

Graphical abstract
Keywords: Pathogen functional mimicry, Pathogen-host protein interaction networks, Human protein-protein interaction networks, Signaling pathways, Transfer learning, GO semantic similarity
Abstract
Pathogen-host protein interactions are fundamental for pathogens to manipulate host signaling pathways and subvert host immune defense. For most pathogens, very few or no experimental studies have been conducted to investigate their signaling cross-talks with host. In this study, we propose a computational framework to validate the biological assumption that human protein–protein interaction (PPI) networks alone are sufficient to infer pathogen-host PPIs via pathogen functional mimicry. Pathogen functional mimicry assumes that a pathogen functionally mimics and substitutes host counterpart proteins in order for the pathogen to get involved in or hijack the host cellular processes. Through pathogen functional mimicry defined via gene ontology (GO) semantic similarity, we first use the known human PPIs as templates to infer pathogen-host PPIs, and the PPIs are further used as training data to build an l2-regularized logistic regression model for novel pathogen-host PPI prediction. Independent tests on the experimental data from human immunodeficiency virus and Francisella tularensis validate the effectiveness of the proposed pathogen functional mimicry technique. Performance comparisons also show that the proposed technique y excels the existing pathogen sequence mimicry approaches and transfer learning methods. The proposed framework provides a new avenue to study the experimentally less-studied pathogens in the worst scenarios that very few or no experimental pathogen-host PPIs are available. As two case studies, we apply the proposed framework to Salmonella typhimurium and Human respiratory syncytial virus to reconstruct the pathogen-host PPI networks and further investigate the interference of these two pathogens with human immune signaling and transcription regulatory system.
1. Introduction
Pathogens are the major etiological agents of human infectious diseases and in some cases are highly associated with human tumorigenesis. For instances, oncogenic virus hepatitis B virus (HBV) is a major cause of hepatocellular carcinoma [1], and oncogenic bacterium Helicobacter pylori results in persistent chronic inflammation and increases the risk of gastric cancer [2]. Protein interactions between pathogen and host are complex and dynamic biological processes that spatiotemporally regulate pathogen infection and host response. Genome-scale reconstruction of pathogen-host protein interaction networks is significant to understand microbe- and virus-associated pathogenesis and to develop therapeutic drugs [3], [4]. At present, pathogen-host PPI networks are built mainly via experimental techniques, e.g. immunoaffinity purification coupled to mass spectrometry (IP-MS), bacterial two-hybrid, yeast two-hybrid (Y2H), liquid chromatography mass spectrometry (LC-MS) and cross-linking coupled with mass spectrometry [3]. To rapidly obtain the global landscape of pathogen-host PPI networks, we need resort to computational modeling.
Recent years have witnessed much progress in computational reconstruction of pathogen-host PPI networks [5], [6], [7], [8]. From data point of view, existing methods are divided into two categories, namely direct and indirect methods. Direct methods [9], [10], [11], [12], [13], [14], [15], [16], [17] are directly built on experimentally verified pathogen-host PPI data of the pathogens themselves, while indirect methods [18], [19], [20], [21], [22] are built on experimental data or interlogs from other species. Direct methods are potentially more credible than indirect methods, but their performance is heavily restricted by the scale of available experimental data. For instance, HIV-1 [11], [12], [13] has accumulated much more experimental data than EBV [16] and HCV [17]. For the vast majority of pathogens, very few or no experimental data are available to computational modeling. In such cases, direct methods are not applicable and indirect methods are often used as alternative solutions [18], [19], [20], [21], [22]. Existing indirect methods are further divided into two subcategories, namely interlog methods [18], [19], [20], [21] and transfer learning methods [22]. Interlog methods are based on the biological assumption that two interacting proteins co-evolve across species to retain interactions between their orthologs. Zhou et al. [18], [19] infer interlogs between M. Tuberculosis H37Rv and Homo sapiens via the known PPIs of other eukaryotic species. Schleker et al. [20] infer protein interactions between Salmonella and human via the experimental PPIs between Salmonella and Arabidopsis. These methods [16], [17], [18], [19], [20] do not build predictive models but infer interlogs via sequence similarities. Mei et al. [21] restrict the interlog inference within the co-evolving pathogen and host, based on which to train a noise-resistance supervised learning model for novel interaction prediction. Comparatively, transfer learning methods use experimental pathogen-host PPIs as training data, but differently to direct methods, the experimental data come from other pathogens. For instance, Kshirsagar et al. [22] use the pathogen-host PPIs from source species as training data to predict pathogen-host PPIs of the target species, e.g. source Salmonella-human PPIs for target Salmonella-mouse PPIs and source Francisella-human PPIs for target Salmonella-human PPIs. However, a potentially large genome gap between species (e.g. Francisella tularensis versus Salmonella typhimurium) may yield a less credible transfer learning model.
Pathogen mimicry of host proteins is a basic strategy for pathogens to subvert host immune pathways and to manipulate host cellular processes [23], [24], [25], [26], [27]. As reviewed in [23], pathogens mimic host proteins at the levels of sequence, structure, motif and interface. Sequence mimicry yields pathogen orthologous proteins mainly through long-term evolution via horizontal gene transfer (HGT). Structure mimicry enables pathogens to take over the host counterparts wherein the pathogen and host proteins do not demonstrate sequence homology. As an economic strategy, interface mimicry only bears local resemblance through mimicry of a surface recognition motif that does not require overall sequence or global structure conservation. Doxey et al. [25] predict virulence-associated mimicries of pathogen via sequence similarity. Guven-Maiorov et al. [26] predict protein interactions between Helicobacter pylori and Homo sapiens via interface mimicry. The existing interlog methods [18], [19], [20], [21] are actually based on the biological evidence of sequence mimicry. However, rapid evolution of pathogen and long co-evolution with host potentially makes sequence mimicry only capture a small percentage of pathogen-host PPIs. Structure or interface mimicry promises to predict more credible pathogen-host PPIs, but is more restrictive in applications because the spatiotemporally dynamic structure or interface information is not easily available. Furthermore, regardless of sequence, structure or interface mimicry, most pathogen mimicries bear only a faint resemblance to the host factors [27]. For this reason, it is necessary to develop a more general strategy of pathogen mimicry for computational modeling to enlarge the coverage of pathogen-host PPIs.
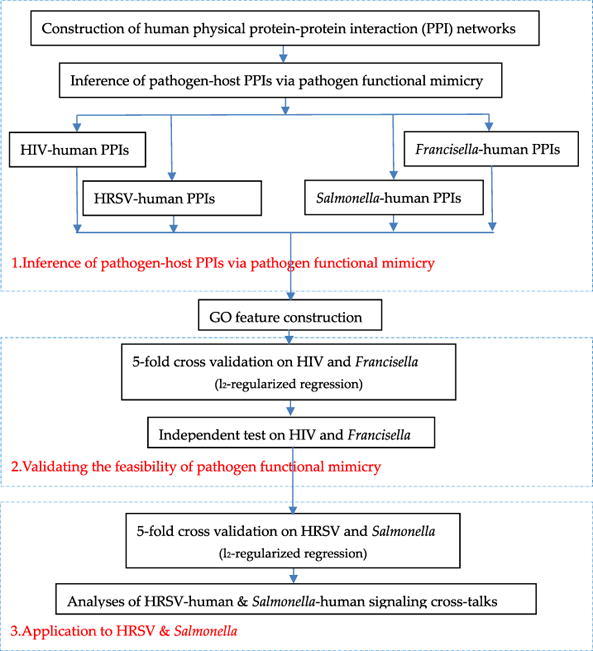
In this study, we propose a computational framework to unravel pathogen-host signaling cross-talks via pathogen functional mimicry and human protein–protein interaction networks. In this framework, a pathogen functionally mimics host counterparts to hijack the corresponding human PPIs into pathogen-host PPIs. The functional mimicry is measured via gene ontology (GO) semantic similarity between pathogen and host proteins. The pathogen-host PPIs inferred via pathogen functional mimicry are used as training data to train an l2-regularized logistic regression model for novel pathogen-host PPI prediction. We use the experimental pathogen-host PPIs of human immunodeficiency virus and Francisella tularensis to validate the effectiveness of pathogen functional mimicry. As two case studies, we apply the proposed framework to Salmonella typhimurium and human respiratory syncytial virus to investigate how the two pathogens interfere with human immune signaling and transcription regulatory system.
2. Methods
2.1. Data preparation
2.1.1. Human physical protein–protein interaction networks
In this study, we assume that human protein–protein interaction (PPI) networks alone are sufficient to infer interactions between pathogen and human host proteins. Behind this assumption is the biological evidence that a pathogen mimics its host counterparts to hijack host PPIs and to manipulate host cellular signaling networks [25], [27]. As reviewed in [27], pathogen and host co-evolve to mutually counteract each other, i.e. a pathogen evolves and mimics host counterparts to subvert host cellular processes, while host counterparts being mimicked might adapt themselves or sometimes adopt reverse mimicry to disfavour the mimicry. In this study, we focus on pathogen mimicry that perturbs host functions.
To date, there are several major databases that have curated a large number of experimentally verified PPIs for the well-studied species Homo sapiens, e.g. HPRD [28], BioGrid [29], IntAct [30], HitPredict [31], etc. From these databases [28], [29], [30], [31], we totally obtain 56,104 physical PPIs, which are associated with 10,839 well-studied human proteins. Here a well-studied gene or gene product is defined as the one that has been annotated with at least one specific GO term of molecular functions or biological processes. The generic GO terms GO:0005575, GO:0008150 and GO:0003674 correspond to the root nodes of cellular component, biological process and molecular function in the GO directed acyclic graph (DAG), respectively. These GO terms provide no specific useful information and thus are removed. According to the criteria, we totally obtain 20,081 well-studied human genes that correspond to 60,126 gene products/proteins from human genome space. All the host counterparts that pathogen mimics are chosen from these well-studied genes and proteins, and non-interacting pathogen-host protein pairs are also sampled from these proteins to construct negative training or independent test data. The reasons that we focus on well-studied human genes are due to the three concerns. First, the proposed pathogen functional mimicry is defined via GO semantic similarity; Second, the proposed framework and its predictions could be biologically well-interpreted; and lastly, since all protein pairs are represented by GO feature vector, well-studied genes do not yield null vectors.
2.1.2. Experimental pathogen-host PPIs as independent test data
In consideration of data size, we choose the pathogen human immunodeficiency virus (HIV) and Francisella tularensis (F. tularensis) to validate the feasibility and effectiveness of inferring pathogen-host PPIs from human PPI networks via pathogen functional mimicry. VirHostNet 2.0 has curated a large number of pathogen-host PPIs from experiments [32], from which we obtain 5018 PPIs between human immunodeficiency virus-1/2 (HIV-1/2) and Homo sapiens. In [33], 1383 PPIs between Francisella tularensis and Homo sapiens are derived via high-throughput yeast two-hybrid (Y2H) assay. Although Y2H technique yields a certain level of false interactions, we use the F. tularensis-human PPIs [33] as independent test data because there is no other source of experimental data available. According to the criteria of well-studied genes and proteins, we totally obtain 3188 PPIs between HIV-1/2 and human from VirHostNet 2.0 [33]. The PPIs are associated with 6 HIV proteins and 2061 human target proteins. From the study [33], we obtain 1382 experimental PPIs between F. tularensis and human, which are associated with 349F. tularensis proteins and 996 human target proteins. These data are used as the positive independent test data. To estimate the risk of model bias, we randomly sample HIV-human and F. tularensis-human protein pairs as the negative independent test data, which is equal in size to and disjoint with the positive independent test data.
As two case studies, we choose Salmonella typhimurium (S. typhimurium) and human respiratory syncytial virus (HRSV) to investigate how they interfere with human signaling and transcriptional activities. Both pathogens are experimentally less-studied in terms of the available pathogen-host PPIs. Based on the criteria of well-studied genes and proteins, we obtain 62 S. typhimurium-human PPIs from [34] involving 25 S. typhimurium proteins and 51 human proteins, and obtain 29 HRSV-human PPIs from [35] involving 4 HRSV proteins and 27 human proteins. The two datasets are used as the positive independent test data and meanwhile are used to co-determine model hyperparameters (e.g. the regularizer C, see the section of “Supervised learning via l2-regularized logistic regression”) with cross validation. Exhaustive hyperparameter tuning via cross validation is prone to lead to model overtraining and performance overestimation. If there is other source of experimental data, independent test is recommended to be used to co-determine the hyperparameters with cross validation. As such, a balance could be achieved between learning and generalization ability. In this study, we use the pathogen-host PPIs inferred via pathogen functional mimicry as the training data and the available experimental data are used as the independent test data. The hyperparameters are co-determined by cross validation and independent test. For the pathogens that have no experimental pathogen-host PPI data, cross validation is the only approach to determine the hyperparameters.
Based on the criteria of well-studied genes and proteins, we totally obtain {490; 2123; 47; 1828} well-studied proteins for HIV, F. tularensis, HRSV and S. typhimurium respectively, from which the negative independent test data are sampled.
2.2. Construction of training data via pathogen mimicry
Different from the study [21] that infers pathogen-host PPIs from the pathogen (e.g. Mycobacterium tuberculosis) PPI networks via pathogen sequence mimicry, this study infers pathogen-host PPIs from human PPI networks via pathogen functional mimicry. Let denote human physical PPI networks, where denotes the set of vertexes representing human proteins and denotes the set of edges representing pairs of interacting proteins. Given a pathogen protein and a human protein , p is assumed to functionally mimic h, denoted as , if the following criteria is satisfied.
| (1) |
where , and denote protein functional similarity scores in terms of biological processes (BP), molecular functions (MF) and cellular components (CC), respectively. The thresholds and are used to determine the number of pathogen-host PPIs inferred via pathogen functional mimicry. The functional similarity scores between proteins (, and ) are calculated via GO semantic similarity scores between the proteins’ GO terms.
Assume that the sets of GO terms for pathogen protein p and human protein h are and , respectively. Let subscript i and j denote the element of GO term set, protein functional similarity is defined as follows.
| (2) |
At present, several methods have been proposed to combine semantic similarities of GO terms into gene semantic similarity, e.g. maximal GO semantic similarities (MAX), averaging GO semantic similarities (AVG), cosine similarity of GO term vectors and Jaccard index between GO term sets [36]. Which of the semantic similarities to choose depends on particular applications. In this study, we choose the MAX strategy to define gene semantic similarity because we give priority to the cases that two genes are involved in identical cellular processes. Because of incompleteness and skewed distribution of annotations between genes, the other methods, i.e. AVG, cosine similarity and Jaccard index, will decrease gene similarity scores even though two genes are annotated with identical GO terms of biological processes. It is noted that the MAX strategy defines gene semantic similarity by the maximal GO semantic similarities and does not require vector representation of GO terms. Now we describe how to calculate the semantic similarity between two GO terms.
For simplicity, the two GO terms of in equation (2) are denoted using A and B, then denotes the semantic similarity between GO terms A and B. If p and h possess some common specific GO terms of cellular components except the generic root GO term GO:0005575, they are assumed to be subcellularly co-localized. The semantic similarity between biological processes and molecular functions GO terms, exclusive of GO:0008150 and GO:0003674, is calculated according to [37], [38]. In [36], the semantic value of a GO term is defined as the aggregate contribution (measured via S-value) of itself and its ancestor GO terms. Given a GO term A and its , where denotes the GO term set that includes A and its ancestor GO terms in GO DAG, and denotes the set of edges. The S-value of any GO term is defined as follows.
| (3) |
where denotes the weight of the edge linking term t to its child term , assuming 0.8 and 0.6 for the “is-a” and “part-of” relations, respectively. The semantic value of GO term A is defined as follows.
| (4) |
Based on Formula (3), (4), the semantic similarity between GO term A and B is defined as follows.
| (5) |
Formula (5) shows that the semantic similarity between two GO terms is determined by their locations in DAG and the semantic relations with their ancestor terms. Substituting Formula (5) into Formula (2), we can obtain protein functional similarity and . Based on pathogen functional mimicry as defined by Formula (2), (3), (4), (5), the pathogen-host PPI networks are derived as follows.
| (6) |
where the superscript denotes pathogen-host associations, denotes well-studied pathogen proteins, and denote well-studied host proteins. Formula (6) indicates that for any human PPI , if pathogen protein functionally mimics one protein, e.g. , i.e., then interacts with its partner .
To reconstruct genome-scale pathogen-host PPI networks, we further use the networks to build a predictive model. To reduce computational complexity, we only sample a specific number of pathogen-host PPIs from the networks as the positive training data () with the constraint . From the pairs of well-studied pathogen and host proteins, we randomly sample the same number of negative training data that are disjoint with the positive training data.
2.3. GO feature construction
Recent studied have shown that gene ontology (GO) is one of the most discriminative features to predict protein-protein interactions [39], [40]. However, GO terms are highly unevenly distributed. For the less-studied or novel genes, sparsity of GO terms will become more serious and in the worst case there are no annotations available for the gene. To tackle this problem, Maetschke et al. [40] aggregate ancestor GO terms into the feature representation. This method enriches the feature information but meanwhile increases the correlations between features. In this framework, we do not incorporate the semantic correlation of ancestor terms in GO DAG, but use the GO terms of homologs to enrich the feature information of the concerned gene. Every pair of proteins is depicted with two instances, namely target instance and homolog instance. The target instance represents the GO terms of the gene/protein concerned, and the homolog instance represents the GO terms of the homologs. Homologs are obtained from SwissProt [41] via PSI-BLAST [42] against all species. To obtain more homologs, we use default E-value = 10. We explicitly consider counteracting the noise introduced from homolog knowledge transfer (see the section of “Supervised learning via l2-regularized logistic regression”). GO terms are retrieved from GOA [43].
For each protein i in the training set , we obtain the homolog set of GO terms and the target set of GO terms . Accordingly, the whole set of GO terms of the training set is defined as follows.
| (7) |
For each protein pair, the feature vectors for the target and homolog instances are formally defined as follows.
| (8) |
For GO term , and denote the feature vector component g of target instance and homolog instance , respectively. The GO termsare discarded. Formula (8) indicates that the component corresponding to GO term g in both feature vectors ( and ) is set as 2, if both the proteins are annotated with the common GO term g; the value is set as 0, if neither protein is annotated with the GO term ; otherwise, the value is set as 1. If any protein is not annotated with GO terms as defined in S and no homologs are found for it or its homologs are also not annotated with GO terms within S, all the protein pairs associated with the protein will be discarded because of null vector representation.
2.4. Supervised learning via l2-regularized logistic regression
Noise resistance and computational complexity are two critical factors for us to choose a proper base learner. In this framework, pathogen functional mimicry and homolog knowledge transfer potentially introduce a certain level of noise. Pathogen functional mimicry may yield functionally associated host counterparts that pathogen does not mimic and hijack. Homologs potentially develop novel molecular functions and thus homolog knowledge transfer may introduce noise into the homolog instances. Meanwhile, the homolog instances double the size of the sampled positive training data (), which initially are very large. In machine learning field, regularization technique could well resist noise and outlier, and regression method could fast fit large training data in a linear time. In this study, we adopt l2-regularized logistic regression [44] that has been implemented in the toolbox LIBLINEAR [45] as the base learner.
Given training data x and labels y that consist of a set of instance-label pairs , the decision function of logistic regression is defined as . L2-regularized logistic regression derives weight vector via solving the prime optimization problem.
| (9) |
where denotes the penalty parameter/regularizer. The second term penalizes potential noise/outliers. The prime optimization problem is solved via its dual form.
| (10) |
where denotes Lagrangian operator and .
For each protein pair , the decision function yields the outputs and for the target instance and the homolog instance, respectively. The two outputs are then combined into one final decision.
| (11) |
where denotes absolute value. The final label for protein pair is defined as follows.
| (12) |
where is used to filter out weak positive predictions and denotes undetermined predictions.
2.5. Experimental setting and model evaluation
Three experimental settings namely combined-instance, homolog-instance and target-instance are designed to validate that homolog knowledge transfer via homolog instance is effective to tackle GO sparsity. The target-instance setting yields the baseline performance. If the homolog-instance setting achieves equivalent or better performance, homolog knowledge transfer is validated to be effective. To simplify the parameter tuning, the regularizer C is chosen from the set , where I denotes the integer set. The threshold as defined in Formula (12) is set .
Five performance metrics are used to evaluate the model performance including Receiver Operating Characteristic AUC (ROC-AUC), sensitivity (SE), precision (PR), Matthews correlation coefficient (MCC), Accuracy and F1 score. ROC-AUC is calculated from the decision values as defined in Formula (11). The other metrics are calculated from a confusion matrix M, in which each element records the counts that class i are classified to class j. From M, we define several intermediate variables as Formula (13), based on which label-specific PRl, SEl and MCCl for each label are further defined in Formula (14). The overall accuracy and MCC are defined by Formula (15).
| (13) |
| (14) |
| (15) |
where L denotes the number of labels and assumes 2 in this study. F1 score is defined as follows.
| (16) |
3. Results
3.1. Validating the effectiveness of inferring pathogen-host PPIs from human PPI networks via pathogen mimicry
We choose the virus human immunodeficiency virus (HIV) and the bacterium Francisella tularensis (F. tularensis) to validate the effectiveness of inferring pathogen-host PPIs from human PPI networks via pathogen functional mimicry. The two pathogens are well studied in terms of pathogen-host protein interactions. The training data are constructed as described in Formula (1), (2), (3), (4), (5), (6) and the independent test data come from experimental data.
3.1.1. Validation against human immunodeficiency virus
The threshold and as defined in Formula (1) are used to achieve a trade-off between data size and data quality. A higher threshold increases the data quality but decreases the scale of the pathogen-host PPI networks as defined by Formula (6), and vice versa a lower threshold decreases the data quality but increases the scale of networks. The number of human counterparts that pathogen mimics directly determines the scale of the pathogen-host PPI networks defined by Formula (6), so that the two thresholds vary with pathogens. How large the networks scale should be is hard to determine and it is hard to design an objective function to optimize the two thresholds, because it is unknown how many pathogen-host PPIs actually exist. We empirically determine these two thresholds so that the pathogen-host PPI networks defined by Formula (6) are not too large (e.g. exceeding 55,000 PPIs) or too small (e.g. fewer than 1000 PPIs) and the computational cost is acceptable. For human immunodeficiency virus, the thresholds are set as and we totally obtain 50,060 positive and 50,060 negative training data (see Supplementary file S1–S2). The positive training data contain 35 HIV proteins and 2757 human proteins, while the negative training data contain 490 HIV proteins and 34,159 human proteins. The ROC curves of 5-fold cross validation on the training data are illustrated in Fig. 1(A). In the three experimental settings, the ROC curves nearly coincide and validate the effectiveness of homolog knowledge transfer via homolog instances. The ROC-AUC scores show that the binary learner of l2-regularized logistic regression well separates the pathogen-host PPIs inferred via pathogen functional mimicry from the randomly sampled pathogen-host protein pairs. The other metrics provided in Table 1, e.g. PR, SE and MCC, further show that the proposed framework is less biased on the training data. It is noted that the model performance varies widely and heavily depends on the regularizer hyperparameter C as defined in Formula (9), so that no error deviation or error bars are provided. In addition, pathogen functional mimicry yields different training data with varying thresholds () and is time-consuming for multiple-round model evaluation. If a training set is fixed, multiple rounds of 5-fold cross validation show little variation. In the case that several factors co-determine the model stability, we report the optimum performance only.
Fig. 1.

ROC curves for 5-fold cross validation performance on human immunodeficiency virus and Francisella tularensis.
Table 1.
Performance estimation of 5-fold cross validation and independent test on HIV and F. tularensis.
| HIV | Size | Combined-instance |
Homolog-instance |
Target-instance |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PR | SE | MCC | PR | SE | MCC | PR | SE | MCC | ||
| Positive | 50,060 | 0.935 | 0.9452 | 0.8856 | 0.9353 | 0.9452 | 0.8854 | 0.9352 | 0.9454 | 0.8812 |
| Negative | 50,060 | 0.9441 | 0.9336 | 0.8854 | 0.9436 | 0.9333 | 0.8851 | 0.9387 | 0.9273 | 0.8797 |
| [Acc; MCC] | [93.95%; 0.8854] | [93.93%; 0.8852] | [93.68%; 0.8808] | |||||||
| [ROC-AUC] | [0.9819] | [0.9818] | [0.9805] | |||||||
| F1 Score | 0.9401 | 0.9402 | 0.9403 | |||||||
|
F. tularensis |
size | Combined-instance |
Homolog-instance |
Target-instance |
||||||
| PR | SE | MCC | PR | SE | MCC | PR | SE | MCC | ||
| Positive | 41,796 | 0.7083 | 0.9683 | 0.684 | 0.708 | 0.972 | 0.687 | 0.7323 | 0.9683 | 0.6972 |
| Negative | 41,796 | 0.9499 | 0.6012 | 0.6493 | 0.9558 | 0.6013 | 0.6531 | 0.9459 | 0.6103 | 0.6587 |
| [Acc; MCC] | [78.47%; 0.6343] | [78.61%; 0.6363] | [79.79%; 0.6540] | |||||||
| [ROC-AUC] | [0.9510] | [0.9541] | [0.9504] | |||||||
| F1 Score | 0.8181 | 0.8193 | 0.834 | |||||||
| Independent test | HIV |
F. tularensis |
||||||||
| (Recognition rate) | Positive | Negative | Positive | Negative | ||||||
| 73.09% | 91.67% | 66.47% | 60.78% | |||||||
However, the performance of 5-fold cross validation is achieved on the training data generated via pathogen functional mimicry instead of experimental data. We have to validate the learned model against experimental pathogen-host PPI data. The performance of independent test on 3188 experimentally verified HIV-human PPIs and 3188 randomly sampled negative independent test data is illustrated in Fig. 1(B) and the details are provided in Table 1. The proposed framework correctly recognizes 73.09% of the experimentally verified HIV-human PPIs and predicts 91.67% of the randomly sampled HIV-human protein pairs as negative. The results show that the model encouragingly well generalizes to unseen experimental data even though it learns from data inferred via pathogen functional mimicry.. The performance of independent test on experimental HIV-human PPIs validates the feasibility of inferring pathogen-host PPIs from human PPI networks via pathogen functional mimicry.
3.1.2. Validation against Francisella tularensis
As a bacterial case, we choose the bacterium Francisella tularensis to test whether the proposed framework is applicable to bacterial pathogens. The thresholds as defined in Formula (1) are also set as and we totally obtain 41,796 positive and 41,796 negative training data (see Supplementary file S3–S4). The positive training data contain 125 F. tularensis proteins and 4025 human proteins, while the negative training data contain 2123 F. tularensis proteins and 30,556 human proteins. Similarly, the ROC curves in the three experimental settings as illustrated in Fig. 1(C) also nearly coincide and validate the effectiveness of homolog knowledge transfer on Francisella tularensis. The ROC-AUC scores are also fairly encouraging. However, the metrics such as PR, SE and MCC show a large bias towards the positive class, e.g. SE 0.9683 positive class versus 0.6012 negative class for the combined-instance setting (see Table 1). These results indicate that the proposed framework potentially take a certain risk of false positive predictions. Nevertheless, the learned model still correctly recognizes 66.47% of the 1382 experimentally verified F. tularensis-human PPIs. Meanwhile, the learned model recognizes 60.78% of the negative independent test data (see Table 1), indicating that bias exists on Francisella tularensis but is still under control. The risk of false positive predictions could be reduced by increasing the thresholdas defined in Formula (12). Here we assume .
3.2. Applications to human respiratory syncytial virus and Salmonella typhimurium
As two case studies, we apply the proposed framework to human respiratory syncytial virus and Salmonella typhimurium. In this section, we study the model performance on the two pathogens. In the next section, we further investigate their interference with human signaling and transcriptional activities.
3.2.1. Performance on human respiratory syncytial virus
Comparatively, the protein functional similarities between HRSV and human proteins as defined by Formula (1), (2), (3), (4), (5) are much lower. To obtain a sufficient number of pathogen-host PPIs, we choose lower thresholds . As a result, we only obtain 1310 HRSV-human PPIs as the positive training data. The positive training data contain 3 HRSV proteins and 1036 human proteins, while the negative training data contain 47 HRSV proteins and 1301 human proteins. The coincident ROC curves in the three experimental settings as illustrated in Fig. 2(A) reaffirm the effectiveness of homolog knowledge transfer. The other metrics are provided in Table 2. It is evident that the proposed framework is less biased on human respiratory syncytial virus, e.g. SE 0.9908 versus 0.9473 in the combined-instance setting. These results, along with the results shown in Fig. 2(B), show that the proposed framework achieves sound and unbiased performance of cross validation and independent test.
Fig. 2.

ROC curves for 5-fold cross validation performance on human respiratory syncytial virus and Salmonella typhimurium.
Table 2.
Performance estimation of 5-fold cross validation and independent test on HRSV and S. typhimurium.
| HRSV | Size | Combined-instance |
Homolog-instance |
Target-instance |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PR | SE | MCC | PR | SE | MCC | PR | SE | MCC | ||
| Positive | 1,310 | 0.9495 | 0.9908 | 0.9409 | 0.9495 | 0.9908 | 0.9408 | 0.9516 | 0.9908 | 0.9403 |
| Negative | 1,310 | 0.9904 | 0.9473 | 0.9407 | 0.9904 | 0.9472 | 0.9407 | 0.9895 | 0.9447 | 0.9399 |
| [Acc; MCC] | [96.91%; 0.9400] | [96.90%; 0.9399] | [96.88%; 0.9395] | |||||||
| [ROC-AUC] | [0.9952] | [0.9952] | [0.9948] | |||||||
| F1 Score | 0.9697 | 0.9697 | 0.9708 | |||||||
|
S. typhimurium |
Size | Combined-instance |
Homolog-instance |
Target-instance |
||||||
| PR | SE | MCC | PR | SE | MCC | PR | SE | MCC | ||
| Positive | 50,000 | 0.8428 | 0.943 | 0.796 | 0.8433 | 0.9431 | 0.796 | 0.8578 | 0.9434 | 0.8032 |
| Negative | 50,000 | 0.9353 | 0.824 | 0.7914 | 0.935 | 0.8236 | 0.7912 | 0.9301 | 0.828 | 0.796 |
| [Acc; MCC] | [88.36%; 0.7891] | [88.36%; 0.7891] | [88.84%; 0.7972] | |||||||
| [ROC-AUC] | [0.9545] | [0.9545] | [0.9543] | |||||||
| F1 Score | 0.8901 | 0.8904 | 0.8986 | |||||||
| Independent test (Recognition rate) | HRSV |
S. typhimurium |
||||||||
| Positive |
Negative |
Positive |
Negative |
|||||||
| 79.31% | 96.55% | 75.81% | ||||||||
As shown in Table 2, the proposed framework correctly recognizes 79.31% of the experimentally verified 29 HRSV-human PPIs and 96.55% of negative independent test data. These results show that the proposed framework is applicable to human respiratory syncytial virus, though the protein functional similarities between HRSV and human proteins are relatively low.
3.2.2. Performance on Salmonella typhimurium
Although the thresholds set as high as, the inferred pathogen-host PPIs as defined by Formula (6) still are very large. To reduce computational complexity, we randomly sample 50,000 Salmonella-human PPIs as the positive training data. The positive training data contain 882 Salmonella proteins and 6319 human proteins, while the negative training data contain 1828 Salmonella proteins and 34,280 human proteins. The ROC curves of 5-fold cross validation are illustrated in Fig. 2(C) and the other performance metrics are provided in Table 2. The results still are fairly encouraging, except for a little bias towards the positive class, e.g. SE 0.9430 versus 0.8240 in the combined-instance setting. Independent test shows that the proposed framework correctly recognizes 75.81% of the 62 experimentally verified Salmonella-human PPIs and 66.13% of the negative independent test data.
The computational results also show that the proposed framework achieves much better performance and less bias on viruses (HIV and HRSV) than on bacteria (F. tularensis and S. typhimurium). The larger bias on bacteria is potentially due to the complex bacterial cell wall that forms a strong permeability barrier to the mutual access of bacterial and host genome [46]. Bactria have to resort to a complex secrete system to transport bacterial proteins to the surface or membrane of bacterial cell or directly inject bacterial proteins into the host cell [23]. As such, random sampling is prone to sample pathogen-host protein pairs that are located in two different cells and are mutually inaccessible to, so as to limit the coverage of negative data. The experimentally verified pathogen-host PPIs take place in between physically accessible pathogen and host proteins, while the negative data are sampled in two isolate cell spaces, which is potentially a factor contributing to the model bias.
3.3. Biological insights into pathogen interference with human immune signaling and transcriptional activities
3.3.1. Genome-scale predictions
Theoretically, the prediction space contains pathogen-host protein pairs for m pathogen proteins and n host proteins. To reduce computational complexity, we only predict 50,000 randomly sampled pathogen-host protein pairs. For human respiratory syncytial virus, the proposed framework predicts 7.15% of the protein pairs as interactions. For Salmonella typhimurium, the prediction set consists of two parts of equal size. One part is sampled from the Salmonella-human PPIs inferred via pathogen functional mimicry, and the other part is randomly sampled from Salmonella-human protein pairs. Both parts contain 25,000 protein pairs. The proposed framework predicts 95.43% of the first part and 25.02% of the second part as interactions. For the convenience of analysis, the positive training data, the positive independent test data and the predicted positive data are merged into 3161 HRSV-human PPIs and 75,062 Salmonella-human PPIs, respectively (see Supplementary file S5–S6).
3.3.2. GO enrichment analyses
In the prediction set, HRSV and S. typhimurium are predicted to target 2739 and 6402 novel human proteins, respectively. For clarity purpose, only 15 top biological processes are illustrated in Fig. 3(A)–(B). As shown in Fig. 3, the two pathogens interfere with some common human cellular processes, e.g. transport (GO:0006810), regulation of transcription, DNA-templated (GO:0006355, protein phosphorylation (GO:0006468), apoptotic process (GO:0006915), positive regulation of transcription by RNA polymerase II (GO:0045944). The overall GO enrichment analyses for the two pathogens are provided in Supplementary file S7–S8. We further take the HRSV protein SH|P69360 and the S. typhimurium protein dnaQ|P0A1G9 for example to analyse their interference with human cellular processes.
Fig. 3.

Fifteen top biological processes interfered with by human respiratory syncytial virus (A) and S. typhimurium (B).
SH|P69360. As illustrated in Fig. 4(A), the HRSV protein SH|P69360 is predicted to target 88 human proteins. GO enrichment analyses show that the human genes targeted by SH|P69360 get involved in the cellular processes of ion transport (GO:0006811), iron ion homeostasis (GO:0055072), secretory granule lumen (GO:0034774), inflammatory response (GO:0006954), negative regulation of apoptotic process (GO:0043066), etc. According to Uniprot (https://www.uniprot.org/uniprot/P69360), the HRSV protein SH|P69360 is a viroporin that forms a homopentameric ion channel with low ion selectivity. SH|P69360 potentially enhances host membrane permeability, disrupts host cellular ion homeostasis and triggers host inflammatory immune response. Meanwhile, SH|P69360 inhibits the host TNFA-mediated signaling pathway and results in delay of apoptosis. We can see that SH|P69360 is involved in some important common cellular processes with its predicted targets. The GO terms are unevenly distributed among SH|P69360 and its host counterparts or target proteins. If we use AVG strategy, cosine similarity or Jaccard index [36], the semantic scores between proteins would be decreased to ignore the identical celluar processes of ion homeostasis and inflammatory response. The MAX strategy we adopt gives priority to the critical cellular processes that pathogen and host counterparts both are involved in.
Fig. 4.

Human proteins predicted to be targeted by the HRSV protein SH|P69360 (A) and the S. typhimurium protein dnaQ|P0A1G9.
dnaQ|P0A1G9. As illustrated in Fig. 4(B), the S. typhimurium protein dnaQ|P0A1G9 is predicted to target 90 human proteins. dnaQ|P0A1G9, named DNA polymerase III subunit epsilon, is a complex of multichain enzyme responsible for most of the replicative synthesis in bacteria, in which the epsilon subunit is a proofreading 3′-5′ exonuclease that performs editing function (https://www.uniprot.org/uniprot/P0A1G9). GO enrichment analyses show that the human genes targeted by dnaQ|P0A1G9 get involved in the cellular processes of DNA replication (GO:0006260), G2/M transition of mitotic cell cycle (GO:0000086), regulation of DNA biosynthetic process (GO:2000278), RNA phosphodiester bond hydrolysis, endonucleolytic (GO:0090502). We can see that the S. typhimurium protein dnaQ|P0A1G9 and its predicted targets participate in some similar and associated cellular processes. These results show that pathogen mimics and substitutes its host counterpart proteins to hijack normal host PPIs. The number of target human proteins seems to be large. Actually, the pathogen-host PPIs inferred via pathogen functional mimicry are not necessarily physical but potentially functional interactions, though the inference templates are human physical PPIs. For bacterial pathogens, the effectors may remain in the bacterial cell or may be secreted and transported to the host cellular organelles to indirectly or directly interfere with host cellular processes.
3.3.3. Pathogen interference with human immune signaling pathways
We further map the pathogen targeted genes onto human immune signaling pathways to study how human respiratory syncytial virus and Salmonella typhimurium interfere with human signaling activities. Human immune signaling pathways are taken from NetPath [47]. For the sake of simplicity, the pathways IL1–IL11 are merged into one IL signaling pathway and we totally obtain 27 pathways. The pathways that the two pathogens target are provided in Supplementary file S9–S10. As shown in Fig. 5(A), the top pathways targeted by most of the HRSV proteins include Tumor necrosis factor alpha (TNF), Epidermal growth factor receptor (EGFR1), T Cell Receptor (TCR), Interleukin-1–11 (IL), Brain-derived neurotrophic factor (BDNF), Receptor activator of nuclear factor kappa-B ligand (RANKL), etc. Furthermore, most HRSV proteins target more than one pathway and 6 HRSV proteins target more than 10 pathways (see Fig. 5(B)).
Fig. 5.

Pathway enrichment analyses. A–B show the number of HRSV proteins that target each specific human immune signaling pathway and the number of human immune signaling pathways that each HRSV protein targets. C–D show the number of S. typhimurium proteins that target each specific human immune signaling pathway and the number of human immune signaling pathways that each S. typhimurium protein targets.
The genome space of Salmonella typhimurium is much larger than that of human respiratory syncytial virus, and thus a human immune signaling pathway is potentially targeted by more S. typhimurium proteins. As shown in Fig. 5(C), the least targeted pathway Thymic stromal lymphopoietin (TSLP) is predicted to be targeted by more than 200 S. typhimurium proteins. The intensively targeted pathways include Tumor necrosis factor alpha (TNF), Androgen receptor (AR), Epidermal growth factor receptor (EGFR1), Transforming growth factor beta receptor (TGFBeta), Interleukin-1–11 (IL), T Cell Receptor (TCR). Furthermore, a S. typhimurium protein is prone to target more pathways. As shown in Fig. 5(D), more than 500 S. typhimurium proteins target more than 15 immune signaling pathways. The interference with human immune signaling pathways is more functional than physical, because subcellularly co-localization as restricted by Formula (1), (2) does not necessarily indicate physical interactions.
3.3.4. Pathogen interference with human cellular transcriptional activities
The computational results show that human respiratory syncytial virus and Salmonella typhimurium are predicted to target 457 and 1697 human genes/proteins that are associated with human cellular transcriptional activities, respectively (see Supplementary file S11–S12). For clarity purpose, only 25 top transcription-associated human proteins are illustrated. Many transcription-associated human proteins are targeted by more than one HRSV protein. For instance, WWP2|O00308, CLU|P10909 and RUVBL2|Q9Y230 are targeted by 4 HRSV proteins and the other transcription-associated human proteins are targeted by 3 HRSV proteins (see Fig. 6(A)). WWP2|O00308 is highly expressed in undifferentiated embryonic stem cells and is involved in regulation of human cellular transcriptional activities, e.g. negative regulation of DNA-binding transcription factor activity (GO:0043433), negative regulation of transcription by RNA polymerase II (GO:0000122), protein ubiquitination (GO:0016567), regulation of ion transmembrane transport (GO:0034765), etc (https://www.uniprot.org/uniprot/O00308). Among the HRSV proteins, only 1–3 HRSV proteins are predicted to target a large number of human transcription-associated proteins (see Fig. 6(B)).
Fig. 6.

Pathogen interference with host cellular transcriptional activities. A–B show the number of HRSV proteins that target each specific human gene/protein associated with gene transcription and the number of human transcriptional genes/proteins that each HRSV protein targets. C–D show the number of S. typhimurium proteins that target each specific human gene/protein associated with gene transcription and the number of human transcriptional genes/proteins that each S. typhimurium protein targets. For clarity, only 25 top transcription associated human proteins are illustrated in (A) and (C).
As shown in Fig. 6(C), the top 25 transcription-associated human proteins are all predicted to be targeted by more than 100 S. typhimurium proteins. These interactions probably take place in two spatially-separated cells and are potentially more functionally than physically. All these human genes targeted by S. typhimurium are involved in human cellular transcription regulation. For instance, ILF2|Q12905 functions predominantly as a heterodimeric complex with ILF3 and potentially regulates the transcription of IL2 gene during T-cell activation (https://www.uniprot.org/uniprot/Q12905). Further GO analyses show that ILF2|Q12905 participates in the cellular processes of positive regulation of transcription, DNA-templated (GO:0045893), transcription, DNA-templated (GO:0006351), neutrophil degranulation (GO:0043312). FOS|P01100 is a nuclear phosphoprotein that forms a tight but non-covalently linked complex with the JUN/AP-1 transcription factor (https://www.uniprot.org/uniprot/P01100). GO analyses show that FOS|P01100 participates in some major transcription-associated cellular processes, e.g. positive regulation of transcription by RNA polymerase II (GO:0045944), positive regulation of pri-miRNA transcription by RNA polymerase II (GO:1902895), transforming growth factor beta receptor signaling pathway (GO:0007179), response to cold (GO:0009409), conditioned taste aversion (GO:0001661), etc. Among these S. typhimurium proteins, only a few proteins are predicted to target a large number of human transcription-associated proteins (see Fig. 6(D)).
3.4. Comparison with existing methods
Most of the existing computational methods directly build predictive models on experimentally verified pathogen-host PPI data and thus are not applicable to the experimentally less-studied pathogens in the context of pathogen-host protein interactions. To infer pathogen-host PPIs for less-studied pathogens, two categories of computational methods have been proposed, namely pathogen mimicry [18], [19], [20], [21], [25], [26] and transfer learning [22]. Pathogen mimicry methods are further divided into pathogen sequence mimicry, i.e. interlog methods [18], [19], [20], [21], [25], and pathogen structural mimicry [26]. Transfer learning method [22] builds a predictive model on the experimental data from other species to predict the pathogen-host PPIs for the pathogen concerned. Transfer learning is also used to build a predictive model on the experimental pathogen-host PPI data from the species itself, e.g. semi-supervised multi-task learning [11] and ensemble transfer learning [13].
In this study, the proposed pathogen functional mimicry is more general and flexible to cover the sequence, motif, structure and interface mimicry, because sequence and structure similarity could ultimately lead to functional similarity. In this section, we compare the proposed framework with a pathogen sequence mimicry method [21] and several transfer learning methods [11], [13], [22]. The transfer learning methods include direct methods that directly build model on the experimental pathogen-host PPIs of the species itself [11], [13] and indirect method that builds model indirectly on the experimental pathogen-host PPIs from other species [22].
3.4.1. Comparison with pathogen sequence mimicry method
Among the pathogen mimicry methods, we only choose to compare with the pathogen sequence mimicry method [21] that is validated against independent experimental data. Mei et al. [21] restrict the interlog inference within the co-evolving M. tuberculosis H37Rv and Homo sapiens. The interlogs are inferred from PPI networks of M. tuberculosis H37Rv alone, which are extracted from STRING [48] and have been reported to be of low quality [49]. In this framework, we use the well-studied human PPI networks as inference template. For comparison purpose, we replace the pathogen M. tuberculosis H37Rv in [21] with human immunodeficiency virus and Francisella tularensis to verify the advantage of pathogen functional mimicry over pathogen sequence mimicry.
Independent test shows that the pathogen sequence mimicry method [21] achieves much worse performance than the proposed framework on the same experimental pathogen-host PPI data from human immunodeficiency virus (see Fig. 7(A)) and Francisella tularensis (see Fig. 7(B)). The pathogen sequence mimicry method [21] recognizes 49.96% and 40.00% of the experimental HIV-human PPIs and F. tularensis-human PPIs respectively, while the proposed framework more encouragingly recognizes 73.09% and 75.81% of the experimental HIV-human and F. tularensis-human PPIs respectively. On the same negative independent test data, the proposed framework achieves 91.67% and 66.13% recognition rate for HIV and F. tularensis respectively, while the pathogen sequence mimicry method [21] only achieves 61.78% and 64.44% recognition rate for HIV and F. tularensis respectively. The proposed pathogen functional mimicry method excels the pathogen sequence mimicry method [21] partly because it covers more types of pathogen mimicries.
Fig. 7.

Performance comparison with the existing methods.
3.4.2. Comparison with cross-species transfer learning method
Kshirsagar et al. [22] builds KMM-SVM based transfer learning models on the pathogen-host PPIs from other pathogens to predict the pathogen-host PPIs of the target pathogen. Take Salmonella typhimurium for example. KMM-SVM trains on experimental Salmonella-mouse PPIs and recognizes 35.5% of experimental Salmonella-human PPIs (see Fig. 7(C)). In addition, KMM-SVM trains on experimental Francisella-human PPIs and recognizes 16.1% of experimental Salmonella-human PPIs (see Fig. 7(D)). The independent test results show that a potentially large genome gap between the source and the target species (e.g. mouse versus human, Francisella versus Salmonella) make cross-species knowledge transfer less effective. However, the proposed framework encouragingly achieves 75.81% independent test performance on the experimental Salmonella-human PPIs. This result shows that pathogen mimicry could evolutionarily narrow the genome gap between co-evolving pathogen and host.
Transfer learning is also used to build predictive models on the experimental pathogen-host PPI data from the species itself. Both methods [11], [13] are built on experimental HIV-human PPIs. However, the semi-supervised multi-task learning [11] only achieves 10% overlap between siRNA screen and predictions, while the ensemble transfer learning [13] only recognizes 55.77% of the largest 1101Tat-associated HIV-human PPIs. The proposed framework encouragingly recognizes 73.09% of the 3188 experimental HIV-human PPIs, though it is trained on the HIV-human PPIs inferred from human PPI-networks via pathogen functional mimicry. The proposed framework excels the existing methods, partly because the experimental data covers very limited pathogen genes/proteins while the proposed pathogen functional mimicry achieves a much larger coverage of genes and obtain much more training data.
4. Discussions
Machine learning modeling of biological problems often requires sufficient experimental data as training data to build credible predictive models. However, in many cases the species concerned are experimentally less-studied. In recent years, transfer learning has been well recognized as an effective technique to achieve cross-species knowledge transfer. Knowledge transferred from well-studied source species is potentially useful to enrich the feature information or augment the training data of the target species. Nevertheless, genome similarity is a critical concern of transfer learning modeling and a large cross-species genome gap is prone to yield less reliable models.
Pathogen mimicry has been recognized as a basic biological mechanism that a pathogen evolves to invade or hijack host cellular processes. Pathogen sequence, structure and interface mimicries have been used to develop computational methods for pathogen-host PPI prediction. From evolutionary point of view, these mimicries requires a long time of evolution and many transient protein interactions may not stringently require structural matches. From computational point of view, these mimicries are biased not to cover the other kinds of mimicries. In this study, we propose a more general and flexible pathogen functional mimicry strategy to infer pathogen-host PPIs from human protein-protein interaction networks alone. Pathogen functional mimicry is defined via GO semantic similarity. In some sense, the purpose of pathogen co-evolving and mimicking host protein sequences and structures is to perform and substitute its host counterpart functions. From this point of view, pathogen functional mimicry actually covers sequence, structure and interface mimicry. As a result, pathogen functional mimicry is less biased to cover more types of mimicries and meanwhile more training data could be obtained. From data point of view, pathogen functional mimicry is less demanding because it does not require the information of protein structure and PPI interface that is not easily available.
Inference template is the second critical concern in pathogen-host PPI prediction. The existing methods generally use the PPIs from third-party species as template to infer pathogen-host PPIs. However, a large genome gap between third-party species and the concerned species is prone to yield less credible results. Pathogen mimicry achieves knowledge transfer across co-evolving pathogen and host, so that the genome gap is reduced. In addition, we use the well-studied host PPI networks (i.e. Homo sapiens) as template, which is more reliable than the bacterial pathogen template [21].
Validation against experimental data is the third critical concern in pathogen-host PPI prediction. In many cases, there are very limited experimental data that can be used as independent test data. In this study, we use all the available experimental data to validate the proposed framework. Independent tests on the experimentally verified pathogen-host PPIs from Human immunodeficiency virus and Francisella tularensis show that the proposed framework encouragingly outperforms the existing methods. As a result, the assumption of pathogen functional mimicry is effectively validated. Nevertheless, performance estimation shows that the proposed framework is somewhat biased towards the positive class. The bias partly results from the negative data sampling. For bacterial pathogens, the bias seems to be more serious, partly because the non-interacting pathogen-host protein pairs are randomly sampled in two different cells and the coverage of negative data is limited, which is similar to the subcellularly restricted sampling method [50]. Negative data sampling is a critical concern in PPI prediction [15], [51], which could be to some extent improved via computational methods, e.g. augmenting the available experimentally derived non-interacting protein pairs via homology [51]. Solution to this problem ultimately depends on the accumulation of experimental data and development of sophisticated computational methods. In addition, the F. tularensis-human PPIs derived via Y2H technique potentially contain a certain level of noise, and thus the independent test performance on Francisella tularensis is not so encouraging.
As two case studies, we apply the proposed framework to human respiratory syncytial virus and Salmonella typhimurium for genome-scale reconstruction of pathogen-host PPI networks. GO enrichment analyses show that the pathogen and predicted human target genes are generally involved in some common cellular processes. Pathway enrichment analyses show that HRSV tends to target Tumor necrosis factor alpha (TNF), Epidermal growth factor receptor (EGFR1), T Cell Receptor (TCR) and Interleukin-1–11 (IL)., while S. typhimurium tends to target Tumor necrosis factor alpha (TNF), Androgen receptor (AR), Epidermal growth factor receptor (EGFR1) and Transforming growth factor beta receptor (TGFBeta). Besides human immune signaling pathways, HRSV and S. typhimurium also interfere with some major human cellular transcriptional activities. For instance, HRSV is predicted to interfere with human protein WWP2 that is highly expressed in undifferentiated embryonic stem cells and is involved in regulation of DNA-binding transcription factor activity. S. typhimurium is predicted to target human protein FOS that forms a tight but non-covalently linked complex with the JUN/AP-1 transcription factor. All the other results are provided in the supplementary files and potentially give biological insights into the signaling cross-talk mechanism between pathogen and host.
According to the comprehensive database VirHostNet 2.0 [32], there are 239 pathogens that are studied in terms of their protein interactions with Homo sapiens. We choose the four pathogens (i.e. human immunodeficiency virus, Francisella tularensis, human respiratory syncytial virus and Salmonella typhimurium) with relative larger experimental data to validate the proposed framework. Development of a web interface to study all the other pathogens is on our further research agenda.
5. Conclusions
In this study, we propose a transfer learning framework based on pathogen functional mimicry to infer pathogen-host PPIs from human PPI networks alone. The performance of independent test on experimental PPI data validates the effectiveness of pathogen functional mimicry, which is more flexible, less biased and less demanding than pathogen sequence and structure mimicries.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
This work is partly supported by the funding from the NIH grants 2U54MD007595, 5P20GM103424-17 and U19AG055373. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author contributions
MS conducted the study and wrote the paper. ZK revised the paper. All authors read and approved the final manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2019.12.008.
Contributor Information
Suyu Mei, Email: meisygle@gmail.com.
Kun Zhang, Email: kzhang@xula.edu.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Vandeven N., Nghiem P. Pathogen-driven cancers and emerging immune therapeutic strategies. Cancer Immunol Res. 2014;2:9–14. doi: 10.1158/2326-6066.CIR-13-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polk D.B., Peek R.M., Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10:403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jean Beltran P.M., Federspiel J.D., Sheng X., Cristea I.M. Proteomics and integrative omic approaches for understanding host-pathogen interactions and infectious diseases. Mol Syst Biol. 2017;13:922. doi: 10.15252/msb.20167062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dix A., Vlaic S., Guthke R., Linde J. Use of systems biology to decipher host-pathogen interaction networks and predict biomarkers. Clin Microbiol Infect. 2016;22:600–606. doi: 10.1016/j.cmi.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Durmuş S., Çakır T., Özgür A., Guthke R. A review on computational systems biology of pathogen-host interactions. Front Microbiol. 2015;6:235. doi: 10.3389/fmicb.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nourani E., Khunjush F., Durmuş S. Computational approaches for prediction of pathogen-host protein-protein interactions. Front Microbiol. 2015;6:94. doi: 10.3389/fmicb.2015.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durmuş Tekir S.D., Ülgen K.Ö. Systems biology of pathogen-host interaction: networks of protein-protein interaction within pathogens and pathogenhuman interactions in the post-genomic era. Biotechnol J. 2013;8:85–96. doi: 10.1002/biot.201200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bandyopadhyay S., Ray S., Mukhopadhyay A., Maulik U. A review of in silico approaches for analysis and prediction of HIV-1-human protein-protein interactions. Brief Bioinform. 2015;16:830–851. doi: 10.1093/bib/bbu041. [DOI] [PubMed] [Google Scholar]
- 9.Mariano R., Wuchty S. Structure-based prediction of host-pathogen protein interactions. Curr Opin Struct Biol. 2017;44:119–124. doi: 10.1016/j.sbi.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Kösesoy İ., Gök M., Öz C. A new sequence based encoding for prediction of host-pathogen protein interactions. Comput Biol Chem. 2019;78:170–177. doi: 10.1016/j.compbiolchem.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Qi Y., Tastan O., Carbonell J.G., Klein-Seetharaman J., Weston J. Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics. 2010;26:i645–i652. doi: 10.1093/bioinformatics/btq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kshirsagar M., Carbonell J., Judith K. Multitask learning for host-pathogen protein interactions. Bioinformatics. 2013;29:i217–i226. doi: 10.1093/bioinformatics/btt245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mei S. Probability weighted ensemble transfer learning for predicting interactions between HIV-1 and human proteins. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eid F.E., ElHefnawi M., Heath L.S. DeNovo: virus-host sequence-based protein protein interaction prediction. Bioinformatics. 2016;32:1144–1150. doi: 10.1093/bioinformatics/btv737. [DOI] [PubMed] [Google Scholar]
- 15.Mei S., Zhu H. A novel one-class SVM based negative data sampling method for reconstructing proteome-wide HTLV-human protein interaction networks. Sci Rep. 2015;5:8034. doi: 10.1038/srep08034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mei S., Zhang K. Computational discovery of Epstein-Barr virus targeted human genes and signalling pathways. Sci Rep. 2016;6:30612. doi: 10.1038/srep30612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emamjomeh A., Goliaei B., Zahiri J., Ebrahimpour R. Predicting protein-protein interactions between human and hepatitis C virus via an ensemble learning method. Mol Biosyst. 2014;10:3147–3154. doi: 10.1039/c4mb00410h. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H., Rezaei J., Hugo W., Gao S., Jin J. Stringent DDI-based prediction of H. Sapiens-M. Tuberculosis H37Rv protein-protein interactions. BMC Syst Biol. 2013;7(Suppl 6):S6. doi: 10.1186/1752-0509-7-S6-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou H., Gao S., Nguyen N.N., Fan M., Jin J. Stringent homology-based prediction of H. Sapiens-M. Tuberculosis H37Rv protein-protein interactions. Biol Direct. 2014;9:5. doi: 10.1186/1745-6150-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schleker S., Garcia-Garcia J., Klein-Seetharaman J., Oliva B. Prediction and comparison of Salmonella-human and Salmonella-Arabidopsis interactomes. Chem Biodivers. 2012;9:991–1018. doi: 10.1002/cbdv.201100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mei S., Flemington E.K., Zhang K. Transferring knowledge of bacterial protein interaction networks to predict pathogen targeted human genes and immune signaling pathways: a case study on M. tuberculosis. BMC Genomics. 2018;19:505. doi: 10.1186/s12864-018-4873-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kshirsagar M., Schleker S., Carbonell J., Klein-Seetharaman J. Techniques for transferring host-pathogen protein interactions knowledge to new tasks. Front Microbiol. 2015;6:36. doi: 10.3389/fmicb.2015.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guven-Maiorov E., Tsai C.J., Nussinov R. Pathogen mimicry of host protein-protein interfaces modulates immunity. Semin Cell Dev Biol. 2016;58:136–145. doi: 10.1016/j.semcdb.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Via A., Uyar B., Brun C., Zanzoni A. How pathogens use linear motifs to perturb host cell networks. Trends Biochem Sci. 2015;40:36–48. doi: 10.1016/j.tibs.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Doxey A.C., McConkey B.J. Prediction of molecular mimicry candidates in human pathogenic bacteria. Virulence. 2013;4:453–466. doi: 10.4161/viru.25180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guven-Maiorov E., Tsai C.J., Ma B., Nussinov R. Prediction of host-pathogen interactions for helicobacter pylori by interface mimicry and implications to gastric cancer. J Mol Biol. 2017;429:3925–3941. doi: 10.1016/j.jmb.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elde N.C., Malik H.S. The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol. 2009;7:787–797. doi: 10.1038/nrmicro2222. [DOI] [PubMed] [Google Scholar]
- 28.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S. Human protein reference database–2009 update. Nucleic Acids Res. 2009;37(Database issue) doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatr-Aryamontri A., Breitkreutz B.J., Oughtred R., Boucher L., Heinicke S. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015;43(Database issue) doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orchard S., Ammari M., Aranda B., Breuza L., Briganti L. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42(Database issue):D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.López Y., Nakai K., Patil A. HitPredict version 4: comprehensive reliability scoring of physical protein-protein interactions from more than 100 species. Database (Oxford) 2015;pii:bav117. doi: 10.1093/database/bav117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guirimand T., Delmotte S., Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015;43(Database issue) doi: 10.1093/nar/gku1121. D583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dyer M.D., Neff C., Dufford M., Rivera C.G., Shattuck D., Bassaganya-Riera J. The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schleker S., Sun J., Raghavan B., Srnec M., Müller N. The current Salmonella-host interactome. Proteomics Clin Appl. 2012;6:117–133. doi: 10.1002/prca.201100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guirimand T., Delmotte S., Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015;43(Database issue):D583–D587. doi: 10.1093/nar/gku1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu M., Thomas P.D. GO functional similarity clustering depends on similarity measure, clustering method, and annotation completeness. BMC Bioinf. 2019;20:155. doi: 10.1186/s12859-019-2752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J.Z., Du Z., Payattakool R., Yu P.S., Chen C.F. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23:1274–1281. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- 38.Yu G., Li F., Qin Y., Bo X., Wu Y., Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26:976–978. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- 39.Qi Y., Bar-Joseph Z., Klein-Seetharaman J. Evaluation of different biological data and computational methods for use in protein interaction prediction. Proteins. 2006;63:490–500. doi: 10.1002/prot.20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maetschke S., Simonsen M., Davis M., Ragan M.A. Gene Ontology-driven inference of protein–protein interactions using inducers. Bioinformatics. 2012;28:69–75. doi: 10.1093/bioinformatics/btr610. [DOI] [PubMed] [Google Scholar]
- 41.Boeckmann B., Bairoch A., Apweiler R., Blatter M.C., Estreicher A. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrell D., Dimmer E., Huntley R.P., Binns D., O'Donovan C. The GOA database in 2009–an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;(37Database issue) doi: 10.1093/nar/gkn803. D396-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu F., Huang F., Lin C. Dual coordinate descent methods for logistic regression and maximum entropy models. Mach Learn. 2011;85:41–75. [Google Scholar]
- 45.Fan R., Chang K., Hsieh C., Wang X., Lin C. LIBLINEAR: a library for large linear classification. Mach Learn Res. 2008;9:1871–1874. [Google Scholar]
- 46.Ben-Kahla I., Al-Hajoj S. Drug-resistant tuberculosis viewed from bacterial and host genomes. Int J Antimicrob Agents. 2016;48:353–360. doi: 10.1016/j.ijantimicag.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Kandasamy K., Mohan S.S., Raju R., Keerthikumar S., Kumar G.S. NetPath: a public resource of curated signal transduction pathways. Genome Biol. 2010;11:R3. doi: 10.1186/gb-2010-11-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue) doi: 10.1093/nar/gku1003. D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou H., Wong L. Comparative analysis and assessment of M. Tuberculosis H37Rv protein-protein interaction datasets. BMC Genomics. 2011;12(Suppl 3):S20. doi: 10.1186/1471-2164-12-S3-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ben-Hur A., Noble W. Choosing negative examples for the prediction of protein-protein interactions. BMC Bioinform. 2006;7:S2. doi: 10.1186/1471-2105-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mei S., Zhang K. Neglog: homology-based negative data sampling method for genome-scale reconstruction of human protein-protein interaction. Networks Int J Mol Sci. 2019;20 doi: 10.3390/ijms20205075. pii: E5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.