1 (G) and TNFα (H) from co-cultured epithelial cells among groups as indicated (n=3). Data represent the mean± SD (*p<0.05, **p<0.01, ***p<0.01).
Discussion
In this study, we examined the effects of Twist1 in the distal nephron and in myeloid cells on renal inflammation and injury following exposure to the toxin aristolochic acid (AA). We find that tubular Twist1 promotes toxin-induced injury not only through direct effects on tubular cells but also by provoking an influx of myeloid cells and stimulating the generation of pro-inflammatory cytokines in macrophages with consequent kidney fibrogenesis. By contrast, despite the known effects of Twist1 in myeloid cells to suppress inflammation, we find that macrophage Twist1 does not impact renal damage, inflammation, or fibrosis following AA exposure.
Renal tubular cells are susceptible to various toxic and metabolic insults that lead to cell death. AA triggers persistent injury including DNA damage in tubular cells, resulting in the production of reactive oxygen species, secretion of chemokines, cell cycle arrest and apoptosis.28 We found that mice with Twist1 deletion from the distal nephron had less tubular injury compared with WT controls, consistent with Twist1’s direct effects in the proximal tubule.13 Moreover, in cultured primary tubular cells, Twist1 deficiency protected against apoptosis after AA exposure, suggesting that Twist1 acts as a pro-apoptotic factor in tubular cells. Prior studies showed tubules with Twist1 deficiency preserve more fatty acid metabolism and β-oxidation, which promote tubular cell survival and dedifferentiation in fibrosing kidneys.13, 29 By contrast, in tumor cells overexpression of Twist1 promotes tumor cell survival and metastasis.30-32 Thus, the effects of Twist1 on cell survival may be context- and cell-dependent.
Our interest in Twist1 stemmed from the finding that the pro-hypertensive hormone angiotensin II induces Twist1 in myeloid cells (data not shown). Angiotensin II drives injury and fibrosis in kidney cells with secondary immune activation, but in myeloid cells can suppress cytokine generation,24, 33 so we explored whether Twist1 similarly has opposing actions in renal tubular and myeloid cells during AA nephropathy. In the current study, ablation of Twist1 in the distal nephron attenuates induction of the macrophage chemokine MCP-1 (CCL2) in the damaged kidney leading to diminished renal accumulation of CD64-expressing macrophages. This finding is broadly consistent with those of Lovisa et al, who reported reduced renal accumulation of CD11b+ myeloid cells in the injured kidneys of animals lacking Twist1 in the proximal tubule.13 Moreover, in our co-culture experiments, macrophages incubated with Twist1-deficient tubular cells express less MCP-1 than those incubated with WT macrophages. Thus, tubular Twist1 can impact both paracrine and autocrine recruitment of myeloid cells. As macrophages are capable of secreting multiple cytokines that mediate renal fibrosis, persistence of macrophage infiltration is strongly associated with renal fibrosis and progressive chronic kidney disease.34, 35 Consistent with the known effects of injured tubular cells to modulate the phenotype of macrophages during acute kidney injury36, we find that AA-treated tubular cells promote macrophage secretion of pro-inflammatory cytokines such as IL1β in co-culture. We and others have seen that IL-1 can drive gene expression programs in renal tubular cells that promote fibrogenesis24 either via direct effects on tubular cells or via paracrine effects on the pericytes that become myofibroblasts.37 In our hands, AA-mediated induction of the TLR ligand fibronectin (FN)38 in tubular cells is a key driver of IL-1β and TNF generation in macrophages. However, these effects of tubular cells on macrophage activation are abrogated by Twist1 deletion in tubular cells. Collectively, these results indicate that Twist1 in the distal nephron directly damages the renal epithelium but also promotes recruitment and activation of myeloid cells during AA nephropathy that can exaggerate the severity of kidney fibrosis via a FN-dependent pathway.
Like angiotensin II, Twist1 in myeloid cells suppresses their generation of pro-inflammatory and fibrotic cytokines.15, 39 Therefore, we posited that mice harboring selective deletion of Twist1 in macrophages would have exaggerated kidney injury and fibrosis compared with WTs after AA treatment. However, deletion of Twist1 directly from myeloid cells in the Twist1 MKO cohort had no impact at 5 weeks on renal tubular injury, interstitial fibrosis, or local inflammation. While prior studies have shown Twist1 regulates the production of several pro-inflammatory cytokines including TNFα, IL-6, IL-8 in vitro15, 39, the complex inflammatory milieu in the AA-injured kidney may have overriding effects on the function and differentiation of infiltrating macrophages that supersede an effect of myeloid Twist1. In this regard, mRNA levels for inflammatory cytokines were similar in WT and MKO kidneys during AA nephropathy, indicating that the capacity of renal parenchymal cells to modulate local cytokine levels in this setting overrides any contribution from Twist1-dependent signals in infiltrating macrophages. Indeed, we see that tubular Twist1 has a profound effect on the local generation of inflammatory cytokines.
In summary, Twist1 in the distal nephron rather than in myeloid cells drives AA-induced kidney injury, chronic inflammation, and interstitial fibrosis. In addition to its direct effects on renal tubular cell injury, tubular Twist1 promotes kidney fibrogenesis by driving renal accumulation of CD64+ macrophages and their FN-dependent production of IL-1β and MCP-1. By contrast, Twist1 in LysM+ infiltrating myeloid cells does not modulate renal inflammation or injury following AA-exposure. These studies illustrate the complex effects of the Twist1 signaling cascade on toxin-induced renal disease and the benefits of ultimately targeting this pathway in a cell-specific manner.
Concise Methods
Animals
All mice were housed and bred in the animal facilities at the Durham Veterans’ Affairs Medical Center according to NIH guidelines. All animal experiments were approved by the Durham Veteran’s Affairs Medical Center Institutional Animal Care and Use Committee. Homozygous Twist1 floxed mice on 129/SvEv background (kind gift from Richard Behringer)17 were crossed with 129/SvEv Ksp-Cre mice40 to generate Ksp-Cre+; Twist1fl/fl (KKO) mice and WT control littermates. Similarly, 129/SvEv Twist1fl/fl mice were bred with 129/SvEv LysM-Cre mice41 to yield LysM-Cre+; Twist1fl/fl (MKO) mice and WT controls. To map the distribution pattern of Cre recombinase expression in AAN model, mT/mG mice from The Jackson Laboratory (Bar Harbor, ME) were crossed with the LysM-Cre recombinase transgenic lines; mT/mG mice normally express red fluorescent protein in all tissues. When Cre is present, the mT cassette is deleted, triggering expression of membrane-targeted eGFP. As male mice experienced high mortality with AA exposure, only female mice, aged 8-12 weeks were used in our experiments.
Chronic Aristolochic Acid Model
Twist1 KKO, Twist1 MKO, and wild-type (WT) female mice were injected with aristolochic acid I (5mg/kg, IP) (Sigmal-Aldrich, A9451) every other day for 12 days. Kidney tissues and blood samples were obtained 5 weeks after the last injection for further analysis.
Quantitative determination of collagen in AA-exposed kidneys.
The kidney tissue collagen content was quantified using Sirius Red/ Fast Green Collagen Staining Kit (Chondrex, cat: 9046) according to the manufacturer’s instructions. Briefly, 5-μm-thick sections of paraffin-embedded kidney tissue were stained with Sirius Red and Fast Green for 30 minutes. After rinsing the stained tissue section with distilled water repeatedly until the water runs clear, the dye was eluted with Dye Extraction buffer. Collect the eluted Dye solution and read the OD values at 540 nm and 605 nm with a spectrophotometer.
RNA Isolation and PCR Analysis
Total RNA was isolated from cells or tissue using RNeasy mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. cDNA was synthesized using the SuperScript II First-Strand Synthesis kit (Invitrogen, Carlsbad, CA). Real-time PCR was perfomed using cDNA, SYBR or Taqman primers. GAPDH was measured as a housekeeping gene. The relative amount of mRNA to GAPDH was calculated using the equation: 2ΔCT, in which ΔCT =CT gene-CTGAPDH.
Histological Analysis and Immunohistochemistry Staining
Mouse kidney samples were fixed in 10% formalin (Sigma-Aldrich) overnight and embedded in paraffin. Sections 5 μm in thickness were used for PAS staining and immunohistochemistry staining. Renal damage including dead tubules, loss of brush borders, tubule dilatation, cast formation, tubular epithelial swelling, and vacuolar degeneration was semi-quantitatively scored. Score 0 represents injury area less than 5%; whereas 1, 2, 3, and 4 exhibits the damage involving 5–25%; 26–50%; 51–75%; >75% of the whole kidney area, respectively. The investigator, who was blinded to experimental conditions, assigned a score to each field based on the degree of kidney injury. At least ten randomly fields were assessed under the microscope (X200), and an average score was calculated for each mouse. To visualize interstitial collagen deposition and macrophage infiltration, sections were stained with anti-type I collagen and F4/80 as previously described, respectively.42
Western Blots
Kidney tissues (20mg) were homogenized in RIPA buffer (Sigmal-Aldrich). Concentration of protein was quantitated using the DC protein assay kit (Bio-Rad laboratories). Equal amounts of sample were subjected to electrophoresis through 4-12% Bis-Tris Gels and transferred to PVDF membranes. After blocking with 5% milk in TBST, the blots were incubated with anti-collagen type I antibody (SouthernBiotech, cat: 1310-01), anti-α-SMA antibody (Sigma, cat: A5228), FN (Abcam, cat: 2413), anti-GAPDH (CST, cat:2118) or anti-cleaved caspase 3 (CST, cat: 9664) overnight in 4°C. The blots were then washed and in cubated for 1 hour at room temperature with individual secondary antibodies accordingly. Bands were detected using an enhanced chemiluminescence detection system. The detected bands were quantified by densitometry through Image J 1.38 for windows.
Kidney Flow Cytometry
Kidney single-cell suspension was prepared by mechanical and enzymatic digestion as described previously.42 The kidney single-cell suspensions were incubated with Fc Block (BioLegend, San Diego, CA) for 30 minutes and treated with anti-CD11b, anti-CD45, anti-CD64, and Near-infrared dead cell indicator (Life Technologies, cat: L34976) for 30 minutes at 4°C. Cells were washed and fixed with Fix/Perm buffer (BD Biosciences, cat: 554655). 20 μl of CountBright absolute counting beads (Invitrogen, cat: C36950) were added to cells, and samples were analyzed on an LSRII flow cytometry (BD). Data were analyzed using FlowJo software version 10.2 (Tree Star, Inc., Ashland, OR). Absolute CD11b+CD64+ cell numbers from each sample were obtained according to CountBright manufacturer’s instructions. The detailed gating strategy is shown in Supplementary Figure S3A.
Annexin V/PI Staining and Flow Cytometric Analysis
WT and Twist1 KO primary tubular cells were seeded on 6 well-plates, then were changed to serum-free media for 16 hrs after reaching 80% confluence in complete media. These cells were treated with AA (10μM) for 24 hrs. 24 hrs later, cells were harvested and stained with Annexin V-FITC/PI staining according to the manufacturer’s protocol (BioLegend, cat: 640914) and then analyzed by flow cytometry. Annexin V+/PI− cells were identified as early apoptotic cells, and Annexin V+/PI+ cells were identified as late apoptotic cells. The detailed gating strategy is shown in Supplementary Figure S3B.
Primary Tubular Cell and Peritoneal Macrophage Culture
To maximize the purity of tubular cells with Twist1 deficiency from KKO, WT or KKO kidney outer cortex was removed and the rest of kidney was kept, minced into small pieces, which were transferred through two layers of nylon sieves (125 μm and 106 μm of pore size, respectively). The tubular pellets were seeded on collagen pre-coated flasks (Corning) with DMEM-Ham’s F-12 medium supplemented with 10% FBS, 1% Insulin-Transferrin-Selenium (Gibico), 1% L-glutamine, and 1% penicillin/streptomycin after sieving. The plate was incubated in a humidified incubator (5% CO2, 37°C). The medium was changed two days later and maintained every other day until the monolayer of cells reached 90% confluence. FN or Scramble siRNA (Integrated DNA Technologies) was transfected into primary renal tubular cells (RTCs) using Lipofectamine 3000 reagent (Thermo Fisher) according to the manufacturer’s instructions.
Primary peritoneal macrophages were obtained from the peritoneal exudates of mice. 1×106 cells ml−1 were first seeded on transwell inserts (BD, cat: 353502) in DMEM (Gibco) supplied with 10% serum (Sigma) and 1% Pen/Strep (Gibco). After three to four hrs at 37°C and 5% CO2, non-adherent cells were removed by washing twice with warm PBS. Macrophage were stimulated with vehicle and FN-III (R&D, cat:3938-FN) at 10μg/ml for 6 hrs and were harvested for IL1β and TNFα expression analysis.
Co-culture Experiments
Serum-starved tubular cells were treated w/o AA (10μM) for 12 hrs. Afterwards, tubular cells were washed twice with warm PBS to remove AA. Meanwhile, adhesive macrophages were seeded into a transwell insert and rested for 6 hrs. Then, these inserts were placed into another companion plate coated with renal tubular cells pre-treated w/o AA. After co-culture for 24 hrs, renal tubular cells and macrophages were harvested for RNA analysis.
Statistical Analyses
All data are expressed as the mean ± SD. For comparisons between groups with normally distributed data, statistical significance was assessed using two tailed unpaired student’s t test. For comparisons between groups with non-normally distributed variables, a Wilcoxon test was employed. Comparison among groups was performed with one-way analysis of variance test.
Supplementary Material
Figure S1. Twist1 deletion in distal nephron does not cause renal abnormalities at baseline. (A) Representative micrographs of PAS staining showing kidney cortex and medulla from WT and KKO mice. Scale bar =100 μm (B) Immunofluorescence staining for Aqp1 and Cdh16 in WT and KKO kidneys. Scale bar =100 μm (C) mRNA expression of Cdh16 and Aqp1 in WT and KKO kidneys (n=3). (D) Western blot analyses of renal expression of Cdh16 and Aqp1 proteins (n=3). (E) Semi-quantification of Cdh16 and Aqp1 from (D). (F) Measurement of BUN in serum from WT and KKO mice (n=3). Cdh16, Cadherin 16; Aqp1, aquaporin-1. Data represent the mean± SD (*p<0.05).
Figure S2. Body weights of WT and KKO mice after aristolochic acid (AA) exposure at 5 weeks (n≥8).
Figure S3. The gating strategy for excluding cell debris and analyzing viable myeloid cells in aristolochic acid (AA)-exposed kidneys.
Figure S4. mRNA expression of Aqp1, Cdh16 and Twist1 in primary renal tubular cells (RTCs) from WT and KKO kidneys (n=3). Data represent the mean ± SD (**p<0.01).
Figure S5. The gating strategy for the in vitro cultured tubular cells.
Figure S6. Knocking down fibronectin (FN) in primary renal tubular cells (RTCs) pretreated with AA blunts IL-1β and MCP-1 mRNA induction in co-cultured macrophages. (A) mRNA expression of FN in primary RTCs transfected with scramble siRNA or FN siRNA (n=3). (B) mRNA expression for IL1β and MCP-1 from macrophages co-cultured with RTCs as indicated (n=3). Data represent the mean ± SD (*p<0.05, ***p<0.001).
Translational Statement.
The current studies demonstrate that Twist1 in the distal nephron instigates damage and myeloid inflammation throughout the kidney following chronic exposure to AA. By contrast, Twist1 in macrophages does not impact the progression of AA-induced nephropathy in our hands. These data argue for selective targeting of Twist1 signals in the renal epithelium for patients with tubulointerstitial nephritis associated with AA exposure, such as Balkan and Chinese herbal nephropathies.
Acknowledgments
Sources of Support. This work was supported by NIH grants DK087893, HL128355; Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893.
Footnotes
Supplementary information is available at Kidney International's website.
Financial disclosures: None
Conflicts of interest: None
References
- 1.Eddy AA. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int Suppl (2011), 4: 2–8, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest, 124: 2299–2306, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol, 7: 684–696, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanherweghem JL, Depierreux M, Tielemans C, et al. Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet, 341: 387–391, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: a worldwide problem. Kidney Int, 74: 158–169, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Depierreux M, Van Damme B, Vanden Houte K, et al. Pathologic aspects of a newly described nephropathy related to the prolonged use of Chinese herbs. Am J Kidney Dis, 24: 172–180, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Vanhaelen M, Vanhaelen-Fastre R, But P, et al. Identification of aristolochic acid in Chinese herbs. Lancet, 343: 174, 1994. [DOI] [PubMed] [Google Scholar]
- 8.Michl J, Ingrouille MJ, Simmonds MS, et al. Naturally occurring aristolochic acid analogues and their toxicities. Nat Prod Rep, 31 : 676–693, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Su T, Li XM, et al. Aristolochic acid nephropathy: variation in presentation and prognosis. Nephrol Dial Transplant, 27: 292–298, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Howard TD, Paznekas WA, Green ED, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet, 15: 36–41, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev, 9: 686–699, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Qin Q, Xu Y, He T, et al. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res, 22: 90–106, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med, 21: 998–1009, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low-Marchelli JM, Ardi VC, Vizcarra EA, et al. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res, 73: 662–671, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharif MN, Sosic D, Rothlin CV, et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med, 203: 1891–1901, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sosic D, Richardson JA, Yu K, et al. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell, 112: 169–180, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Chen YT, Akinwunmi PO, Deng JM, et al. Generation of a Twist1 conditional null allele in the mouse. Genesis, 45: 588–592, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Pham D, Walline CC, Hollister K, et al. The transcription factor Twist1 limits T helper 17 and T follicular helper cell development by repressing the gene encoding the interleukin-6 receptor alpha chain. J Biol Chem, 288: 27423–27433, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KW, Lee NK, Ham S, et al. Twist1 is essential in maintaining mesenchymal state and tumor-initiating properties in synovial sarcoma. Cancer Lett, 343: 62–73, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Xue G, Restuccia DF, Lan Q, et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov, 2: 248–259, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Cosyns JP, Dehoux JP, Guiot Y, et al. Chronic aristolochic acid toxicity in rabbits: a model of Chinese herbs nephropathy? Kidney Int, 59: 2164–2173, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Debelle FD, Nortier JL, Husson CP, et al. The renin-angiotensin system blockade does not prevent renal interstitial fibrosis induced by aristolochic acids. Kidney Int, 66: 1815–1825, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Scarpellini A, Huang L, Burhan I, et al. Syndecan-4 knockout leads to reduced extracellular transglutaminase-2 and protects against tubulointerstitial fibrosis. J Am Soc Nephrol, 25: 1013–1027, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang JD, Patel MB, Griffiths R, et al. Type 1 angiotensin receptors on macrophages ameliorate IL-1 receptor-mediated kidney fibrosis. J Clin Invest, 124: 2198–2203, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansieau S, Bastid J, Doreau A, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell, 14: 79–89, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Bildsoe H, Loebel DA, Jones VJ, et al. Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev Biol, 331: 176–188, 2009. [DOI] [PubMed] [Google Scholar]
- 27.Mudaliar H, Pollock C, Panchapakesan U Role of Toll-like receptors in diabetic nephropathy. Clin Sci (Lond), 126: 685–694, 2014. [DOI] [PubMed] [Google Scholar]
- 28.Romanov V, Whyard TC, Waltzer WC, et al. Aristolochic acid-induced apoptosis and G2 cell cycle arrest depends on ROS generation and MAP kinases activation. Arch Toxicol, 89: 47–56, 2015. [DOI] [PubMed] [Google Scholar]
- 29.Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med, 21: 37–46, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eckert MA, Lwin TM, Chang AT, et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell, 19: 372–386, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li QQ, Xu JD, Wang WJ, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res, 15: 2657–2665, 2009. [DOI] [PubMed] [Google Scholar]
- 32.Beck B, Lapouge G, Rorive S, et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell, 16: 67–79, 2015. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Rudemiller NP, Patel MB, et al. Competing Actions of Type 1 Angiotensin II Receptors Expressed on T Lymphocytes and Kidney Epithelium during Cisplatin-Induced AKI. J Am Soc Nephrol, 27: 2257–2264, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huen SC, Cantley LG. Macrophages in Renal Injury and Repair. Annu Rev Physiol, 79: 449–469, 2017. [DOI] [PubMed] [Google Scholar]
- 35.Rogers NM, Ferenbach DA, Isenberg JS, et al. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol, 10: 625–643, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol, 22: 317–326, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol, 176: 85–97, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem, 276: 10229–10233, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Zheng S, Hedl M, Abraham C Twist1 and Twist2 Contribute to Cytokine Downregulation following Chronic NOD2 Stimulation of Human Macrophages through the Coordinated Regulation of Transcriptional Repressors and Activators. J Immunol, 195: 217–226, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol, 13: 1837–1846, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Clausen BE, Burkhardt C, Reith W, et al. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res, 8: 265–277, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Rudemiller NP, Patel MB, Zhang JD, et al. C-C Motif Chemokine 5 Attenuates Angiotensin II-Dependent Kidney Injury by Limiting Renal Macrophage Infiltration. Am J Pathol, 186: 2846–2856, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Twist1 deletion in distal nephron does not cause renal abnormalities at baseline. (A) Representative micrographs of PAS staining showing kidney cortex and medulla from WT and KKO mice. Scale bar =100 μm (B) Immunofluorescence staining for Aqp1 and Cdh16 in WT and KKO kidneys. Scale bar =100 μm (C) mRNA expression of Cdh16 and Aqp1 in WT and KKO kidneys (n=3). (D) Western blot analyses of renal expression of Cdh16 and Aqp1 proteins (n=3). (E) Semi-quantification of Cdh16 and Aqp1 from (D). (F) Measurement of BUN in serum from WT and KKO mice (n=3). Cdh16, Cadherin 16; Aqp1, aquaporin-1. Data represent the mean± SD (*p<0.05).
Figure S2. Body weights of WT and KKO mice after aristolochic acid (AA) exposure at 5 weeks (n≥8).
Figure S3. The gating strategy for excluding cell debris and analyzing viable myeloid cells in aristolochic acid (AA)-exposed kidneys.
Figure S4. mRNA expression of Aqp1, Cdh16 and Twist1 in primary renal tubular cells (RTCs) from WT and KKO kidneys (n=3). Data represent the mean ± SD (**p<0.01).
Figure S5. The gating strategy for the in vitro cultured tubular cells.
Figure S6. Knocking down fibronectin (FN) in primary renal tubular cells (RTCs) pretreated with AA blunts IL-1β and MCP-1 mRNA induction in co-cultured macrophages. (A) mRNA expression of FN in primary RTCs transfected with scramble siRNA or FN siRNA (n=3). (B) mRNA expression for IL1β and MCP-1 from macrophages co-cultured with RTCs as indicated (n=3). Data represent the mean ± SD (*p<0.05, ***p<0.001).