

Abstract
The skin is the largest organ of the body. The establishment of immunological memory in the skin is a crucial component of the adaptive immune response. Once naive T cells are activated by antigen-presenting cells, a small fraction of them differentiate into precursor memory T cells. These precursor cells ultimately develop into several subsets of memory T cells, including central memory T (TCM) cells, effector memory T (TEM) cells, and tissue resident memory T (TRM) cells. TRM cells have a unique transcriptional profile, and their most striking characteristics are their long-term survival (longevity) and low migration in peripheral tissues, including the skin. Under physiological conditions, TRM cells that reside in the skin can respond rapidly to pathogenic challenges. However, there is emerging evidence to support the vital role of TRM cells in the recurrence of chronic inflammatory skin disorders, including psoriasis, vitiligo, and fixed drug eruption, under pathological or uncontrolled conditions. Clarifying and characterizing the mechanisms that are involved in skin TRM cells will help provide promising strategies for reducing the frequency and magnitude of skin inflammation recurrence. Here, we discuss recent insights into the generation, homing, retention, and survival of TRM cells and share our perspectives on the biological characteristics of TRM cells in the recurrence of inflammatory skin disorders.
Keywords: Tissue-resident Memory T cells, Skin inflammatory disorders, Recurrence, Psoriasis
Subject terms: Immunological memory, Chronic inflammation
Introduction
Our body is covered by barrier tissues, such as the skin and the external mucosa. The entry of pathogens through these barrier tissues stimulates dendritic cells (DCs) that underlie the mucosal epithelium and the skin. DCs capture incoming antigens and subsequently migrate to local draining lymph nodes for antigen presentation to naive T cells.1 Once activated, naive T cells proliferate and transform into effector T cells that migrate to B cell areas or to inflamed tissues. Among activated naive T cells, a small fraction differentiate into the memory T cell precursors. Based on effector function, proliferative capacity, and migration potential, these precursor memory cells ultimately develop into several subsets of memory T cells, including central memory T (TCM) cells, effector memory T (TEM) cells, and tissue resident memory T (TRM) cells.2 In addition to peripheral barrier tissues, nonbarrier tissues, such as the brain, sensory ganglia, liver, bone marrow, and adipose tissue, have also been reported to contain memory T cells.3–5
While TRM cells have only been described for two decades, our understanding of them and interest in them are increasing. TRM cells are not just memory T cells that are located in peripheral tissues. They have a unique transcriptional profile that differs from that of TCM cells or TEM cells. For instance, TRM cells do not express CD62L (L-selectin) or CCR7 (a lymph node homing receptor). They express C-type lectin CD69, CD103 (an E-cadherin receptor), and CD49a depending on the TRM subset and peripheral tissues.6 Moreover, in contrast to recirculating memory T cells, skin TRM cells show strong enhancement of genes that facilitate extracellular free fatty acid (FFA) acquisition/metabolism and mitochondrial oxidative metabolism,7–9 which suggests their prominent ability to adapt to the local skin microenvironment.
Apart from long-term survival (longevity), low migration in peripheral tissues is likewise a striking characteristic of TRM cells. TCM cells target and survey the lymph node and then egress and return to the blood after infection.10 TEM cells survey nonlymphoid peripheral tissues and egress from peripheral tissues to blood vessels via the lymphatic system, while TRM cells do not. Generally, TRM cells never recirculate through the blood once they take up residence in peripheral tissues, and a recent study of TRM cells in secondary lymphoid organs indicated that TRM cells in the skin or mucosa can give rise to TRM cells within draining lymph nodes.11–13
TRM cells are heterogeneous and can be divided into CD8+ and CD4+ subsets. CD8+ TRM cells play an irreplaceable role in peripheral tissues, in which they enhance immune responses. For instance, CD8+ TRM cells in skin lesions in psoriasis, a common chronic inflammatory skin disorder, have been demonstrated to generate IL-17 to promote local skin inflammation.14 The features of CD4+ TRM cells are much more unclear, but recent evidence has indicated that they play a critical role in in situ protective immunity against skin infections (e.g. Leishmania and Candida albicans infections) and in mucosal sites, such as the lung, small intestine, and female reproductive tract.15–19
TRM cells are known to act as rapid on-site security alarms and provide immune protection against pathogen infections.20 In addition, evidence has suggested that TRM cells also develop after sensitization to otherwise harmless environmental antigens or self-antigens.21 Given their biology and behavior (long-term survival and low migration), aberrantly activated TRM cells have been strongly implicated in the recurrence of chronic inflammatory skin diseases22, including psoriasis,23 fixed drug eruption (FDE),24 mycosis fungoides (MF),21 vitiligo,25 and allergic contact dermatitis (ACD).26 Here, we discuss recent insights into the generation, homing, retention, and survival of TRM cells. In fact, these points are not stand-alone processes of TRM cells. We discuss them separately for the purpose of better understanding. We also share our perspectives regarding the biological characteristics of skin TRM cells in the recurrence of inflammatory skin disorders.
Generation of TRM cells
The generation of antigen-specific TRM cells is essential for rapid and long-lasting immunological protection. It is now known that the commitment to the memory lineage occurs early after infection,27,28 when a fraction of naive T cells activated by local DCs differentiate into memory T cell precursors. These precursors can be divided into distinct subsets according to the expression of the receptors CD127 and KLRG1 (killer cell lectin-like receptor subfamily G member 1).
CD127 is an IL-7 receptor α-chain. In humans, the IL-7/CD127 interaction promotes the differentiation, survival, and homeostasis of T cells. It is enhanced in rheumatoid arthritis, inflammatory bowel disease, and inflammatory skin diseases, including psoriasis and atopic dermatitis.29 CD127 is considered a marker of memory precursor cells; however, it has been indicated that the IL-7/CD127 interaction alone is not sufficient for the formation of CD8+ memory T cell precursors.30,31 Another receptor, KLRG1, is an inhibitory cell surface receptor expressed on subsets of NK cells and memory T cells. It inhibits immune responses by regulating the senescence and development of NK and T cells.32 In humans, E-cadherin, a calcium-dependent adhesion molecule on skin keratinocytes and DCs, is the ligand for KLRG1. The inhibition of KLRG1 function by blocking E-cadherin has been shown to result in a significant enhancement of Akt phosphorylation and T-/cell receptor (TCR)-induced proliferative activity in highly differentiated CD8+ T cells.33
It has been shown that KLRG1dim/CD127low and KLRG1dim/CD127high cells give rise to long-lived T cells, while short-lived T cells are derived from KLRG1high/CD127− cells.1,34 TRM cells, like long-lived TCM cells have been demonstrated to be derived from KLRG1dim cells, while TEM cells are derived from KLRG1high cells.34,35 Moreover, recent studies in humans and mice with ACD have demonstrated that skin TRM cells and lymph node TCM cell clones share overlapping TCR complementarity determining region 3 (CDR3) sequences.26,36 This was supported by an in vivo experiment on the generation of TRM cells in human skin-engrafted mice. The experiment demonstrated that, compared with other memory subsets, injected TCM cells enter grafted skin in larger numbers, giving rise to more TRM cells.2 In addition, CD8+ TCM cells have been demonstrated to differentiate into functional CD69+CD103− TRM cells following viral clearance in the skin to act as the major tissue-resident population.37 These data suggest that at least some TRM cells and TCM cells are derived from a common naive T cell precursor after skin immunization. They also suggest that TRM cells are probably generated after successful tissue homing and tissue residency.
Published results have indicated that specific priming signals from DCs may differentially affect these two identical TCR CDR3 memory T cell precursors to generate TCM and TRM cells. For instance, a study showed that optimal TRM cell generation can be promoted by dendritic cell natural killer lectin group receptor 1 (DNGR-1)+ DCs during viral infection and skin immunization. DNGR-1+ DCs provide unique signals associated with IL-15 and the transcription factor T-bet, and favor longer cell retention in the lymph nodes.38
In addition to the expression of CD127 and KLRG1, the expression of the chemokine receptor CX3CR1 has been recently reported by Gerlach and colleagues to define distinct memory CD8+ T cell subsets following viral infection; TCM cells are CX3CR1− cells, TEM cells are CX3CR1hi cells, and TRM cells are CX3CR1−/low cells.10 The authors also identified another distinct memory subset called TPM (CX3CR1int) that predominantly surveys peripheral tissues with unique phenotypic, homeostatic, and migratory properties.
At present, several signaling pathways, including TCR stimulation affinity, the mTOR pathway, and IL-15 signaling, have been suggested to control the differentiation fate of naive CD8+ T cells into short-lived effector cells or memory precursor T cells.
The fine changes in the magnitude of affinity between TCR and major histocompatibility complex (MHC) can lead to markedly different downstream consequences in memory T cell generation and function.39,40 There is strong evidence that TRM precursor cells receive TCR signals with a different affinity from that of other subsets.41 It has been shown that TCR stimulation affinity is inversely associated with the establishment of functional CD8+ TRM cells in the brain. Lower stimulation confers greater functionality to brain TRM cells.42 Moreover, the transcriptome of lung TRM cells has been demonstrated to depend on the TCR-MHC interaction.43 However, whether this process is the same for TRM cells in the skin and the molecular mechanisms by which TCR-MHC induces events to optimize skin TRM cell formation require further exploration.
The mammalian target of rapamycin (mTOR) pathway is a central regulator that links diverse environmental stimuli, immune signals, and nutrient metabolism.44 It is clear that the effect of rapamycin on mTORC1/2 is magnitude-dependent. mTORC1 is sensitive to routine rapamycin inhibition, and prolonged rapamycin treatment inhibits mTORC2 assembly.45 The mTOR signaling pathway is activated in the early phase of the immune response and shut down or turned off as time passes. It has been suggested to play a key role in the generation of TRM cells.46,47 It has been demonstrated that rapamycin enhances the formation of CD8+ memory T cells in the circulation and secondary lymphoid tissues,47 but it inhibits the formation of CD8+ TRM cells in the intestinal and vaginal mucosa.46 The inhibition of functional CD8+ TRM cells in the mucosa by rapamycin can protect a mice from lethal CD8+ T cell-mediated intestinal autoimmunity.46 These findings suggest an opposing role of the mTOR pathway in the generation of resident a d nonresident CD8+ memory T cells, most likely due to the various levels of rapamycin in different microenvironments.
IL-15 is an important homeostatic cytokine, and many types of cells in various peripheral tissues, including the skin, can produce IL-15. Therefore, TRM cells have the advantage of receiving IL-15 signals more frequently and more robustly than circulating T cells.47 IL-15 has been closely linked to the generation of TRM cells, but it is not indispensable.47,48 Within CD8+ memory T cell populations, both IL-15-dependent and IL-15-independent (e.g., in the lung) populations have been described. IL-15 activates mTOR signaling, and the inhibition of mTOR can lead to a predominantly IL-15-independent memory population.49
Despite this progress, we still have not completely uncovered the link(s) between the phenotype of TRM cells and the corresponding signals, especially the possible crosstalk among TCR, mTOR, and IL-15. Therefore, information regarding the signals that regulate the generation of TRM cells is not sufficient, and there is still a long road ahead.
Homing of TRM cells
Distinct subsets of mature memory T cells are recruited to different tissues. TCM cells are mainly recruited to lymphoid tissues by the expression of the lymph node homing receptors CD62L and CCR7. TEM cells are spread throughout diverse peripheral tissues by homeostasis. TRM cells are recruited to peripheral tissues, where they initially encounter pathogens or attacks.12
Evidence has indicated that the homing of TRM cells to peripheral target tissues is a chemokine-dependent but antigen-independent process within tissue-draining lymph nodes.50–52 Depending on the expression of tissue-specific receptors (Table 1), subsets of TRM cells can home to specific tissues. For instance, in patients with psoriasis, atopic dermatitis, or ACD, most TRM cells that infiltrate skin lesions express CCR10, a skin homing receptor,53,54 TRM cells that are recruited to the gut express integrin α4β7, CXCR3, and CCR9,55 TRM cells that enter the kidney have enhanced E/P-selectin expression promoted by TGF-β,56,57 BLT-1, CCR6, and α1β1 integrin (VLA-1) are lung homing addressins,58 and CCR5 and CXCL10 are brain homing addressins.59,60 However, this is not always the case. For instance, integrin α4β7+ TRM cells have also been found in certain skin contact hypersensitivity responses;61 P-selectin is likewise important for the homing of CD4+ TRM cells to the gut, and CXCR3 also plays a key role in the homing of TRM cells to the HSV-infected skin and vagina.62
Table 1.
The phenotype, tissue distribution, and other immune characteristics of TCM, TEM, and TRM cells
| Subset | Phenotype | Tissue distribution | Migration | Proliferation | Cytokine | Ref. |
|---|---|---|---|---|---|---|
| TCM | CD62L+, CCR7+, CD69+/−, CD103 (Integrin αE)−, CD11a+/−, KLRG1−, CD127 (IL-7R)+, CX3CR1− | Lymph nodes, Spleen, Blood | High | Prompt | Poor | 10,34,35,76,77 |
| TEM |
CD62L−, CCR7−, CD69+/−, CD103 (Integrin αE)−, CD11a+/−, KLRG1+, CD127 (IL-7R)+/−, CX3CR1+ Skin: CLA+, CCR4+, CCR8+, CCR10+ Gastrointestinal tract: CCR9+, Integrin α4β7+ |
Spleen, Lymph nodes, Blood, Gastrointestinal tract, Lung, Liver, Skin, Reproductive tract | High | Slow | Prompt | 10,34,35,76,77 |
| TRM |
CD62L−, CCR7−, CD69+, CD103 (Integrin αE)+/−, CD11a+, KLRG1−, CD127 (IL-7R)+/−, CX3CR1+/−, CD49a (Integrin α1) +/− Skin: CLA+, CCR4+, CCR8+, CCR10+ Gastrointestinal tract: CCR9+, Integrin α4β7+ Lung: CCR6+, BLT-1+, Integrin α1β1+ |
Skin, Gastrointestinal tract, Lung, Liver, Reproductive tract | Low | Poor | Prompt | 1,10,34,35,53–62,76,77 |
In addition to epithelial cells in the skin and mucosa, other types of cells (e.g., neutrophils) are also capable of producing chemokines for T cell recruitment. It has been demonstrated that, during influenza virus infection, neutrophils that are recruited to the infected sites early on leave behind long-lasting chemokine CXCL12-containing trails that are critical for virus-specific CD8+ T cell recruitment.63 In psoriasis, a chronic inflammatory skin condition, neutrophils have been suggested to be involved in skin lesion initiation.64–66 It is now clear that the skin influx of neutrophils is the early stage of psoriatic initialization. Neutrophil influx results in the formation of Munro’s microabscesses (psoriasis vulgaris, Fig. 1) in the epidermis stratum corneum or spongiform pustules of Kogoj in the stratum spinosum (pustular psoriasis).67 It seems that there is a link between the homing of TRM cells to psoriatic lesions and early neutrophil influx. However, whether neutrophils in psoriatic lesions leave behind chemokine-containing trails for TRM cell homing similar to generated during influenza virus infection and the type of chemokine(s) involve require more studies.
Fig. 1.

Epidermal influx (① → ② → ③) of neutrophils from the peripheral blood in the early stage of psoriatic lesion initialization. Neutrophils form Munro’s microabscesses (③) when they reach the epidermis stratum corneum. The dotted line indicates the border between the epidermis and the dermis. Routine hematoxylin-eosin staining of psoriatic lesions at ×400 magnification are shown
It has been demonstrated that repeated skin infections can lead to the progressive accumulation of protective TRM cells not only in infection sites but also in uninvolved (normal-appearing) skin.12 In addition to infections, physical stimuli can also provoke memory T cell accumulation and even reactivation. The Koebner phenomenon is an example in dermatological practice. It represents the development of isomorphic inflammatory skin lesions (e.g., psoriasis) in the uninvolved (normal-appearing) skin following various traumas (e.g., surgical incision, tattooing, insect bites, tape stripping, and needle acupuncture, Fig. 2).68,69 These isomorphic lesions are always historically identical to those that arise spontaneously. Although the specific mechanism has not been completely elucidated, extensive studies have shown that nonlesional skin in inflammatory dermatosis is clearly distinct from normal skin with respect to the increased expression of immune-related genes,70,71 and certain T cell-related cytokines and adhesion molecules have been suggested to be involved in the Koebner phenomenon in the skin.72,73 In general, the time required for koebnerization is several days. Thus, it is rational to speculate that there may be a potential link between the homing/accumulation of TRM cells in normal-appearing skin following those stimuli and the Koebner phenomenon, which requires further investigation.
Fig. 2.

Clinical images of the Koebner phenomenon. Psoriatic lesions develop in uninvolved (normal-appearing) skin at the site traumas. The accumulation and reactivation of memory T cells at these sites have been suggested to occur. Left, new linear psoriatic lesions occurred along scratches on the abdomen; Middle, new psoriatic lesions occurred on a scar from abdominal surgery; Right: psoriatic lesions occurred at the site of tattoo removal by medical laser on the leg. Asterisks, original psoriatic lesions; arrows, psoriatic lesions induced by trauma
All of these data and observations further enhance our understanding of memory T cell homing. However, the factors that promote aberrant chemokine production for TRM cell homing to peripheral skin tissues need to be elucidated and would likely helpful for relieving the recurrence of chronic inflammatory skin diseases.
Retention of TRM cells
Once TRM cells are taken up by peripheral tissues, they do not recirculate back to the lymph nodes and; like TEM cells, they equilibrate in the blood. TRM cells can persist for months (e.g., in the lung) or years (e.g., in the skin), even though the pathogens are undetectable.12,74 Although the molecular mechanisms responsible for the retention of TRM cells are not fully understood, CD69, integrins (CD49a, VLA-1, CD103, αvβ6, and αvβ8), CCR7, the CCL27-CCR10 axis, and aryl hydrocarbon receptor (AhR) have been suggested as key factors in the process.
In humans and mice, CD69 is expressed by the majority of CD4+ and CD8+ TRM cells in multiple sites, including the skin.75 It is an early marker of TCR-mediated activation. Effector T cells and circulating memory T cells express CD69 upon activation, but its expression is downregulated afterwards; however, TRM cells in peripheral tissues express CD69 constitutively.76,77 These distinct CD69 expression patterns distinguish tissue residents from circulating memory populations. Human CD69+ TRM cells are transcriptionally and phenotypically distinct from CD69−TRM cells. They exhibit a core gene signature including homing, residency, proliferative turnover, and activation elements with key homology with mouse TRM cells. This core signature associated with the CD69+ subset (e.g., PD-1, CRTAM, CXCR6, DUSP6, and CD101) is conserved across CD4+ and CD8+ TRM lineages and multiple peripheral tissues.75
The main mechanism by which CD69 participates in the retention of TRM cells is the inhibition of the cell-surface expression and function of S1PR1 (sphingosine 1-phosphate receptor type 1).77,78 S1P is a downstream target of Kruppel-like factor 2 (KLF2) and mTOR, and it is a vital lipid second messenger involved in T cell egress from peripheral tissues.28,79 It has been demonstrated that CD8+ TRM cells lack S1PR1 expression, and the forced expression of S1PR1 or genetic deletion of CD69 prevents the retention of TRM cells in peripheral tissues.41,77
CD49a (integrin α1) and VLA-1 (α1β1 integrin heterodimer) are involved in collagen binding and can mediate the retention of TRM cells in mucosal tissues via attachment to the extracellular matrix. Blocking antibody treatment or genetic deficiency of VLA-1 decreases the number of TRM cells in the lung.80–82
CD103 (integrin αE) is paired with integrin β7, and it is often enhanced in inflammatory diseases, particularly in those in which T cells infiltrate epithelial tissues.83 In the skin, it has been verified that CD103+ TRM cells reside in both the epidermis and dermis, while CD103− TRM cells prefer the dermis.2,7 Similar to the CD49a-collagen interaction, CD103 confers substrate specificity for cell adhesion to E-cadherin (epithelial cadherin), as does KLRG1 (Fig. 3). However, the CD103 binding site on E-cadherin has been suggested to be distinct from the KLRG1 binding site.84 Moreover, KLRG1-E-cadherin inhibits effector T cell function, whereas the binding of CD103 to E-cadherin enhances cell–cell interaction and adhesion.85 The expression of CD103 and KLRG1 has been demonstrated to be mutually exclusive. TGF-β may play certain roles in this process, as it is known to induce CD103 but downregulate KLRG1 expression on CD8+ T cells.85,86
Fig. 3.
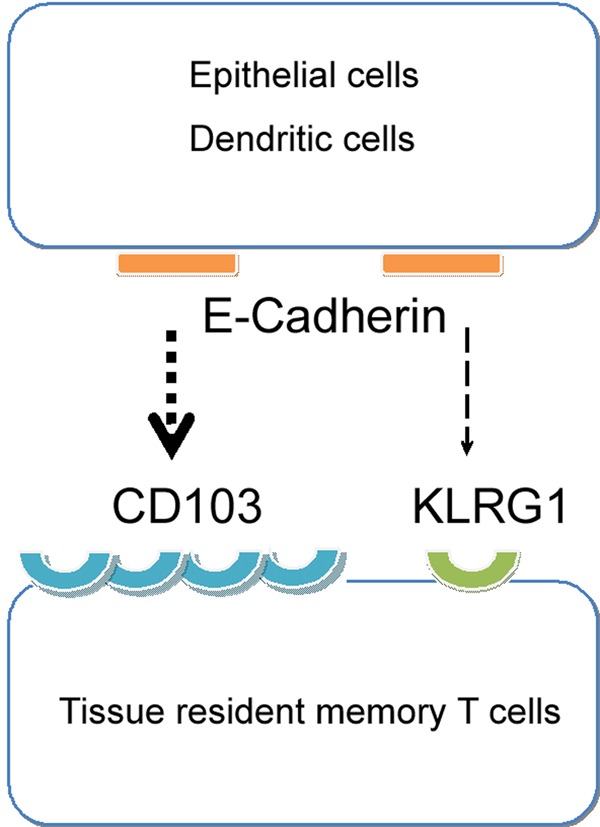
CD103 on TRM cells is the main receptor for E-cadherin from epithelial cells or DCs. The interaction between CD103 and E-cadherin enhances cell–cell adhesion and the residence of TRM cells
CD103 and CD49a are not merely biomarkers of certain subsets of TRM cells. They confer substrate specificity for cell adhesion and define different subsets of TRM cells in certain tissues.6,14,87 Their relationship, especially whether they can compensate for one another, is unknown, and more research is needed. For instance, CD103+ TRM cells can reside in the tissues of the adult central nervous system, in which E-cadherin is not expressed.11 This suggests that E-cadherin is likely not the sole ligand of CD103 in TRM cells or that there are other compensatory molecules responsible for the residence of TRM cells in nonepithelial tissues.
Other integrins (e.g., αvβ6 and αvβ8) that are expressed by interfollicular keratinocytes in the skin have also been reported to maintain the residence of TRM cells in the skin by activating latent TGF-β. The treatment of the skin with ultraviolet irradiation (a therapeutic regimen against chronic skin inflammation) can decrease their expression on keratinocytes and that of the active form of TGF-β, which leads to reduced skin retention of TRM cells and skin inflammation.88
Chemokines have also been suggested to regulate the tissue retention of memory T cells. The lymph node homing receptor CCR7 is involved in egress from the skin, and its cooperation with S1PR1 in accelerating the egress of TRM cells has been suggested.1,41 Therefore, the downregulation of CCR7 may be an additional mechanism by which TRM cells are retained in the skin. CCR10 and its ligand CCL27 is the most skin-specific chemokine receptor/ligand pair implicated in inflammatory skin diseases. This pair is thought to regulate not only homing (as mentioned above) but also the retention of skin TRM cells.57 It has been reported that the level of CCL27 is not only increased during skin inflammation but also remains high several weeks after allergen skin challenges. In parallel with increased CCL27 expression, large numbers of CD4+CCR10+ TRM cells exist in ACD lesions that have returned to normal clinically weeks after challenge.89 These results suggest that the CCL27-CCR10 axis is one of the main mechanisms of the retention of TRM cells in ACD.
Aryl hydrocarbon receptor (AhR), a member of the helix-loop-helix transcription factor family, is also associated with the retention of TRM cells in the skin.90 AhR is thought to mediate the toxic reaction of chemical substances at barrier sites, such as the skin and gut. It is clear that AhR participates in crucial biological processes, including innate and adaptive immune responses, in vivo.91 The activation of AhR can inhibit the inflammatory reaction, and its antagonism promotes this process in skin lesions.92–95 An animal model of psoriasis with AhR deficiency exhibits more severe pathological characteristics than those of wild-type animals.93 Studies have shown that CD8+ TRM cells express a higher level of AhR than TCM and TEM cells.96 In a mouse model of dinitrofluorobenzene-induced skin inflammation, AhR knockout in CD8+ cells does not alter the number of CD8+ cells recruited to the skin lesions, but they disappear only a few days later.90,97 This suggests that AhR contributes to the skin retention or survival of TRM cells, which is similar to the roles of AhR in the maintenance of liver-resident natural killer cells.98 In addition, it has been demonstrated that treatment with the potent exogenous AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin can transiently increase CD69 expression.99 However, the exact role of AhR and the biochemical mechanism underlying AhR-mediated TRM cell retention require further investigation.
Overall, the above studies show that the distinct regulators of TRM cell retention may be promising intervention targets for TRM cells and suggest that they are probably tissue-specific. However, a recent investigation identified the transcription factors Hobit and Blimp1 as universal central regulators that instruct the retention of TRM cells in the skin, gut, liver, and kidneys of mice.100 These findings are suggestive of the complexity of TRM cell retention regulation.
Survival of TRM cells
There is limited understanding of the molecular mechanisms and signaling processes that regulate TRM cell survival and apoptosis. It has been suggested that TRM cell survival in skin tissues is mainly regulated by the local microenvironment, even in the absence of antigen presentation.101 A combination of retention in the skin and local inflammatory signaling by IL-7, IL-15, or TGF-β has been suggested to be required for the survival and homeostasis of skin TRM cells.101–103 Tissue-specific instruction is involved in the survival of TRM cells. Skin keratinocytes, DCs, and fibroblasts may play critical roles in this process because they sense environmental danger signals (e.g., microbial agents) and are the major source of IL-7, IL-15, TGF-β, and IL-17 polarizing cytokines.104,105
It has been demonstrated that the local proliferation of TRM cells in the skin in response to local antigen challenge can maintain the survival of a stable pool of tissue-resident memory T cells.106 Multiple interrelated signaling pathways, including the Notch, JAK/STAT5, PI3K/Akt, and Wnt signaling pathways, have been suggested to be involved in the survival of TRM cells in peripheral tissues.
Notch is a transcriptional regulator involved in T cell development and the formation of memory T cells. It controls the survival of TCM cells by regulating Akt phosphorylation and glucose uptake.107 Recent genetic and pharmacological experiments have provided compelling evidence that Notch signaling is also required for the survival of TRM cells. Notch-deficient mice have decreased expression of CD103 on CD8+ TRM cells in their airways.108 Moreover, the activator of Notch signaling, Delta-like ligand, can induce the expression of IFN-γ mRNA in TRM cells but not in TEM cells. Although most TRM cell-specific gene expression is not dependent on Notch signaling, the inhibition of Notch can affect the expression of a few TRM cell-specific genes, including Itgae, Acer2, Tmem37, Rplp2, and Arl5c.108,109 Among these specific genes, Itgae encodes CD103, and its downregulation by Notch inhibition leads to the lower surface intensity of CD103 on TRM cells; Acer2 hydrolyzes the sphingolipid ceramide into sphingosine and FFAs. Exogenous FFA consumption has been demonstrated to be critical for the long-term survival of skin TRM cells. The T cell-specific deletion of the lipid transport molecules fatty-acid-binding proteins 4 and 5 (FABP4/5) impairs exogenous FFA uptake by skin CD8+ TRM cells and greatly decreases their long-term survival in vivo, while having no effect on the survival of TCM cells.8,9 These findings suggest that Notch signaling controls the survival of TRM cells at least in part by regulating their metabolic functions.
In response to IL-7 and/or IL-15, long-lived memory T cells rapidly activate JAK/STAT5, as do effector T cells. However, the phosphorylation levels of STAT5 are much higher than the levels of STAT5.110 Constitutive STAT5 activation has been demonstrated to profoundly enhance antiapoptotic Bcl-2 expression and memory T cell survival.111,112 The forced expression of the active form of STAT5 prolongs the survival of what would otherwise be short-lived terminally differentiated effector T cells.113 PI3K/Akt signaling has been shown to be activated more robustly in long-lived memory precursor T cells than in short-lived effector cells, and the functional suppression of PI3K/Akt can lead to defective CD8+ memory T cell formation in vivo.111,114 However, constitutive PI3K/Akt activation does not enhance memory CD8+ T cell survival but rather represses IL-7 and IL-15 receptor expression, STAT5 phosphorylation, and Bcl-2 expression (Fig. 4).111,115,116 This suggests that the survival of memory T cells likely depends on optimally balanced PI3K/Akt signaling, the maintenance of JAK/STAT5 signaling, and their complex interplay. However, whether this is true for the survival of skin TRM cells requires further investigation.
Fig. 4.

The balance of PI3K/Akt signaling, the maintenance of JAK/STAT5 signaling, and their complex interplay in the survival of TRM cells. The activation of both PI3K/Akt signaling and JAK/STAT5 signaling support TRM cell survival (left). However, the constitutive activation of PI3K/Akt represses IL-7/IL-15 receptor expression and STAT5 phosphorylation (right)
The Wnt pathway is evolutionarily conserved and promotes stem cell self-renewal. It has been suggested to be involved in the maintenance of the longevity of mature memory CD8+ T cells via downstream transcription factors (e.g., TCF-1).117,118 TCF-1 is highly expressed in naive T cells, downregulated in effector T cells, and upregulated in TCM cells. TCF-1-deficient memory CD8+ T cells are progressively lost over time and show decreased levels of antiapoptotic Bcl-2 and Eomes. The forced expression of Eomes can partly rescue TCF-1-deficient CD8+ TCM cells from time-dependent fade.118–120 These findings suggest that canonical Wnt signaling plays a critical role in TCM cells in vivo.
However, in TRM cells, the role of Wnt signaling seems to not be identical to that in TCM cells. It has been demonstrated that the downregulation of T-box transcription factors (Eomes and T-bet) is crucial for IL-15 and TGF-β signaling in CD8+ TRM cell survival,121 which is supposed to be different from that of TCM cells. In particular, Eomes should be nearly eliminated for optimal CD8+ TRM cell development, and residual T-bet expression is sufficient for IL-15-mediated long-term survival in a range of different tissues.122 TGF-β has been shown to promote the development of pulmonary TRM cells via a signaling pathway that does not require downstream Smad4.123 It plays an important role in the downregulation of Eomes and T-bet.41 Furthermore, the TGF-β and Notch pathways undergo intensive crosstalk, and along with optimally balanced PI3K/Akt and/or JAK/STAT5 signaling, they most likely constitute the integrated mechanisms involved in the survival of TRM cells.
TRM cells in the recurrence of inflammatory skin disorders
The skin is the largest organ of the body. It contains a large number of T cells, reaching up to 2 × 1010 cells or twice the number in the blood.124 Common T cell subsets, e.g., Th17 and Th22 cells, are associated with the severity of inflammatory skin diseases.125–127 The inhibition these cells has been proven to be an effective treatment temporarily, but it fails to prevent disease recurrence. This suggests that other T cell subsets may be directly responsible for the recurrence of inflammatory skin disorders.
It has long been known that the recurrence of cutaneous chronic inflammation, especially psoriasis and FDE, frequently occurs in previously affected sites (Fig. 5). Therefore, immunological memory has been proposed to be involved in flare-up reactivity and the chronicity of inflammatory disorders.128 With respect to the striking characteristics of TRM cells (long-term survival and low migration in peripheral tissues), it has been suggested that skin TRM cells may actively participate in the recurrence of inflammatory skin disorders.21
Fig. 5.

The recurrence of cutaneous chronic inflammation frequently occurs in previously resolved sites. Therefore, immunological memory is proposed to be involved. Left, psoriatic lesions on the trunk recurred at previously resolved sites; Middle, recurrent fixed drug eruption on the buttock; Right, recurrent vitiligo lesions on the upper extremity. Arrow, recurrent lesions; asterisks, previously resolved areas
Psoriasis
Psoriasis is a common chronic inflammatory skin disorder that usually appears on the skin as red patches covered with white scales. Genetic predisposition and environmental factors that affect the immune system (e.g., stressful events and skin trauma) are involved in triggering psoriasis.129
The primary onset of psoriatic lesions is often followed by recurrence in previously resolved sites, and the recurrence rate is approximately 90% (unpublished data, Zhu Shen). The immune memory maintained by local resident memory T cells has been suggested to play major roles in its development and flare-ups.130 For instance, T cells in lesional skin but not peripheral blood maintain the psoriatic phenotype in an SCID xenotransplantation mouse model.131 Moreover, immunodeficient mice can develop psoriatic skin lesions when normal-appearing skin is grafted from psoriatic patients.130 CD8+ T cells in psoriatic lesions are highly activated and express large amounts of CD69 and CD103. In contrast, few T cells constitutively express these proteins in the peripheral blood.23,132 Furthermore, it is clear that TEM cells interact with the vascular addressin E-selectin and are trafficked to the skin during infection or attack. However, the inhibition of TEM cell infiltration by blocking E-selectin is not an effective treatment for psoriasis.133 More importantly, recent studies have shown that TCRαβ+ resident T cells accumulate in psoriatic resolved sites, even in normal-appearing skin, and that they are capable of producing IL-17 and IFN-γ to trigger psoriasiform responses.104,134,135 These findings support the important role of lesion-resident T cells in psoriasis development.
It has been demonstrated that psoriatic TRM cells are retained in resolved lesions even for months after effective treatment with methotrexate (MTX), anti-TNF-α, or IL-12/23 biotherapy.23,136 For instance, after routine MTX treatment (12.5 mg/week) for 8 weeks, the level of inflammatory infiltrates is reduced; however, the persistence of CD8+ T cells that express CD69 in resolved skin lesions has been observed.136 Our unpublished data have also shown that a high number of CD8+CD69+ TRM cells remains in resolved psoriatic lesions (Fig. 6). In addition to its function in TRM cell retention, CD69 has been demonstrated to control the secretion of IL-22, which contributes to psoriasis development.137 Furthermore, CD69 has been demonstrated to be coexpressed with FABP4 and FABP5, two vital molecules highly expressed in psoriatic lesions, by CD8+ TRM cells enriched in human psoriatic lesions. FABP4 and FABP5 are involved in exogenous FFA acquisition/metabolism and long-term TRM cell survival, as discussed above.8,9,138 These results demonstrate that a large number of TRM cells remain in resolved sites of psoriasis.
Fig. 6.

Immunofluorescence staining of sections from resolved psoriatic areas. (a) DAPI staining; (b) DAPI and CD8 staining; (c) DAPI and CD69 staining; (d) DAPI, CD8 and CD69 staining. The yellow in the merged image indicates CD8+CD69+ TRM cells in the resolved psoriatic areas (arrows)
Psoriatic TRM cells are heterogeneous and functional. It has been reported that psoriatic CD8+ TRM cells can produce IL-17A upon ex vivo stimulation, and CD4+ TRM cells respond with IL-22 production for as long as 6 years after TNF-α inhibition.23 Both CD8+CD103+ and CD8+CD49a− subsets of TRM cells from psoriatic lesions predominantly generate IL-17 responses.14,23 Furthermore, our unpublished data have demonstrated that the intradermal injection of culture supernatant from stimulated CD8+CD69+ TRM cells isolated from clinically resolved psoriatic tissues into a mouse model of imiquimod-induced psoriasiform dermatitis can significantly reactivate psoriasis-like histological phenotypes (e.g., inflammatory infiltrates and epidermic hyperplasia). These data demonstrate that TRM cells in resolved skin lesions are functional and capable of producing cytokines that are known to be critical for psoriasis development, which further supports the role of TRM cells in psoriasis recurrence.
The CCL27-CCR10 axis has been suggested to be pivotal for the retention of TRM cells. However, it does not seem to be vital for the retention of TRM cells in psoriasis recurrence because the CCR10 level is sharply reduced in resolved sites compared with neighboring psoriatic lesions. Moreover, CCL27 expression is lower in psoriatic lesions than in perilesional skin (unpublished data, Zhu Shen),139,140 most likely because of the negative feedback of IL-17, INF-γ, or TNF-α in the lesions.141,142
Similar to those of CCR10, the levels of IL-7 and its receptor CD127 are sharply reduced (over 10-fold) in resolved sites compared with neighboring psoriatic lesions (unpublished data, Zhu Shen). IL-15 is an important proinflammatory cytokine and has important roles in psoriasis.143,144 It has been demonstrated to be required for the formation and maintenance of CD103+ TRM cells in the skin epidermis following HSV infection.7 Our unpublished data have shown that the IL-15 level in resolved sites remains as high as that in neighboring recurrent psoriatic lesions and can be upregulated by experimental Koebner phenomenon. In addition, MTX plus a neutralizing IL-15 antibody can decrease CD69 expression and the number of CD8+CD69+ TRM cells in psoriatic organ cultures (unpublished data, Zhu Shen). This suggests the important role of IL-15 in the survival of TRM cells in psoriasis recurrence.
These findings demonstrate the persistence of immunological memory in psoriatic resolved sites, and TRM cells are one of the vital subsets of resident memory T cells in psoriatic lesion recurrence. This suggests that targeting TRM cells is a novel potential therapeutic strategy for decreasing psoriasis recurrence; however, more evidence, especially from psoriatic animal modes, is needed.
Fixed drug eruption
Adverse reactions to medications are common in dermatological practice. FDE is a localized variant of systemic medication-induced cutaneous adverse reactions. It usually presents with a single or a small number of erythematous or violaceous plaques, even those that are bullous or necrotic. FDE is characterized by rapid recurrence months or even years later in susceptible patients when a medication of the same or similar structure is taken again.145 It occurs in exactly the same location as the first instance (Fig. 5) or in previously traumatized sites, such as insect bite, burn scars, and venipuncture sites (the Koebner phenomenon discussed above).24,146
In resolved FDE lesions, histological staining has shown the predominance of an intraepidermal population of CD8+ memory T cells that is capable of producing IFN-γ and TNF-α upon activation.21,24,147,148 These T cells constitutively express the cutaneous lymphocyte-associated antigens CD11a, CD69, and CD103 but not CD62L or CCR7.24,147,149 Moreover, the rate of production of IFN-γ is much faster (3 h after challenge) than that of their peripheral counterparts.24,149
The clinical and pathologic features observed in FDE lesions can be explained by the presence of CD8+ TRM cells. For instance, these T cells can transiently acquire a natural killer-like phenotype upon clinical challenge with the causative medication and release cytotoxic granules that lead to the epidermal damage seen in FDE lesions;150 the influx of regulatory T cells into the epidermis in fully evolved lesions has been suggested to limit the harmful immune reactions of these CD8+ TRM cells.147 Moreover, IL-15 derived from lesional keratinocytes can maintain the survival of these TRM cells, even without antigenic stimulus over a prolonged period.24,150 The above evidence suggests that aberrantly activated TRM cells are one of the major effector cells in FDE recurrence.
Drug eruptions represent a wide spectrum of cutaneous reactions. Unlike mild FDE, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are potentially life-threatening and at the extremely severe end of the drug eruption spectrum. Skin TRM cells have been suggested to play a key role in the pathogenesis of SJS and TEN;151 however, more direct evidence is needed.
Vitiligo
Vitiligo is a chronic skin disorder characterized by skin depigmentation (Fig. 5), and it is clinically characterized by pale to white patches on the skin. Psychological stress, skin damage (the Koebner phenomenon mentioned above), a family history of vitiligo, and other concomitant autoimmune conditions are triggers. Vitiligo prominently recurs in the same site after treatment is discontinued, suggesting the involvement of immunological memory.25
In vitiligo, an abnormal immune reaction has been demonstrated to attack the melanocytes of the skin. Recently, melanocyte-specific autoreactive TRM cells in vitiligo lesions have been described, and TRM cells have been suggested to cooperate with TCM cells to maintain vitiligo in a mouse model.25,152 Research has demonstrated that, in skin lesions of patients with vitiligo, CD8+CD49a+ TRM cells constitutively express perforin and granzyme B and thus exhibit a strong cytotoxic phenotype.14 More recent studies have shown that stable and active perilesional skin in vitiligo is enriched with a subset of CD8+ TRM cells that express CD69, CD103, and CXCR3. They are functional and exhibit the increased production of IFN-γ and TNF-α and moderate cytotoxic activity.153
Likewise, IL-15 has been reported to promote the survival of TRM cells in vitiligo. IL-15-deficient mice reportedly exhibit the impaired formation of TRM cells. Targeting IL-15 signaling with an antibody against CD122 (subunit of IL-15 receptor) can reverse established vitiligo in mice by inhibiting the production of IFN-γ by TRM cells or even depleting TRM cells in skin lesions.154 These studies not only emphasize the involvement of TRM cells in vitiligo recurrence but also provide novel potential therapeutic strategies for diseases involving TRM cells.
Atopic dermatitis
Atopic dermatitis (AD), also known as atopic eczema, is a chronic pruritic inflammatory skin condition. AD often involves scaly and red rashes on the cheeks, scalp, or flexion of the arms and legs. It is characterized clinically by waves of recurrence in the above areas. Both genetic and stimulating factors, e.g., scratching, infections, dry skin, colds, and stress, play important roles in AD development.155
AD recurrence is accompanied by uncontrolled immune activation, and immune memory has been suggested to be involved.138,156 It has been shown that TCR diversity is similar in lesional and nonlesional skin. Most top expanded lesional T cell clones are also present in nonlesional skin, and they are largely maintained even after months of treatment with topical glucocorticoids. This suggests the presence of potentially pathogenic TRM cells in lesional and nonlesional skin in AD.157
Recently, thymic stromal lymphopoietin (TSLP), an triggering factor for AD, has been reported to increase CD69 expression and the number of CD69+ TRM cells in AD.158 It has been demonstrated that, as in psoriasis, CD69+ TRM cells in AD skin lesions express considerable molecules related to tissue residency. Moreover, both CD4+CD69+ and CD8+CD69+ TRM cells are enriched with various potentially pathologic cytokine genes, e.g., IL-4, IL-13, IL-17, and IL-22, which indicates that these multifunctional TRM cells might be the main cause of AD recurrence.159
Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a classic example of a T cell-mediated hypersensitivity reaction in the skin. Red, pruritic, swollen erythema or blisters occur 24 to 48 h after the skin comes in contact with an irritant or allergen. The lesions may persist for weeks after exposure stops and will recur upon future exposure.160,161
The damage to skin cells caused by ACD depends on the rapid activation of a subtype of specific T cells. There is emerging evidence to suggest the contribution of TRM cells to ACD flare-ups.162,163 It has been shown the response to ACD challenge in both mice and humans sensitized to contact allergens (2,4-dinitrofluorobenzene and nickel) is dramatically increased at sites previously challenged by these allergens. Furthermore, the response magnitude is correlated with the accumulation of TRM cells that are capable of producing IL-17A and IFN-γ in the skin.163 It has been demonstrated that a large number of CD4+CCR10+ TRM cells exist in ACD lesions that have clinically returned to normal 21 days after allergen challenge. This is likely associated with local skin immunological memory and rapid recall responses in previously challenged sites.89
Mycosis fungoides
Mycosis fungoides (MF) is a skin-limited variant of cutaneous T cell lymphoma. It may be clinically and histologically manifested as an inflammatory skin disorder in its early stages. MF usually begins as eczematous patches and proceeds to plaques and nodules that are infiltrated by T cells. These early inflammatory lesions are often stable for years and are therapeutically responsive to topical glucocorticoids, phototherapy, and low-dose irradiation. However, they usually recur in previously affected sites once therapy is withdrawn.164
It has been demonstrated that immune cells that are trafficked to the skin play crucial roles in MF. Recent studies have suggested that MF is a malignancy of distinct memory T cell subsets.21 T cells isolated from MF skin lesions do not express CD62L (L-selectin) or CCR7 (a lymph node homing receptor), but they strongly express CCR4 and a cutaneous lymphocyte-associated antigen, which is a phenotype that is suggestive of skin TRM cells.165 This is consistent with the formation of fixed patches or plaques on the skin in MF. However, the factors that drive these skin TRM cells into malignancy are not well known.
Perspectives
The long-term survival and low migration of skin TRM cells combined with their potent effector functions are evidence of their potential roles in the recurrence of inflammatory skin disorders. However, more studies regarding the direct contribution of TRM cells to skin inflammation recurrence are needed. Moreover, further investigations are needed to uncover more mechanisms underlying the biology of TRM cells, including common mechanisms and distinct mechanisms. At present, there are several vital points that require more attention.
First, TRM cells are heterogeneous, and subsets of TRM cells that are involved in the recurrence of inflammatory skin disorders are most likely distinct in the expression of surface markers and biological behaviors. For instance, the main TRM cells involved in vitiligo are CD8+CD49a+ TRM cells that constitutively express perforin and granzyme B; however, TRM cells in psoriasis are CD8+CD49a− and predominantly generate IL-17.14 Moreover, special anatomical niches, e.g., the hair follicles on the scalp and the folds of the inframammary region, are likely other important determinants of the diversity of subsets in the skin. TRM cells are a crucial component of the adaptive immune response. To avoid the complete collapse of the immune defense system against infections, disease specificity and/or anatomical position should be considered when intervening with the biological functions of distinct subsets of TRM cells. Therefore, the identification of distinct subsets of skin TRM cells will be helpful for determining precise intervention strategies in the future.
Second, attention should be paid to the crosstalk between subsets of TRM cells from different skin compartments (the dermis and epidermis) in the development of skin inflammation. Are they cooperative or antagonistic? In addition, recent studies have suggested that γδ T cells and innate lymphocyte cells can also form long-lived resident memory-like subsets upon local inflammation or infection.166–168 Is there crosstalk between these unconventional TRM cells and the conventional αβ ones we reviewed here? The clarification of this crosstalk will contribute to our understanding of the overall immunological balance and lay the foundation for the development of therapeutics on the whole.
Third, attention should also be paid to the interactions between the skin microbiome (bacteria, fungi, viruses, archaea and skin mites) and the training of skin TRM cells. Skin TRM cells are exposed to the skin microbiome and their antigens for life. How the composition of the skin microbiome and whether certain species of the skin microbiome impact the training of skin TRM cells are not known, but much can be learned from the gastrointestinal tract.156 This may open up new avenues for topical therapeutic strategies.
Next, skin TRM cells have the prominent ability to adapt to the local environment, at least through FFA consumption and mitochondrial oxidative metabolism. We know that the majority of chronic skin inflammation is concomitant with metabolism-related disorders (e.g., diabetes mellitus). Do skin TRM cells obtain more robust survival energy under such conditions? Are there any differences between skin TRM cells in skin inflammation only and those in metabolic disorders? The answers to these questions will provide new clinical therapeutic strategies from a new perspective of cell survival energy.
Finally, further studies with animal models are needed to establish whether the manipulation of TRM cells or their molecular pathways involved in recruitment, retention, and long-term survival may help to control inflammatory disease continuation and/or recurrence. In fact, manipulating TRM cells is a challenging task because it is difficult to deplete TRM cells in animal models with antibodies against surface markers. Encouragingly, the exploration of the manipulation of molecular pathways involved in the biological behavior of TRM cells is already underway. For example, MTX plus a neutralizing IL-15 mAb has been demonstrated to manipulate CD69 expression and CD8+CD69+ TRM cells in psoriatic organ cultures, although MTX alone failed (unpublished data); S1PR1 modulators are being developed to control autoimmune and inflammatory diseases including psoriasis and inflammatory bowel disease.169,170
In conclusion, skin TRM cells have been suggested to play vital roles in the recurrence of inflammatory skin disorders. Clarifying and characterizing the mechanisms underlying the roles of skin TRM cells will provide new perspectives for controlling chronic inflammation and promising strategies for reducing the frequency and magnitude of skin inflammation recurrence.
Acknowledgements
In view of space limitation, we apologize to the colleagues whose contributions to the field could not be cited. The National Natural Science Foundation of China (no. 81573054, 81371729, 81771783), the Clinical Research and Translation Key Project of Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital (no. 2016LZ02), and the Sichuan Science and Technology Program (no. 2019JDTD0027).
Competing interests
The authors declare no competing interests.
References
- 1.Sheridan BS, Lefrancois L. Regional and mucosal memory T cells. Nat. Immunol. 2011;12:485–491. doi: 10.1038/ni.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watanabe R, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 2015;7:279ra39. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou J, et al. CD4+ T cells memorize obesity and promote weight regain. Cell Mol. Immunol. 2017;15:630–639. doi: 10.1038/cmi.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landrith TA, et al. CD103+ CD8 T cells in the toxoplasma-infected brain exhibit a tissue-resident memory transcriptional profile. Front Immunol. 2017;8:335. doi: 10.3389/fimmu.2017.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun H, Sun C, Xiao W, Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol. Immunol. 2019;16:205–215. doi: 10.1038/s41423-018-0192-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topham DJ, Reilly EC. Tissue-resident memory CD8+ T cells: from phenotype to function. Front Immunol. 2018;9:515. doi: 10.3389/fimmu.2018.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mackay LK, et al. The developmental pathway for CD103 (+) CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 8.Stolley JM, Masopust D. Tissue-resident memory T cells live off the fat of the land. Cell Res. 2017;27:847–848. doi: 10.1038/cr.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan Y, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543:252–256. doi: 10.1038/nature21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlach C, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 2015;21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang X, et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beura LK, et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T Cells. Immunity. 2018;48:327–338.e5. doi: 10.1016/j.immuni.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheuk S, et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity. 2017;46:287–300. doi: 10.1016/j.immuni.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilk MM, et al. Lung CD4 tissue-resident memory T cells mediate adaptive immunity induced by previous infection of mice with bordetella pertussis. J. Immunol. 2017;199:233–243. doi: 10.4049/jimmunol.1602051. [DOI] [PubMed] [Google Scholar]
- 16.Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science. 2014;346:93–98. doi: 10.1126/science.1257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glennie ND, Volk SW, Scott P. Skin-resident CD4+ T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PLoS Pathog. 2017;13:e1006349. doi: 10.1371/journal.ppat.1006349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park CO, et al. Staged development of long-lived T-cell receptor αβ TH17 resident memory T-cell population to Candida albicans after skin infection. J. Allergy Clin. Immunol. 2018;142:647–662. doi: 10.1016/j.jaci.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen QP, Deng TZ, Witherden DA, Goldrath AW. Origins of CD4+ circulating and tissue-resident memory T-cells. Immunology. 2019;157:3–12. doi: 10.1111/imm.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 2013;14:509–513. doi: 10.1038/ni.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark RA. Resident memory T cells in human health and disease. Sci. Transl. Med. 2015;7:269rv1. doi: 10.1126/scitranslmed.3010641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho AW, Kupper TS. T cells and the skin: from protective immunity to inflammatory skin disorders. Nat. Rev. Immunol. 2019;19:490–502. doi: 10.1038/s41577-019-0162-3. [DOI] [PubMed] [Google Scholar]
- 23.Cheuk S, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J. Immunol. 2014;192:3111–3120. doi: 10.4049/jimmunol.1302313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizukawa Y, Shiohara T. Fixed drug eruption: a prototypic disorder mediated by effector memory T cells. Curr. Allergy Asthma Rep. 2009;9:71–77. doi: 10.1007/s11882-009-0011-8. [DOI] [PubMed] [Google Scholar]
- 25.Richmond JM, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J. Invest Dermatol. 2019;139:769–778. doi: 10.1016/j.jid.2018.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaide O, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat. Med. 2015;21:647–653. doi: 10.1038/nm.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lefrançois L, Obar JJ. Once a killer, always a killer: from cytotoxic T cell to memory cell. Immunol. Rev. 2010;235:206–218. doi: 10.1111/j.0105-2896.2010.00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obar JJ, Lefrançois L. Early events governing memory CD8+ T-cell differentiation. Int Immunol. 2010;22:619–625. doi: 10.1093/intimm/dxq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willis CR, et al. Interleukin-7 receptor blockade suppresses adaptive and innate inflammatory responses in experimental colitis. J. Inflamm. (Lond.) 2012;9:39. doi: 10.1186/1476-9255-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klonowski KD, Williams KJ, Marzo AL, Lefrançois L. Cutting edge: IL-7-independent regulation of IL-7 receptor alpha expression and memory CD8 T cell development. J. Immunol. 2006;177:4247–4251. doi: 10.4049/jimmunol.177.7.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc. Natl Acad. Sci. USA. 2007;104:11730–11735. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhat J, Kabelitz D. Multilayer epigenetic analysis reveals novel transcription factor networks in CD8 T cells. Cell Mol. Immunol. 2018;15:199–202. doi: 10.1038/cmi.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henson SM, et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood. 2009;113:6619–6628. doi: 10.1182/blood-2009-01-199588. [DOI] [PubMed] [Google Scholar]
- 34.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat. Immunol. 2014;15:1104–1115. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gebhardt T, Carbone FR. Unpleasant memories: tissue-embedded T cell memory drives skin hypersensitivity. Nat. Med. 2015;21:551–552. doi: 10.1038/nm.3874. [DOI] [PubMed] [Google Scholar]
- 37.Osborn JF, et al. Central memory CD8+ T cells become CD69+ tissue-residents during viral skin infection independent of CD62L-mediated lymph node surveillance. PLoS Pathog. 2019;15:e1007633. doi: 10.1371/journal.ppat.1007633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iborra S, et al. Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1+ dendritic cells. Immunity. 2016;45:847–860. doi: 10.1016/j.immuni.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krummey SM, et al. Low-affinity memory CD8+ T cells mediate robust heterologous immunity. J. Immunol. 2016;196:2838–2846. doi: 10.4049/jimmunol.1500639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krummey SM, et al. Enhanced requirement for TNFR2 in graft rejection mediated by low-affinity memory CD8+ T cells during heterologous immunity. J. Immunol. 2016;197:2009–2015. doi: 10.4049/jimmunol.1502680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takamura S. Persistence in temporary lung niches: a survival strategy of lung-resident memory CD8+ T cells. Viral Immunol. 2017;30:438–450. doi: 10.1089/vim.2017.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maru S, Jin G, Schell TD, Lukacher AE. TCR stimulation strength is inversely associated with establishment of functional brain-resident memory CD8 T cells during persistent viral infection. PLoS Pathog. 2017;13:e1006318. doi: 10.1371/journal.ppat.1006318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshizawa A, et al. TCR-pMHC encounter differentially regulates transcriptomes of tissue-resident CD8 T cells. Eur. J. Immunol. 2018;48:128–150. doi: 10.1002/eji.201747174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012;249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarbassov DD, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell. 2006;22:159–268. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 46.Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J. Immunol. 2014;193:2067–2071. doi: 10.4049/jimmunol.1400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sowell RT, Marzo AL. Resident-memory CD8 T cells and mTOR: generation, protection, and clinical importance. Front Immunol. 2015;6:38. doi: 10.3389/fimmu.2015.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker TC, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med. 2002;195:1541–1548. doi: 10.1084/jem.20020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, et al. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity. 2011;34:541–553. doi: 10.1016/j.immuni.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J. Exp. Med. 2016;213:951–966. doi: 10.1084/jem.20151855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, Fuhlbrigge RC, Karibian K, Tian T, Kupper TS. Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity. 2006;25:511–520. doi: 10.1016/j.immuni.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 52.Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 2002;195:135–141. doi: 10.1084/jem.20011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Homey B, et al. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat. Med. 2002;8:157–165. doi: 10.1038/nm0202-157. [DOI] [PubMed] [Google Scholar]
- 54.Xia M, et al. regulates balanced maintenance and function of resident regulatory and effector T cells to promote immune homeostasis in the skin. J. Allergy Clin. Immunol. 2014;134:634–644.e10. doi: 10.1016/j.jaci.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masopust D, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010;207:553–564. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCully ML, et al. Epidermis instructs skin homing receptor expression in human T cells. Blood. 2012;120:4591–4598. doi: 10.1182/blood-2012-05-433037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma C, Mishra S, Demel EL, Liu Y, Zhang N. TGF-β controls the formation of kidney-resident T cells via promoting effector T cell extravasation. J. Immunol. 2017;198:749–756. doi: 10.4049/jimmunol.1601500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teijaro JR, et al. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein RS, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glass WG. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohmatsu H, et al. α4β7 Integrin is essential for contact hypersensitivity by regulating migration of T cells to skin. J. Allergy Clin. Immunol. 2010;126:1267–1276. doi: 10.1016/j.jaci.2010.08.048. [DOI] [PubMed] [Google Scholar]
- 62.Haddad W, et al. P-selectin and P-selectin glycoprotein ligand 1 are major determinants for Th1 cell recruitment to nonlymphoid effector sites in the intestinal lamina propria. J. Exp. Med. 2003;198:369–377. doi: 10.1084/jem.20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim K, et al. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science. 2015;349:aaa4352. doi: 10.1126/science.aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, et al. Characterization and biological significance of IL-23-induced neutrophil polarization. Cell Mol. Immunol. 2018;15:518–530. doi: 10.1038/cmi.2017.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toichi E, Tachibana T, Furukawa F. Rapid improvement of psoriasis vulgaris during drug-induced agranulocytosis. J. Am. Acad. Dermatol. 2000;43:391–395. doi: 10.1067/mjd.2000.103264. [DOI] [PubMed] [Google Scholar]
- 66.Lin AM, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 2011;187:490–500. doi: 10.4049/jimmunol.1100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chowaniec O, et al. Earliest clinical and histological changes in psoriasis. Dermatologica. 1981;163:42–51. doi: 10.1159/000250139. [DOI] [PubMed] [Google Scholar]
- 68.Camargo CM, Brotas AM, Ramos-e-Silva M, Carneiro S. Isomorphic phenomenon of Koebner: facts and controversies. Clin. Dermatol. 2013;31:741–749. doi: 10.1016/j.clindermatol.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 69.Diani M, Cozzi C, Altomare G. Heinrich Koebner and his phenomenon. JAMA Dermatol. 2016;152:919. doi: 10.1001/jamadermatol.2015.6015. [DOI] [PubMed] [Google Scholar]
- 70.Suárez-Fariñas M, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011;127:954–964. doi: 10.1016/j.jaci.2010.12.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, et al. Identification of unique proteomic signatures in allergic and non-allergic skin disease. Clin. Exp. Allergy. 2017;47:1456–1467. doi: 10.1111/cea.12979. [DOI] [PubMed] [Google Scholar]
- 72.Baker BS, Powles AV, Lambert S, Valdimarsson H, Fry L. A prospective study of the Koebner reaction and T lymphocytes in uninvolved psoriatic skin. Acta Derm. Venereol. 1988;68:430–434. [PubMed] [Google Scholar]
- 73.Sagi L, Trau H. The Koebner phenomenon. Clin. Dermatol. 2011;29:231–236. doi: 10.1016/j.clindermatol.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 74.Casey KA, et al. Antigen- independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumar BV, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017;20:2921–2934. doi: 10.1016/j.celrep.2017.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sathaliyawala T, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skon CN, et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mackay LK, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015;194:2059–2063. doi: 10.4049/jimmunol.1402256. [DOI] [PubMed] [Google Scholar]
- 79.Pérès M, Montfort A, Andrieu-Abadie N, Colacios C, Ségui B. S1P: the elixir of life for naive T cells. Cell Mol. Immunol. 2018;15:657–659. doi: 10.1038/cmi.2017.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang N, Bevan MJ. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39:687–696. doi: 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chapman TJ, Topham DJ. Identification of a unique population of tissue-memory CD4+ T cells in the airways after influenza infection that is dependent on the integrin VLA-1. J. Immunol. 2010;184:3841–3849. doi: 10.4049/jimmunol.0902281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ray SJ, et al. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity. 2004;20:167–179. doi: 10.1016/s1074-7613(04)00021-4. [DOI] [PubMed] [Google Scholar]
- 83.Kilshaw PJ. Alpha E beta 7. Mol. Pathol. 1999;52:203–207. doi: 10.1136/mp.52.4.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y, et al. Structure of natural killer cell receptor KLRG1 bound to E-cadherin reveals basis for MHC-independent missing self recognition. Immunity. 2009;31:35–46. doi: 10.1016/j.immuni.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schwartzkopff S, et al. TGF-β downregulates KLRG1 expression in mouse and human CD8(+) T cells. Eur. J. Immunol. 2015;45:2212–2217. doi: 10.1002/eji.201545634. [DOI] [PubMed] [Google Scholar]
- 86.Yu CI, et al. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-β. Immunity. 2013;38:818–830. doi: 10.1016/j.immuni.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schön MP, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J. Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- 88.Mohammed J, et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat. Immunol. 2016;17:414–421. doi: 10.1038/ni.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moed H, et al. Increased CCL27- CCR10 expression in allergic contact dermatitis: implications for local skin memory. J. Pathol. 2004;204:39–46. doi: 10.1002/path.1619. [DOI] [PubMed] [Google Scholar]
- 90.Zaid A, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl. Acad. Sci. USA. 2014;111:5307–5312. doi: 10.1073/pnas.1322292111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Hsueh-Chun, Wong Tzu-Hsuan, Wang Li-Ting, Su Hsiang-Han, Yu Hsiu-Yueh, Wu Ai-Hsuan, Lin Yu-Chun, Chen Hua-Ling, Suen Jau-Ling, Hsu Shih-Hsien, Chen Li-Chen, Zhou Yufeng, Huang Shau-Ku. Aryl hydrocarbon receptor signaling promotes ORMDL3-dependent generation of sphingosine-1-phosphate by inhibiting sphingosine-1-phosphate lyase. Cellular & Molecular Immunology. 2018;16(10):783–790. doi: 10.1038/s41423-018-0022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Colonna M. AHR: Making the Keratinocytes Thick Skinned. Immunity. 2014;40:863–864. doi: 10.1016/j.immuni.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 93.Di Meglio P, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity. 2014;40:989–1001. doi: 10.1016/j.immuni.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leavy O. Mucosal immunology: The ‘AHR diet’ for mucosal homeostasis. Nat. Rev. Immunol. 2011;11:806. doi: 10.1038/nri3115. [DOI] [PubMed] [Google Scholar]
- 95.Li Y, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–640. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 96.Hijnen D, et al. CD8+ T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J. Invest Dermatol. 2012;133:973–979. doi: 10.1038/jid.2012.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hooper LV. You AhR what you eat: linking diet and immunity. Cell. 2011;147:489–491. doi: 10.1016/j.cell.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 98.Zhang LH, Shin JH, Haggadone MD, Sunwoo JB. The aryl hydrocarbon receptor is required for the maintenance of liver-resident natural killer cells. J. Exp. Med. 2016;213:2249–2257. doi: 10.1084/jem.20151998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horras CJ, Lamb CL, King AL, Hanley JR, Mitchell KA. Consequences of TCDD treatment on intra-hepatic lymphocytes during liver regeneration. J. Immunotoxicol. 2012;9:359–367. doi: 10.3109/1547691X.2012.664577. [DOI] [PubMed] [Google Scholar]
- 100.Mackay LK, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 101.Mackay LK, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 103.Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 104.Gallais Sérézal, I. et al. A skewed pool of resident T cells triggers psoriasis-associated tissue responses in never-lesional skin from patients with psoriasis. J. Allergy Clin. Immunol.143, 1444–1454 (2019). [DOI] [PubMed]
- 105.Martini E, et al. Dynamic changes in resident and infiltrating epidermal dendritic cells in active and resolved psoriasis. J. Invest Dermatol. 2017;137:865–873. doi: 10.1016/j.jid.2016.11.033. [DOI] [PubMed] [Google Scholar]
- 106.Park SL, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018;19:183–191. doi: 10.1038/s41590-017-0027-5. [DOI] [PubMed] [Google Scholar]
- 107.Maekawa Y, et al. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat. Med. 2015;21:55–61. doi: 10.1038/nm.3758. [DOI] [PubMed] [Google Scholar]
- 108.Hombrink P, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016;17:1467–1478. doi: 10.1038/ni.3589. [DOI] [PubMed] [Google Scholar]
- 109.Wijeyesinghe S, Masopust D. Resident memory T cells are a Notch above the rest. Nat. Immunol. 2016;17:1337–1338. doi: 10.1038/ni.3617. [DOI] [PubMed] [Google Scholar]
- 110.Riou C, et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J. Exp. Med. 2007;204:79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hand TW, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl Acad. Sci. USA. 2010;107:16601–11606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology. 2010;130:418–426. doi: 10.1111/j.1365-2567.2009.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grange M, et al. Control of CD8 T cell proliferation and terminal differentiation by active STAT5 and CDKN2A/CDKN2B. Immunology. 2015;145:543–557. doi: 10.1111/imm.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quigley M, Huang X, Yang Y. Extent of stimulation controls the formation of memory CD8 T cells. J. Immunol. 2007;179:5768–5777. doi: 10.4049/jimmunol.179.9.5768. [DOI] [PubMed] [Google Scholar]
- 115.Li Y, et al. Persistent antigen and prolonged AKT-mTORC1 activation underlie memory CD8 T cell impairment in the absence of CD4 T cells. J. Immunol. 2015;195:1591–1598. doi: 10.4049/jimmunol.1500451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim EH, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J. Immunol. 2012;188:4305–4314. doi: 10.4049/jimmunol.1103568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gattinoni L, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou X, et al. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao DM, et al. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J. Immunol. 2010;184:1191–1199. doi: 10.4049/jimmunol.0901199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jeannet G, et al. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl Acad. Sci. USA. 2010;107:9777–9782. doi: 10.1073/pnas.0914127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pallett LJ, et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017;214:1567–1580. doi: 10.1084/jem.20162115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mackay LK, et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity. 2015;43:1101–1111. doi: 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 123.Hu Y, Lee YT, Kaech SM, Garvy B, Cauley LS. Smad4 promotes differentiation of effector and circulating memory CD8 T cells but is dispensable for tissue-resident memory CD8 T cells. J. Immunol. 2015;194:2407–2414. doi: 10.4049/jimmunol.1402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J. Invest Dermatol. 2009;130:362–370. doi: 10.1038/jid.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Song X, He X, Li X, Qian Y. The roles and functional mechanisms of interleukin-17 family cytokines in mucosal immunity. Cell Mol. Immunol. 2016;13:418–431. doi: 10.1038/cmi.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Giacomin PR, et al. Epithelial-intrinsic IKKα expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J. Exp. Med. 2015;212:1513–1528. doi: 10.1084/jem.20141831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013;34:174–181. doi: 10.1016/j.it.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scheper RJ, et al. Induction of immunological memory in the skin. Role local T cell Retent. Clin. Exp. Immunol. 1983;51:141–148. [PMC free article] [PubMed] [Google Scholar]
- 129.Carey W, et al. Relapse, rebound, and psoriasis adverse events: an advisory group report. J. Am. Acad. Dermatol. 2006;54:S171–S181. doi: 10.1016/j.jaad.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 130.Boyman O, et al. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J. Exp. Med. 2004;199:731–736. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gilhar A, David M, Ullmann Y, Berkutski T, Kalish RS. T-lymphocyte dependence of psoriatic pathology in human psoriatic skin grafted to SCID mice. J. Invest Dermatol. 1997;109:283–288. doi: 10.1111/1523-1747.ep12335758. [DOI] [PubMed] [Google Scholar]
- 132.Teraki Y, Shiohara T. Preferential expression of alphaEbeta7 integrin (CD103) on CD8+ T cells in the psoriatic epidermis: regulation by interleukins 4 and 12 and transforming growth factor-beta. Br. J. Dermatol. 2002;147:1118–1126. doi: 10.1046/j.1365-2133.2002.05005.x. [DOI] [PubMed] [Google Scholar]
- 133.Bhushan M, et al. Anti-E-selectin is ineffective in the treatment of psoriasis: a randomized trial. Br. J. Dermatol. 2002;146:824–831. doi: 10.1046/j.1365-2133.2002.04743.x. [DOI] [PubMed] [Google Scholar]
- 134.Gallais Sérézal I, et al. Resident T cells in resolved psoriasis steer tissue responses that stratify clinical outcome. J. Invest Dermatol. 2018;138:1754–1763. doi: 10.1016/j.jid.2018.02.030. [DOI] [PubMed] [Google Scholar]
- 135.Matos TR, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J. Clin. Invest. 2017;127:4031–4041. doi: 10.1172/JCI93396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Torres-Alvarez B, Castanedo-Cazares JP, Fuentes-Ahumada C, Moncada B. The effect of methotrexate on the expression of cell adhesion molecules and activation molecule CD69 in psoriasis. J. Eur. Acad. Dermatol Venereol. 2007;21:334–339. doi: 10.1111/j.1468-3083.2006.01916.x. [DOI] [PubMed] [Google Scholar]
- 137.Cibrian D, et al. CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat. Immunol. 2016;17:985–996. doi: 10.1038/ni.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pan Y, Kupper TS. Metabolic reprogramming and longevity of tissue-resident memory T cells. Front Immunol. 2018;9:1347. doi: 10.3389/fimmu.2018.01347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nomura I, et al. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J. Allergy Clin. Immunol. 2003;112:1195–1202. doi: 10.1016/j.jaci.2003.08.049. [DOI] [PubMed] [Google Scholar]
- 140.Gudjonsson JE, et al. Assessment of the psoriatic transcriptome in a large sample: additional regulated genes and comparisons with in vitro models. J. Invest Dermatol. 2010;130:1829–1840. doi: 10.1038/jid.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Karakawa M, et al. CCL27 is downregulated by interferon gamma via epidermal growth factor receptor in normal human epidermal keratinocytes. J. Cell Physiol. 2014;229:1935–1945. doi: 10.1002/jcp.24643. [DOI] [PubMed] [Google Scholar]
- 142.Kanda N, Koike S, Watanabe S. IL-17 suppresses TNF-alpha-induced CCL27 production through induction of COX-2 in human keratinocytes. J. Allergy Clin. Immunol. 2005;116:1144–1150. doi: 10.1016/j.jaci.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 143.Villadsen LS, et al. Resolution of psoriasis upon blockade of IL-15 biological activity in a xenograft mouse model. J. Clin. Invest. 2003;112:1571–1580. doi: 10.1172/JCI18986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Duffin KC, Krueger GG. Genetic variations in cytokines and cytokine receptors associated with psoriasis found by genome-wide association. J. Invest Dermatol. 2009;129:827–833. doi: 10.1038/jid.2008.308. [DOI] [PubMed] [Google Scholar]
- 145.Korkij W, Soltani K. Fixed drug eruption. A brief review. Arch. Dermatol. 1984;120:520–524. [PubMed] [Google Scholar]
- 146.Mizukawa Y, Shiohara T. Trauma-localized fixed drug eruption: involvement of burn scars, insect bites and venipuncture sites. Dermatology. 2002;205:159–161. doi: 10.1159/000063892. [DOI] [PubMed] [Google Scholar]
- 147.Teraki Y, Shiohara T. IFN-gamma-producing effector CD8+ T cells and IL-10-producing regulatory CD4+ T cells in fixed drug eruption. J. Allergy Clin. Immunol. 2003;112:609–615. doi: 10.1016/s0091-6749(03)01624-5. [DOI] [PubMed] [Google Scholar]
- 148.Shiohara T, Mizukawa Y, Teraki Y. Pathophysiology of fixed drug eruption: the role of skin-resident T cells. Curr. Opin. Allergy Clin. Immunol. 2002;2:317–323. doi: 10.1097/00130832-200208000-00005. [DOI] [PubMed] [Google Scholar]
- 149.Mizukawa Y, et al. Direct evidence for interferon-gamma production by effector-memory-type intraepidermal T cells residing at an effector site of immunopathology in fixed drug eruption. Am. J. Pathol. 2002;161:1337–1347. doi: 10.1016/s0002-9440(10)64410-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br. J. Dermatol. 2008;158:1230–1238. doi: 10.1111/j.1365-2133.2008.08516.x. [DOI] [PubMed] [Google Scholar]
- 151.Iriki H, et al. Toxic epidermal necrolysis in the absence of circulating T cells: a possible role for resident memory T cells. J. Am. Acad. Dermatol. 2014;71:e214–e216. doi: 10.1016/j.jaad.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 152.Willemsen M, Linkutė R, Luiten RM, Matos TR. Skin-resident memory T cells as a potential new therapeutic target in vitiligo and melanoma. Pigment Cell Melanoma Res. 2019;32:612–622. doi: 10.1111/pcmr.12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Boniface K, et al. Vitiligo Skin Is Imprinted with Resident Memory CD8 T Cells Expressing CXCR3. J. Invest Dermatol. 2018;138:355–364. doi: 10.1016/j.jid.2017.08.038. [DOI] [PubMed] [Google Scholar]
- 154.Richmond Jillian M., Strassner James P., Zapata Lucio, Garg Madhuri, Riding Rebecca L., Refat Maggi A., Fan Xueli, Azzolino Vincent, Tovar-Garza Andrea, Tsurushita Naoya, Pandya Amit G., Tso J. Yun, Harris John E. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Science Translational Medicine. 2018;10(450):eaam7710. doi: 10.1126/scitranslmed.aam7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387:1109–1122. doi: 10.1016/S0140-6736(15)00149-X. [DOI] [PubMed] [Google Scholar]
- 156.Patra V, Laoubi L, Nicolas JF, Vocanson M, Wolf P. Perspective on the interplay of ultraviolet-radiation, skin microbiome and skin resident memory TCRαβ+ cells. Front Med (Lausanne) 2018;5:166. doi: 10.3389/fmed.2018.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Brunner PM, et al. Nonlesional atopic dermatitis skin shares similar T-cell clones with lesional tissues. Allergy. 2017;72:2017–2025. doi: 10.1111/all.13223. [DOI] [PubMed] [Google Scholar]
- 158.Kim S, et al. Multicytokine-producing tissue resident memory (TRM) cells in atopic dermatitis patient. J. Invest Dermatol. 2016;136:S9. [Google Scholar]
- 159.Kim S, Kim J, Park C, Kupper T, Lee K. Distinct transcriptome signature of skin-resident memory T cells and migratory memory T cells in atopic dermatitis. J. Invest Dermatol. 2018;138:S4. [Google Scholar]
- 160.Mowad CM, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J. Am. Acad. Dermatol. 2016;74:1029–1040. doi: 10.1016/j.jaad.2015.02.1139. [DOI] [PubMed] [Google Scholar]
- 161.Torchia, D., Capretti, C., Pizzo, B. & Francalanci, S. Patch test triggering recurrence of distant dermatitis: the flare-up phenomenon. CMAJ. 179, 341 (2008). [DOI] [PMC free article] [PubMed]
- 162.Gamradt P, et al. Inhibitory checkpoint receptors control CD8+ resident memory T cells to prevent skin allergy. J. Allergy Clin. Immunol. 2019;143:2147–2157. doi: 10.1016/j.jaci.2018.11.048. [DOI] [PubMed] [Google Scholar]
- 163.Schmidt JD, et al. Rapid allergen-induced interleukin-17 and interferon-γ secretion by skin-resident memory CD8+ T cells. Contact Dermat. 2017;76:218–227. doi: 10.1111/cod.12715. [DOI] [PubMed] [Google Scholar]
- 164.Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. Prognosis, management, and future directions. J. Am. Acad. Dermatol. 2014;70:223.e1–17. doi: 10.1016/j.jaad.2013.08.033. [DOI] [PubMed] [Google Scholar]
- 165.Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116:767–771. doi: 10.1182/blood-2009-11-251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Khairallah C, Chu TH, Sheridan BS. Tissue adaptations of memory and tissue-resident gamma delta T cells. Front Immunol. 2018;9:2636. doi: 10.3389/fimmu.2018.02636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hunter S, et al. Human liver infiltrating γδ T cells are composed of clonally expanded circulating and tissue-resident populations. J. Hepatol. 2018;69:654–665. doi: 10.1016/j.jhep.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol. Rev. 2018;283:54–76. doi: 10.1111/imr.12650. [DOI] [PubMed] [Google Scholar]
- 169.D’Ambrosio D, Freedman MS, Prinz J. Ponesimod, a selective S1P1 receptor modulator: a potential treatment for multiple sclerosis and other immune-mediated diseases. Ther. Adv. Chronic Dis. 2016;7:18–33. doi: 10.1177/2040622315617354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Blankenbach KV, Schwalm S, Josef P, Dagmar MZH. Sphingosine-1-phosphate receptor-2 antagonists: therapeutic potential and potential risks. Front Pharm. 2016;7:167. doi: 10.3389/fphar.2016.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]