

We report the isothermal digital detection of microRNAs using DNA circuits encoding a background-free amplification mechanism.
Abstract
MicroRNAs, a class of transcripts involved in the regulation of gene expression, are emerging as promising disease-specific biomarkers accessible from tissues or bodily fluids. However, their accurate quantification from biological samples remains challenging. We report a sensitive and quantitative microRNA detection method using an isothermal amplification chemistry adapted to a droplet digital readout. Building on molecular programming concepts, we design a DNA circuit that converts, thresholds, amplifies, and reports the presence of a specific microRNA, down to the femtomolar concentration. Using a leak absorption mechanism, we were able to suppress nonspecific amplification, classically encountered in other exponential amplification reactions. As a result, we demonstrate that this isothermal amplification scheme is adapted to digital counting of microRNAs: By partitioning the reaction mixture into water-in-oil droplets, resulting in single microRNA encapsulation and amplification, the method provides absolute target quantification. The modularity of our approach enables to repurpose the assay for various microRNA sequences.
INTRODUCTION
MicroRNAs are endogenous short noncoding RNA strands involved in posttranscriptional regulation of gene expression (1). Discovered 20 years ago, microRNAs are emerging as attractive diagnostic, prognostic, or predictive biomarkers because, first, accumulating clinical evidence suggests that dysregulation of microRNAs is closely related to various diseases, including cancers, neuronal or heart diseases, and diabetes (2), as well as to resistance to chemotherapy or radiotherapy (3). Second, they are present in bodily fluids, making them accessible from minimally invasive liquid biopsies (serum, plasma, urine, etc.) (4). Circulating biomarkers can be assessed readily, potentially enabling a regular follow-up of treatment efficacy, detection of relapses, early diagnostic, or large-scale population screening (5). Last, these nucleic acids with known sequences are amenable to rational and rapid design of generic detection tests.
The detection of circulating microRNAs in blood is still a challenging task, as they are highly diluted. Besides, disease-related microRNAs must be accurately quantified to leverage their use in clinical procedures. Hence, the precise measurement of microRNA concentrations remains the critical bottleneck, encouraging the development of sensitive, specific, and quantitative detection technologies. The current gold standard for sensitive microRNA detection is the reverse transcription quantitative polymerase chain reaction (RT-qPCR) (6). Despite the high sensitivity of RT-qPCR, the technique has inescapable drawbacks: (i) the RT step is known to introduce substantial bias in the quantification (7–9); (ii) primers and probe must be designed for each target and rely on sophisticated design due to the short length of the target; (iii) the thermocycling protocol should be optimized for each assay; (iv) the target itself is amplified and is therefore a hazardous source of carryover contamination (10); and (v) PCR is known to be inhibited by biological samples (11). In addition, the procedure relies on a real-time tracking of the amplification and requires standard calibration, thus giving only access to a relative estimate of the amount of microRNA.
This last issue can, in principle, be addressed with a digital quantification [such as droplet PCR (12)] based on the isolation of single nucleic acid molecules in microcompartments. Although droplet PCR emerges as a powerful method for precise and absolute quantification of nucleic acids, it retains the weaknesses of RT-qPCR for microRNA assessment (13). Isothermal alternatives (14) have been proposed [EXPAR (15, 16), LAMP (17), RCA (18), HCR (19), etc.] that rely on simpler one-step protocols and free themselves from the reverse transcription reaction. Among these isothermal strategies, EXPAR (exponential amplification reaction) has gained attention due to its simplicity and competing sensitivity. However, the exponential nature of the reaction renders it prone to leaky reactions, eventually triggering the production of amplified products from spurious reactions (20, 21). These nonspecific amplifications are always observed with a short delay after the target-triggered reaction. This issue makes such amplification chemistry ill-adapted to a digital readout where only the target-containing compartments are expected to produce a positive signal (fig. S1). Preheating separately the substrates [templates and deoxyribonucleotide triphosphate (dNTP)] and the enzymes (polymerase and endonuclease) may delay target-independent amplification; however, a digital readout would still require a very precise control over the incubation time to limit the occurrence of false-positive compartments (16). Hairpin-shaped templates have been shown to improve the specificity over homologous sequences, although target-independent amplification is not delayed (22). Several additives such as graphene oxide (23), tetramethylammonium chloride (24), or single-strand binding protein (24, 25) have been proposed to decrease these spurious reactions and increase assay sensitivity. Recently, Urtel et al. (26) successfully suppressed background amplification using a template lacking one of the four nucleotides (dT-free template), a three-letter nicking site, and 2′-deoxyriboadenosine 5′-triphosphate (dATP)–free amplification mixture. Similarly, locked nucleic acid and peptide nucleic acid modification of the 3′ domain of EXPAR template have been used to reduce background amplification (25, 27). Nevertheless, no biosensing application has been validated yet with these last two approaches (25–27).
Besides enzyme-based amplification reaction, enzyme-free systems have been reported using toehold-mediated strand displacement reactions. These reactions are also prone to leak, coming from the spurious release of output strands, which is detrimental for the sensitivity in sensing applications (28). Many strategies have been described to mitigate the leaks, encompassing clamped (29–31) or long (32) domains, the introduction of mismatches (33), or using ultrapure DNA complexes (28, 31, 32). However, in comparison with the enzyme-powered approaches, most enzyme-free circuits display limited sensitivity as a consequence of lower signal amplification efficiency.
Here, we propose a background-free isothermal amplification technique relying on a DNA-based molecular program specifically designed to catalyze a target-triggered signal amplification while avoiding nonspecific amplification reactions. The amplification system is coupled to a catalytic degradation mechanism that continuously absorbs products stemming from leaky reaction. Applied to the detection of microRNAs, we demonstrate the high sensitivity of this amplification chemistry down to femtomolar concentrations. The suppression of nonspecific amplification reaction allows the robust droplet digital detection of microRNA targets and their absolute endpoint quantification.
RESULTS
Molecular program designed for microRNA detection
Molecular programming is an emerging discipline that involves the design of artificial biomolecular circuits performing information-processing tasks. It uses the predictable Watson and Crick base pairing of synthetic DNA oligonucleotides to enable the rational design of molecular circuits (34, 35). We use here a versatile molecular programming language named PEN-DNA toolbox (Polymerase Exonuclease Nickase-Dynamic Network Assembly) (36, 37). It uses a set of short oligonucleotides (templates) encoding the topology of the network and a mixture of enzymes that catalyze the production and degradation of activator/inhibitor strands. Using this set of reaction modules, we designed a generic molecular program dedicated to the detection of microRNAs.
Figure 1 presents the connectivity of the circuit (cf. also fig. S2 for a more detailed mechanism). The universal signal amplification system is composed of two templates: an autocatalytic template (aT) and a pseudotemplate (pT) (38). The aT is made of a dual-repeat sequence catalyzing the exponential replication of a 12-mer oligonucleotide [signal strands (s-strands)] via a polymerization/nicking cycles. It is well known that such exponential reactions are prone to nonspecific amplification, meaning that even in the absence of the input s-strand, the autocatalysis eventually occurs due to spurious reactions, also known as leak (20). The pT catalyzes the deactivation of the s-strand produced from nonspecific reactions and is therefore essential to avoid unwanted amplification (38). The molecular system aT:pT acts as a bistable switch (fig. S3): In the absence of the s-strand input, the leak is absorbed by the pT and amplification is not observed. If the concentration of s-strand exceeds a certain threshold (tuned by the ratio aT/pT), then the pT-mediated degradation path is saturated and amplification occurs. To make this bistable switch responsive to the microRNA target, a converter template (cT) is connected upstream to the aT: Upon hybridization of the microRNA to the input part of the cT, the latter linearly produces trigger strands (by polymerization/nicking reactions), which, in turn, bind and activate the autocatalytic reaction on the aT. Downstream to the aT, a reporting template (rT) captures the amplified s-strands to produce a fluorescence signal. The dynamical nature of the molecular circuit is preserved by the presence of an exonuclease that degrades all produced strands, keeping the system responsive and avoiding the poisoning of the pT.
Fig. 1. A molecular program dedicated to the detection of microRNA.
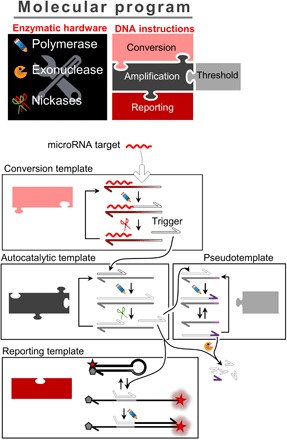
A four-template DNA circuit encodes the connectivity of the molecular program, whose reactions are catalyzed by a set of enzymes (polymerase, exonuclease, and endonucleases): The cT converts the target microRNA to a generic s-strand sequence; the aT exponentially amplifies the s-strand sequence; to avoid target-independent amplification, the pT drives the deactivation of the small amount of triggers originating from leaky reactions; the rT translates the s-strand sequences into a fluorescence signal.
Background-free detection of the microRNA Let-7a
Figure 2 evaluates the sensitivity of this approach for the real-time detection of Let-7a, one of the first identified and most studied microRNA regulators (39). After optimizing the experimental conditions (fig. S4), the four templates were mixed together with the enzymatic processor and spiked with a concentration of the synthetic RNA target Let-7a ranging from 0 to 1 nM (Fig. 2A). The fluorescence of the rT is monitored in real time with a PCR thermocycler set at a constant temperature of 50°C. The negative control (no target) does not produce a positive signal for more than 20 hours (the experiment duration). The limit of detection of the assay is around 1 fM, and the dynamic range spans six orders of magnitude from 1 fM to 100 pM (cf. also extended data fig. S5). The absence of amplification when the cT is missing (Fig. 2C) validates the specificity of the target-triggered amplification. As expected, in the absence of the pT (Fig. 2B), the sensitivity is negatively affected with a limit of detection of 3.7 pM (estimated from 3 SDs from the mean amplification time of the negative control). These results demonstrate the importance of this active leak absorption mechanism for controlling the amplification threshold and thus eliminating background amplification. However, the active consumption of s-strands considerably increases the time of the assay. For instance, it takes 16 hours to trigger the amplification reaction from 1 fM of Let-7a target, which is too slow for most routine procedures.
Fig. 2. In-solution detection of the microRNA Let7a.
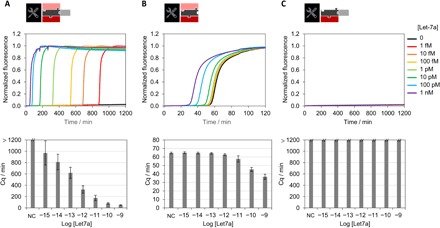
(A) Detection with the four-template molecular program and (B) the molecular program without the pT or (C) without the cT. The amplification reaction is monitored in real time, and the amplification time (Cq) is plotted as a function of the Let-7a concentration.
Droplet digital detection for absolute microRNA quantification
To circumvent this issue, we converted the analog readout, based on relative measurements, to a digital readout where single molecules are detected after being isolated in independent compartments (Fig. 3) (12, 40). Using a flow-focusing microfluidic device, the master mix spiked with a known concentration of Let-7a was partitioned in picoliter-sized water-in-oil droplets (Fig. 3A). The resulting monodisperse emulsion was incubated at 50°C for 200 min, after which the droplets were imaged by fluorescence microscopy (Fig. 3B) and the concentration of the target was computed from the Poisson statistics (fig. S6). We observe a linear correlation between the spiked microRNA concentration and the concentration computed according to the Poisson law (Fig. 3D). The average recovery rate (measured/theoretical concentration) is 110 ± 19%, demonstrating the high accuracy of the quantification over five orders of magnitude. By taking into account the observed limit of blank (fraction of ON droplets in the negative control), concentrations of Let-7a as low as 2.7 fM were successfully detected (cf. also fig. S7). It is important to note that another benefit of the partitioning is that the time necessary to amplify the signal from a single molecule depends only on the volume of the compartments, which sets the concentration of a single target per compartment. For droplets of 0.5 pl (~10 μm in diameter), the concentration of a single molecule corresponds to 3 pM, thus decreasing the incubation time down to ~3 hours (Fig. 2A). The modularity of the DNA circuit enables us to repurpose the molecular program by redesigning solely the cT to the miRNA target of choice (e.g., miR-92a or cel-miR-39 in Fig. 3E), while the other reaction components (aT, pT, and rT) remain unchanged.
Fig. 3. Digital detection of microRNAs.
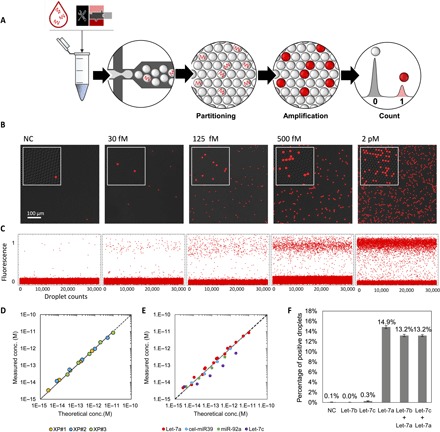
(A) The sample is mixed with the molecular program and partitioned into monodisperse droplets resulting in the random distribution of the microRNA targets among the compartments. After incubation, the droplets are sandwiched between two glass slides and imaged by fluorescence microscopy. The droplets having received at least one target exhibit a positive fluorescence signal (1), while the others remain negative (0), allowing absolute concentration calculation using Poisson statistics. (B) Fluorescence snapshots of emulsified samples spiked with increasing concentrations of Let-7a after amplification. (C) Analysis of 30,000 droplets. (D) Plot showing the measured versus the expected (theoretically spiked) target concentration. The data were obtained from three independent experiments “XP,” and the measured concentration corresponds to the concentration calculated from the Poisson statistics (cf. fig. S6) corrected by the limit of blank (cf. fig. S7). The dotted line is the diagonal. (E) By adapting the cT, the assay is repurposable for other microRNAs. (F) The assay specificity is evaluated from the cross-reactivity of Let-7a over Let-7c (single mismatch) and Let-7b (two mismatches) and in mixtures (1 pM each). Error bars represent the 95% confidence interval over digital quantification.
A major concern related to the quantification of microRNAs is the high sequence homology between microRNA sequences. We thus evaluated the specificity of the current detection method over the Let-7 family. Figure 3F shows a clear discrimination between the Let-7a sequence and analog sequences containing a single (in the case of Let-7c) or two (in the case of Let-7b) mismatched bases. The selectivity of the current method, estimated to be superior to 97%, is among the best ratios described in the literature for microRNA digital assays (41–43). The high selectivity can be explained by the dynamics of the system that needs to produce a concentration of trigger exceeding a given threshold to initiate the amplification. While the production speed of s-strand is maximum for the full matching duplex cT/microRNA, it is considerably reduced with single-base modified targets (fig. S8). The dynamical threshold set by the pT deactivation mechanism acts as an enhancer of the specificity because only activated cT producing triggers at a high rate will eventually initiate the amplification.
Background-free amplification ensures robust digital endpoint analysis
As digital assays rely on an endpoint analysis, it is crucial to have a time window large enough to discriminate the target-containing droplets (exhibiting a positive signal) from the target-free droplets. As mentioned above, most isothermal nucleic acid amplification techniques cannot be transposed to a digital format due to nonspecific reactions that eventually trigger the amplification in all microcompartments, irrespective of the presence of the target (20, 41). Figure 4 shows that in the absence of pT, the droplets turn ON in less than an hour, independently of the presence of the target, the frequency of false-positive droplets (red curve) reaching almost 50%. This result is consistent with a previously described EXPAR system (41). Between 10 and 13 nM of pT, we observe a significant decrease of the false-positive fraction: The pT absorbs part of the leak, but the residual leak is strong enough to unspecifically trigger the amplification in some droplets, resulting in an increase over time of the false-positive ratio. By setting the pT concentration above 16 nM and thus raising the amplification threshold, the self-start was completely suppressed. It is important to note that from 16 to 22 nM of pT, the quantification of Let7a concentration is not affected (green curves). This suggests that the digital assay is tolerant to small variations of the molecular program composition. Because of this amplification threshold, which avoids unwanted amplification, the present method is robust with respect to incubation time, with a time window spanning several hours.
Fig. 4. The elimination of nonspecific amplification reaction allows robust digital detection of microRNA targets.
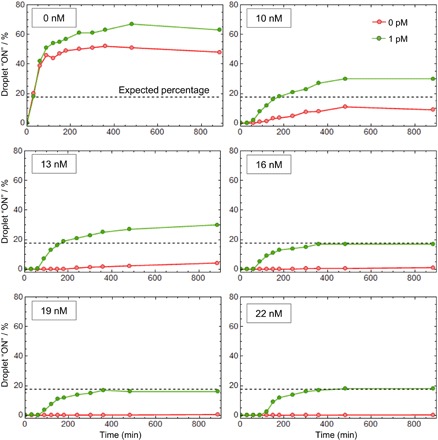
The molecular program with 0 to 22 nM of pT is spiked with 0 or 1 pM of synthetic Let-7a before partitioning. The droplets are incubated at 50°C and imaged by fluorescence at different time points to extract the fraction of positive droplets. The dashed line represents the expected percentage given a target concentration of 1 pM and a droplet diameter of ~9 μm.
Test and validation with total RNA extracts
We then evaluated the possibility to detect endogenous microRNAs from human cells. We quantified the microRNA Let-7a from human colon total RNA using the isothermal digital assay. Figure 5A shows a linear relationship between the total RNA concentration (ranging from 0 to 8 ng/μl) and the measured Let-7a concentration (R2 > 0.99). Similarly, the quantification of Let-7a using RT-qPCR is linearly correlated to the initial extract concentration. Although the quantification of the Let7a microRNA is consistent between both techniques, the measured concentration is significantly higher with the qPCR assay. The quantification by qPCR is estimated from the amplification time (Cq), which is compared to the ones of standard concentrations (fig. S9), assuming the amplification efficiency is the same for both the samples and the standards. However, it has been previously highlighted that imprecision in the PCR calibration or modification of the sample composition significantly affects the quantification (44). This is arguably the origin of the overestimation of the concentration of Let-7a observed here using RT-qPCR. We calculated the precision of our method to be >76% (56% for RT-qPCR, calculated from the mean value over the three extract concentrations tested; Fig. 5B). This is in accordance with previous studies showing the improved accuracy of digital PCR in comparison to qPCR (43–45). As a negative control experiment, the digital quantification of the nonhuman microRNA cel-miR-39ce using the cognate DNA circuit (presented in Fig. 3E) shows the absence of this target in the human extract. Overall, this demonstrates the accuracy of the assay in quantifying endogenous microRNAs.
Fig. 5. microRNA detection from human colon total RNA extract.
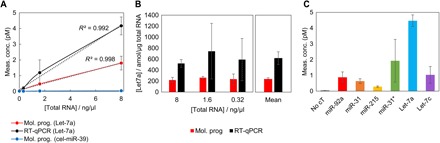
(A) Comparative quantification of microRNAs (Let-7a or miR-39ce) using the proposed molecular program (MP)–based digital assay and RT-qPCR (the error bars are calculated from three independent experiments). (B) The Let-7a concentration (in attomoles per nanogram of total RNA) is calculated for each total RNA concentration, allowing us to determine the precision of the quantification (corresponding to the SD over three replicates of three extract concentrations). (C) Multiple microRNAs are quantified from total RNA extract (10 ng/μl).
DISCUSSION
In conclusion, we present a molecular program–based isothermal amplification strategy that is able to abolish background amplification, thereby enabling digital quantification. The present assay provides sensitive, specific, and quantitative measurement of microRNAs. On the basis of a one-step procedure, it reduces sample manipulations and therefore the risk of carryover contamination. Contamination issues are further reduced by the fact that the system relies on a signal amplification mechanism rather than on the direct replication of the target sequence, as in PCR-based approaches.
Our versatile DNA-based circuit can be theoretically repurposed for any microRNA of interest through the simple redesign of a single oligonucleotide. All other circuit components are identical for all targeted microRNAs, thus eliminating the need for primers and probe design and reducing the assay cost and optimization. We demonstrated the accurate quantification of synthetic target models as well as endogenous microRNA extracted from colon tissues. Preliminary results indicate the possibility to detect these biomolecules directly from plasma samples (fig. S10). However, further investigations are needed to achieve the detection of endogenous circulating biomarkers.
It is of note that the leak absorption mechanism makes the amplification trigger slower compared to some reported EXPAR approaches. In the future, it should be possible to combine it with other leak-reducing amplification strategies [using, for instance, modified templates (27) and specific nickases (26) or by engineering the DNA polymerase to be less leaky (46)]. This would decrease the required concentration of pT needed to absorb the leak and thus speed up the amplification. Nevertheless, the digital format already significantly decreases the assay time. Digitalization also decouples the assay duration from the target concentration, with a typical time down to 3 hours in the current implementation.
Because of its sensitivity and quantitativity, we anticipate that this digital isothermal amplification method may be a promising tool in academic and clinical research.
MATERIALS AND METHODS
Chemicals
Oligonucleotides (templates and synthetic microRNAs) were purchased from Biomers (Germany). The sequences were purified by high-performance liquid chromatography (HPLC) and checked by matrix-assisted laser desorption/ionization mass spectrometry. Templates were designed according to the rules previously described (23, 25). All template sequences (aT, pT, and rT) were protected from the degradation by the exonuclease by 5′ phosphorothioate backbone modifications. A 3′ blocking moiety (phosphate group for aT, pT, and cT and quencher for rT) was used to avoid nonspecific polymerization. Table S1 recapitulates all the sequences used throughout the study. Nb.BsmI and Nt.BstNBI nicking enzymes, Vent(exo-) DNA polymerase, and bovine serum albumin (BSA) were purchased from New England Biolabs (NEB). A 10-fold dilution of Nt.BstNBI was prepared by dissolving the stock enzyme in diluent A (NEB) supplemented with 0.1% Triton X-100. The exonuclease ttRecJ was expressed and purified by chromatography according to a previously published protocol (47). The enzyme was stored at 1.53 μM in diluent A + 0.1% Triton X-100. All the proteins were stored at −20°C. Human colon total RNA (Thermo Fisher Scientific) was aliquoted at 13 μg/ml and stored at −20°C.
RT-qPCR procedure
Quantification was performed by real-time PCR using the Bio-Rad C1000 Touch Thermocycler and CFX96 Real-Time System. A two-step RT-qPCR protocol was performed according to Life Technologies TaqMan MicroRNA Assays instructions and using primers for Let-7a (assay ID, 000377). The reverse transcription program consists of 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. The PCR cycling program consists of 1× at 95°C for 10 min, and 60× cycles of 15 s at 95°C and 1 min at 60°C, for denaturing/annealing plus extensions. Default threshold settings were used as threshold cycle (Ct) data. Ct is the fractional cycle number at which the fluorescence passes the fixed threshold. Ct is presented as mean ± SD for three replicate experiments.
Reaction mixture assembly
All reaction mixtures were assembled at 4°C in 200-μl PCR tubes: The templates were mixed with the reaction buffer [20 mM tris-HCl (pH 7.9), 10 mM (NH4)2SO4, 40 mM KCl, 10 mM MgSO4, 50 μM each dNTP, 0.1% (w/v) Synperonic F 104, 2 μM netropsin, all purchased from Sigma-Aldrich] and the BSA (200 μg/ml). Unless otherwise stated, the template concentration was as follows: aTα, 50 nM; pTα, 15 nM; rTα, 50 nM; cTα, 0.5 nM. After homogenization, the enzymes were added [Nb.BsmI (300 U/ml), Nt.BstNBI (10 U/ml), Vent(exo-) (80 U/ml), ttRecJ (23 nM)]. Each sample was spiked with the microRNA target, serially diluted in 1× tris-EDTA buffer (Sigma-Aldrich) using low-binding DNA tips (Eppendorf). The samples (bulk or emulsion) were incubated at 50°C in a qPCR thermocycler (CFX96 Touch, Bio-Rad), and the fluorescence was recorded in real time. For bulk experiments, the time traces were normalized and Cq (amplification starting times) was determined as 10% of the maximum fluorescence signal.
Droplet generation
A two-inlet (one for the oil, one for the aqueous sample) flow-focusing microfluidic mold was prepared with standard soft lithography techniques using SU-8 photoresist (MicroChem Corp., MA, USA) patterned on a 4-inch silicon wafer. A 10:1 mixture of Sylgard 184 polydimethylsiloxane (PDMS) resin (40 g)/cross-linker (4 g) (Dow Corning, MI, USA) was poured on the mold, degassed under vacuum, and baked for 2 hours at 70°C. After curing, the PDMS was peeled off from the wafer and the inlet and outlet holes of 1.5 mm diameter were punched with a biopsy punch (Integra Miltex, PA, USA). The PDMS layer was bound onto a 1-mm-thick glass slide (Paul Marienfeld GmbH & Co. K.G., Germany) immediately after oxygen plasma treatment. Last, the chip underwent a second baking at 200°C for 5 hours to make the channels hydrophobic (32). The microfluidic chip details are presented in fig. S11. The aqueous sample phase and the continuous phase composed of fluorinated oil (Novec-7500, 3M) containing 1% (w/w) fluorosurfactant (RAN Biotechnologies, MA, USA) were mixed on chip using a pressure pump controller (MFCS-EZ, Fluigent, France) and 200-μm-diameter polytetrafluoroethylene (PTFE) tubing (C.I.L., France) to generate 0.5 pl of droplets by hydrodynamic flow focusing.
Droplet imaging
After incubation, the droplets were imaged by fluorescence microscopy. The bottom slide (76 mm by 52 mm by 1 mm glass slide) was spin-coated with 200 μl of Cytop CTL-809M (Asahi Glass) and dried at 180°C for 2 hours. The emulsion was sandwiched with a 0.17-mm-thick coverslip treated with Aquapel. Ten-micrometer polystyrene particles (Polysciences Inc., PA, USA) were used as spacer to sustain the top slide and avoid the compression of the emulsion. The imaging chamber was finally sealed with an epoxy glue (Sader), and images were acquired using a Nikon Eclipse Ti epifluorescence microscope equipped with a motorized XY stage (Nikon), a Nikon DS-Qi2 camera, an apochromatic 10× (numerical aperture, 0.45) (Nikon), and a CoolLed pE-4000 illumination source. Composite images were generated with the open source ImageJ software. Quantitative data were extracted from the microscopy images using the Mathematica software (Wolfram), following the procedure detailed in fig. S6. The average number of microRNA targets per droplet λ was calculated from the frequency of positive droplets (Fpos) using the Poisson law
This allows the computation of the microRNA concentration
where NA is the Avogadro number and V is the volume of the droplets (in liters). The 95% confidence interval is given by the uncertainty on a binomial proportion
where Fpos and Fneg correspond to the frequency of positive and negative droplets, respectively, and ndrop is the total number of droplets analyzed.
Supplementary Material
Acknowledgments
We thank the reviewers of the 25th International Conference on DNA Computing and Molecular Programming for their insight and advices to improve the quality of this manuscript. Funding: This research was supported by the Université de Recherche Paris Sciences et Lettres (PSL), the ESPCI-Paris, the Université Paris-Descartes, the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM), the Ligue Nationale Contre le Cancer (LNCC, Program “Equipe labelisée LIGUE”; no. EL2016.LNCC/VaT), and the European Research Council (grants 647275 ProFF and 780519 DeepMiR). R.M. received a fellowship from ITMO Cancer within the Frontiers in Life Science PhD program (FdV). We thank the SIRIC CARPEM and the Physicancer program (no. PC201423) fundings. Author contributions: G.G. and R.M. designed the study, performed experiments, and analyzed the data. K.N. and A.-S.K. carried out the optimization of experimental conditions. V.T. provided expertise on droplet microfluidics and microRNA markers and supervised this work. Y.R. conceived, designed, and supervised the study. G.G. and Y.R. wrote the manuscript. All authors discussed the results and commented on the manuscript. Competing interests: Y.R., G.G., R.M., and V.T. are inventors on a patent application related to this work (no. EP 19305669.4, filed 29 May 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaay5952/DC1
Fig. S1. Basic EXPAR system with hot start.
Fig. S2. Detailed chemical reaction network of the molecular program for the detection of microRNAs.
Fig. S3. Bistable amplification switch.
Fig. S4. Experimental condition optimizations.
Fig. S5. Extended data from Fig. 2A.
Fig. S6. Droplet analysis.
Fig. S7. Extended data from Fig. 3D.
Fig. S8. Specificity of the trigger production.
Fig. S9. RT-qPCR calibration.
Fig. S10. Detection of cel-miR-39 in plasma sample.
Fig. S11. Microfluidic chip design.
Table S1. Oligonucleotide sequences used throughout the study.
REFERENCES AND NOTES
- 1.Almeida M. I., Reis R. M., Calin G. A., MicroRNA history: Discovery, recent applications, and next frontiers. Mutat. Res. 717, 1–8 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Wang J., Chen J., Sen S., MicroRNA as biomarkers and diagnostics. J. Cell Physiol. 231, 25–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magee P., Shi L., Garofalo M., Role of microRNAs in chemoresistance. Ann. Transl. Med. 3, 332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosaka N., Iguchi H., Ochiya T., Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 101, 2087–2092 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fesler A., Jiang J., Zhai H., Ju J., Circulating microRNA testing for the early diagnosis and follow-up of colorectal cancer patients. Mol. Diagn. Ther. 18, 303–308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C., Ridzon D. A., Broomer A. J., Zhou Z., Lee D. H., Nguyen J. T., Barbisin M., Xu N. L., Mahuvakar V. R., Andersen M. R., Lao K. Q., Livak K. J., Guegler K. J., Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res. 33, e179 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benes V., Castoldi M., Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available. Methods 50, 244–249 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Raabe C. A., Tang T.-H., Brosius J., Rozhdestvensky T. S., Biases in small RNA deep sequencing data. Nucleic Acids Res. 42, 1414–1426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustin S., Dhillon H. S., Kirvell S., Greenwood C., Parker M., Shipley G. L., Nolan T., Variability of the reverse transcription step: Practical implications. Clin. Chem. 61, 202–212 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Aslanzadeh J., Preventing PCR amplification carryover contamination in a clinical laboratory. Ann. Clin. Lab. Sci. 34, 389–396 (2004). [PubMed] [Google Scholar]
- 11.Opel K. L., Chung D., McCord B. R., A study of PCR inhibition mechanisms using real time PCR. J. Forensic Sci. 55, 25–33 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Vogelstein B., Kinzler K. W., Digital PCR. Proc. Natl. Acad. Sci. U.S.A. 96, 9236–9241 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein E. V., Duewer D. L., Farkas N., Romsos E. L., Wang L., Cole K. D., Steps to achieve quantitative measurements of microRNA using two step droplet digital PCR. PLOS ONE 12, e0188085 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y., Chen F., Li Q., Wang L., Fan C., Isothermal amplification of nucleic acids. Chem. Rev. 115, 12491–12545 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Ness J. V., Ness L. K. V., Galas D. J., Isothermal reactions for the amplification of oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 100, 4504–4509 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia H., Li Z., Liu C., Cheng Y., Ultrasensitive detection of microRNAs by exponential isothermal amplification. Angew. Chem. Int. Ed. 49, 5498–5501 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Du W., Lv M., Li J., Yu R., Jiang J., A ligation-based loop-mediated isothermal amplification (ligation-LAMP) strategy for highly selective microRNA detection. Chem. Commun. 52, 12721–12724 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Harcourt E. M., Kool E. T., Amplified microRNA detection by templated chemistry. Nucleic Acids Res. 40, e65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao X., Ning X., Li Z., Cheng Z., Sensitive detection of miRNA by using hybridization chain reaction coupled with positively charged gold nanoparticles. Sci. Rep. 6, 32358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan E., Erwin B., Dames S., Ferguson T., Buechel M., Irvine B., Voelkerding K., Niemz A., Specific versus nonspecific isothermal DNA amplification through thermophilic polymerase and nicking enzyme activities. Biochemistry 47, 9987–9999 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian J., Ferguson T. M., Shinde D. N., Ramírez-Borrero A. J., Hintze A., Adami C., Niemz A., Sequence dependence of isothermal DNA amplification via EXPAR. Nucleic Acids Res. 40, e87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J., Zhou X., Ma Y., Lin X., Dai Z., Zou X., Asymmetric exponential amplification reaction on a toehold/biotin featured template: An ultrasensitive and specific strategy for isothermal microRNAs analysis. Nucleic Acids Res. 44, e130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Zou B., Rui J., Song Q., Kajiyama T., Kambara H., Zhou G., Exponential amplification of DNA with very low background using graphene oxide and single-stranded binding protein to suppress non-specific amplification. Microchimica Acta 182, 1095–1101 (2015). [Google Scholar]
- 24.Mok E., Wee E., Wang Y., Trau M., Comprehensive evaluation of molecular enhancers of the isothermal exponential amplification reaction. Sci. Rep. 6, 37837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid M. S., Paliwoda R. E., Zhang H., Le X. C., Reduction of background generated from template-template hybridizations in the exponential amplification reaction. Anal. Chem. 90, 11033–11039 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Urtel G., Van Der Hofstadt M., Galas J.-C., Estevez-Torres A., rEXPAR: An isothermal amplification scheme that is robust to autocatalytic parasites. Biochemistry 58, 2675–2681 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komiya K., Komori M., Noda C., Kobayashi S., Yoshimura T., Yamamura M., Leak-free million-fold DNA amplification with locked nucleic acid and targeted hybridization in one pot. Org. Biomol. Chem. 17, 5708–5713 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Olson X., Kotani S., Padilla J. E., Hallstrom N., Goltry S., Lee J., Yurke B., Hughes W. L., Graugnard E., Availability: A metric for nucleic acid strand displacement systems. ACS Synth. Biol. 6, 84–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin P., Choi H. M. T., Calvert C. R., Pierce N. A., Programming biomolecular self-assembly pathways. Nature 451, 318–322 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Seelig G., Soloveichik D., Zhang D. Y., Winfree E., Enzyme-free nucleic acid logic circuits. Science 314, 1585–1588 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Qian L., Winfree E., Scaling up digital circuit computation with DNA strand displacement cascades. Science 332, 1196–1201 (2011). [DOI] [PubMed] [Google Scholar]
- 32.C. Thachuk, E. Winfree, D. Soloveichik, DNA computing and molecular programming, in Lecture Notes in Computer Science, A. Phillips, P. Yin, Eds. (Springer International Publishing, 2015), pp. 133–153. [Google Scholar]
- 33.Jiang Y. S., Bhadra S., Li B., Ellington A. D., Mismatches improve the performance of strand-displacement nucleic acid circuits. Angew. Chem. Int. Ed. 53, 1845–1848 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Padirac A., Fujii T., Rondelez Y., Nucleic acids for the rational design of reaction circuits. Curr. Opin. Biotechnol. 24, 575–580 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Li F., Xiao M., Pei H., DNA-based chemical reaction networks. Chembiochem 20, 1105–1114 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Montagne K., Plasson R., Sakai Y., Fujii T., Rondelez Y., Programming an in vitro DNA oscillator using a molecular networking strategy. Mol. Syst. Biol. 7, 466 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baccouche A., Montagne K., Padirac A., Fujii T., Rondelez Y., Dynamic DNA-toolbox reaction circuits: A walkthrough. Methods 67, 234–249 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Montagne K., Gines G., Fujii T., Rondelez Y., Boosting functionality of synthetic DNA circuits with tailored deactivation. Nat. Commun. 7, 13474 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su J.-L., Chen P.-S., Johansson G., Kuo M.-L., Function and regulation of Let-7 family microRNAs. MicroRNA 1, 34–39 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Hindson C. M., Chevillet J. R., Briggs H. A., Gallichotte E. N., Ruf I. K., Hindson B. J., Vessella R. L., Tewari M., Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 10, 1003–1005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K., Kang D.-K., Ali M. M., Liu L., Labanieh L., Lu M., Riazifar H., Nguyen T. N., Zell J. A., Digman M. A., Gratton E., Li J., Zhao W., Digital quantification of miRNA directly in plasma using integrated comprehensive droplet digital detection. Lab Chip 15, 4217–4226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen L., Hartman M. R., Amardey-Wellington A., Walt D. R., Digital direct detection of microRNAs using single molecule arrays. Nucleic Acids Res. 45, –e137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian H., Sun Y., Liu C., Duan X., Tang W., Li Z., Precise quantitation of MicroRNA in a single cell with droplet digital PCR based on ligation reaction. Anal. Chem. 88, 11384–11389 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Campomenosi P., Gini E., Noonan D. M., Poli A., D’Antona P., Rotolo N., Dominioni L., Imperatori A., A comparison between quantitative PCR and droplet digital PCR technologies for circulating microRNA quantification in human lung cancer. BMC Biotechnol. 16, 60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conte D., Verri C., Borzi C., Suatoni P., Pastorino U., Sozzi G., Fortunato O., Novel method to detect microRNAs using chip-based QuantStudio 3D digital PCR. BMC Genomics 16, 849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghadessy F. J., Ong J. L., Holliger P., Directed evolution of polymerase function by compartmentalized self-replication. Proc. Natl. Acad. Sci. U.S.A. 98, 4552–4557 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamagata A., Masui R., Kakuta Y., Kuramitsu S., Fukuyama K., Overexpression, purification and characterization of RecJ protein from Thermus thermophilus HB8 and its core domain. Nucleic Acids Res. 29, 4617–4624 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaay5952/DC1
Fig. S1. Basic EXPAR system with hot start.
Fig. S2. Detailed chemical reaction network of the molecular program for the detection of microRNAs.
Fig. S3. Bistable amplification switch.
Fig. S4. Experimental condition optimizations.
Fig. S5. Extended data from Fig. 2A.
Fig. S6. Droplet analysis.
Fig. S7. Extended data from Fig. 3D.
Fig. S8. Specificity of the trigger production.
Fig. S9. RT-qPCR calibration.
Fig. S10. Detection of cel-miR-39 in plasma sample.
Fig. S11. Microfluidic chip design.
Table S1. Oligonucleotide sequences used throughout the study.