

Abstract
The oxidized platinum (Pt) can exhibit better electrocatalytic activity than metallic Pt0 in the hydrogen evolution reaction (HER), which has aroused great interest in exploring the role of oxygen in Pt-based catalysts. Herein, we select two structurally well-defined polyoxometalates Na5[H3Pt(IV)W6O24] (PtW6O24) and Na3K5[Pt(II)2(W5O18)2] (Pt2(W5O18)2) as the platinum oxide model to investigate the HER performance. Electrocatalytic experiments show the mass activities of PtW6O24/C and Pt2(W5O18)2/C are 20.175 A mg−1 and 10.976 A mg−1 at 77 mV, respectively, which are better than that of commercial 20% Pt/C (0.398 A mg−1). The in situ synchrotron radiation experiments and DFT calculations suggest that the elongated Pt-O bond acts as the active site during the HER process, which can accelerate the coupling of proton and electron and the rapid release of H2. This work complements the knowledge boundary of Pt-based electrocatalytic HER, and suggests another way to update the state-of-the-art electrocatalyst.
Subject terms: Electrocatalysis, Inorganic chemistry, Hydrogen fuel, Electrocatalysis
While converting water to H2 with a catalyst offers a renewable means to produce carbon-neutral fuels, understanding the catalytic active sites has proven challenging. Here, authors show a structurally well-defined model complex with Pt-O bonding to enable efficient H2 evolution electrocatalysis.
Introduction
Platinum (Pt) is generally considered as a state-of-the-art electrocatalyst for the hydrogen evolution reaction (HER)1–8. In recent decades, enormous efforts have been made to design Pt-based catalysts to boost the utilization and catalytic efficiency of Pt through the composition, morphology, and crystal phase-engineering strategies9–13. Most of these studies have revealed the inherent catalytic activity of Pt0 metal14–23; however, some interesting phenomena involved in oxidized platinum are still far from being studied. For example, when the metal Pt is oxidized, its HER activity can be obviously better than that of metal element Pt0 (Supplementary Figs. 1 and 2). The reason has not been clearly clarified so far. Recently, great efforts have been made to prepare platinum oxide models such as PtOx/TiO224 and MoSx-O-PtOx25 and confirmed that the presence of oxygen in Pt catalysts did possess superior HER catalytic performance comparable to that of commercial Pt/C and even superior to that of MoSx-Pt. These pioneering works inspired chemists to find more suitable and distinct platinum oxide models so as to reveal the role of oxygen in Pt-based electrocatalysts and develop new efficient electrocatalysts superior to commercial Pt/C.
Considering that polyoxometalates (POMs) are a unique type of nanoscale metal-oxo clusters with definite structures26–29 and can be used to simulate the surface of metal oxides30,31, the Pt-containing POMs could be a readily available and ideal platinum oxide model to investigate the electrocatalytic HER. Therefore, we selected two structurally well-defined Pt-containing POMs Na5[H3Pt(IV)W6O24] (abbr. PtW6O24) and Na3K5[Pt(II)2(W5O18)2] (abbr. Pt2(W5O18)2) as model catalysts to investigate their HER performance. Electrochemical experiments show that PtW6O24/C and Pt2(W5O18)2/C electrocatalysts with 1 wt% Pt content exhibit superior catalytic activities. The overpotentials of PtW6O24/C and Pt2(W5O18)2/C with 1 wt% Pt content are 22 and 26 mV at 10 mA cm−2, and their mass activities are 20.175 and 10.976 A mg−1 at an overpotential of 77 mV, respectively, which are better than that of commercial 20% Pt/C (0.398 A mg−1). A series of control experiments, in situ synchrotron radiation experiments, and density functional theory (DFT) calculations suggest that the Pt-O bond in POMs should be the active site for HER. Specifically, Pt is mainly an electron-obtaining center, while its coordinated O atoms are proton-capturing centers. During the HER process, when more electrons and protons were injected, the elongated Pt-O bond accelerates the coupling of protons and electrons, which leads to the rapid release of H2 from the Pt-O bond.
Results
Structure and electronic property of Na5[H3PtW6O24] (PtW6O24)
PtW6O24 compound was synthesized according to the literature32. As depicted in Fig. 1a and Supplementary Figs. 3 and 4, the structure of PtW6O24 is constructed by a {PtO6} octahedron connected to six {WO6} octahedra in an edge-sharing mode. The center Pt atom is surrounded by six O atoms forming the {PtO6} octahedron with a Pt-O bond length of 2.005–2.020 Å. As the pH decreases during synthesis, the number of protons on PtW6O24 cluster increases (Supplementary Table 1). Furthermore, PtW6O24 exhibits excellent stability in a wide pH range (0–6) and under different potentials at room temperature (Supplementary Figs. 5 and 6). The electrospray-ionization mass spectra (EIS) and capillary electrophoresis (CE) show that PtW6O24 compound has reversible redox-active property and good stability (Supplementary Figs. 7 and 8).
Fig. 1. Structure and electronic properties of PtW6O24.

a The ball and stick representation of PtW6O24 with the corresponding semitransparent map of molecular electrostatic potential (MEP). b, c High-resolution XPS spectra of Pt and O for PtW6O24. d 3D representation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for [H6PtW6O24] and [H6PtW6O24]2e. Additional reduced states are given in Supplementary Fig. 14. e Full and one-half views of the MEP distribution for [H6PtW6O24] and [H6PtW6O24]2e: the red and blue or green identify more and less nucleophilic regions for H+. Supplementary Table 6 discusses the proton distributions.
X-ray photoelectron spectroscopy (XPS) also shows the definite oxidized platinum feature (Fig. 1 and Supplementary Fig. 9). In the high-resolution XPS spectra of Pt (Fig. 1b), peaks for Pt 4f7/2 and Pt 4f5/2 are located at 73.9 and 77.3 eV, respectively, which are in accordance with the presence of Pt(IV) as reported in the literature. It is noteworthy that there are no signals at 71.4 and 74.7 eV, indicating the absence of metallic Pt33. The XPS spectra of O 1s is depicted in Fig. 1c, the peaks at 529.6 and 530.7 eV belong to W=O and W-O-W bonds, respectively. The peaks belong to 532.1 and 533.5 eV can be attributed to the protonation of the Pt-OH-W bond and crystalline H2O, respectively34. These experiments further demonstrate that PtW6O24 can be a platinum oxide model to carry out the electrochemical study.
First, the electrochemical cyclic voltammetry (CV) experiments of PtW6O24 were studied in acetonitrile and 0.5 M H2SO4 aqueous solution, respectively35,36. As depicted in Supplementary Fig. 10a, PtW6O24 exhibits a series of obvious redox peaks in the range of −1.5 to 2.0 V in acetonitrile. Specifically, the two redox peaks at 0.68 and 1.02 V are ascribed to the stepwise Pt(III) → Pt(II) and Pt(IV) → Pt(III) processes, respectively (Supplementary Figs. 10–13). The redox peak at −1.2 V is attributed to the reduction of tungsten. The CV results were further simulated by the DFT calculation, which showed that the injection of the initial two electrons mainly occurs on the Pt center (Fig. 1d and Supplementary Fig. 14b), leading to a decrease of the oxidation state from PtIV to PtII. The DFT calculation further suggests that after PtW6O24 species were 2e-reduced, the main contribution to the LUMO became the d orbitals of the surrounding W centers, meaning that the third-reduction step will occur on W instead of Pt. Both CV experiments and DFT calculation reveal that the Pt center in PtW6O24 can more easily obtain electrons than W in PtW6O24, but no metallic Pt0 is generated in the whole electrochemical process. The CV curve of PtW6O24 measured in 0.5 M H2SO4 is obviously different from that in acetonitrile (Supplementary Fig. 10b). Only the redox peaks of platinum can be detected, and then the hydrogen evolution signal arises and covers the redox peak region of tungsten. Moreover, the molecular electrostatic potential (MEP, see Fig. 1e) maps suggest O atoms on PtW6O24 species should be the main proton-capturing centers (because the red area of O atoms in Fig. 1e represents the most basic positions), which may serve as proton transfer stations to continuously supply H2 generation. All above results imply that PtW6O24 may function as an efficient HER catalyst.
Electrocatalytic HER performance of PtW6O24/C
The HER performance of PtW6O24 was assessed by fabricating a PtW6O24/C composite electrocatalyst. Its preparation method and physical characterization are shown in Supplementary Figs. 15–21. The inductively coupled plasma-atom emission spectrometry (ICP-AES) (Supplementary Table 2) demonstrated a 1 wt% Pt content in PtW6O24/C. The electrocatalytic activity of 1% PtW6O24/C for the HER was evaluated and compared to commercial 20% Pt/C in N2-saturated 0.5 M H2SO4. The polarization curves in Fig. 2a and Supplementary Fig. 22 show that 1% PtW6O24/C exhibits excellent HER activity with overpotentials of 22, 55, and 65 mV at current densities of 10, 70, and 100 mA cm−2, respectively, which are better than those of commercial 20% Pt/C (33, 90, and 118 mV) and 1% Pt/C (68, 269, and 357 mV), and exceed most of the reported Pt-based catalysts (Supplementary Fig. 23 and Table 7). As a comparison, series of PtW6O24/C with different loading amount of Pt have been prepared. As described in Supplementary Fig. 24, the HER performance of catalysts was obviously enhanced with the increase of Pt loading. However, when the loading amount of Pt reaches 5%, the electrocatalytic activity did not increase significantly. It is presumed that the partial aggregation of PtW6O24 species results in a decrease in the utilization of the catalyst. In addition, as described in Fig. 2b and Supplementary Fig. 25, the Tafel slope of 1% PtW6O24/C is 29.8 mV dec−1, which is consistent with the Volmer–Tafel mechanism, in which the recombination of chemisorbed H atoms and ions is the rate-determining step37. Furthermore, the 1% PtW6O24/C exhibits an exchange current density of 1.65 mA cm−2 (Supplementary Fig. 26), which means a superior intrinsic electrocatalytic activity. The mass activity and specific activity were normalized by the mass loading and the electrochemical surface area (ECSA) of Pt. As depicted in Fig. 2c, at an overpotential of 77 mV, PtW6O24/C displays a mass activity of 20.175 A mg−1, while the mass activity of 20% Pt/C is 0.398 A mg−1. Furthermore, 1% PtW6O24/C displays a specific activity of 35.266 mA cm−2 at 50 mV, and the value of 20% Pt/C is 0.132 mA cm−2 under the same condition. As shown in Supplementary Fig. 27, the turnover frequencies (TOFs) displays a near linear increase with the overpotential. At an overpotential of 100 mV, 1% PtW6O24/C exhibits a high TOFs of 33.35 s−1, which is a 58.5-fold increase over 20% Pt/C (0.57 s−1). Figure 2d indicates the relationship between the current density, the overpotential and the Pt content, demonstrating that the current density of 1% PtW6O24/C is obviously better than that of 20% Pt/C at all overpotentials. Even compared with Pt/C with a higher Pt content, its value is also better in most of the overpotentials.
Fig. 2. HER performance of the PtW6O24 catalyst.

a The polarization curves of 1% PtW6O24/C and 20% Pt/C in N2-saturated 0.5 M H2SO4. b Tafel slope of 1% PtW6O24/C and 20% Pt/C. c Mass activity of PtW6O24/C and 20% Pt/C at 77 mV (left), the specific activity of 1% PtW6O24/C and 20% Pt/C at 50 mV (right). d The relationship between the current density, the overpotential, and the Pt content. The blue area indicates that the current density of PtW6O24/C and 20% Pt/C is the same. The yellow area indicates that the current density of PtW6O24/C is greater than Pt/C, and the gray area indicates that the current density of PtW6O24/C is lower than Pt/C. e The polarization curves of 1% PtW6O24/C before and after 1000 and 3000 cycles at scan rate of 5 mV s−1. Inset: Time-dependent current density current of 1% PtW6O24/C and 20% Pt/C within 24 h (left). The locally enlarged plot of the time-dependent current density current for the first 0.5 h (right). f In situ XANES spectra of the 1% PtW6O24/C sample at Pt L3-edge. Feature A is attributed to Pt L3-edge and feature B is attributed to W L2-edge.
Electrochemical impedance spectroscopy (EIS) are depicted in Supplementary Figs. 28 and 29 and suggest that 1% PtW6O24/C possesses an extremely low charge transfer resistance (Rct = 6.7 Ω), which is obviously lower than that of 20% Pt/C (Rct = 46.8 Ω), demonstrating the fast Faradaic process between the interface of the catalysts and electrolyte. The Faradic efficiency of 1% PtW6O24/C is nearly 100% for the HER, resulting in the high evolution efficiency of H2 (Supplementary Fig. 30). As shown in Supplementary Fig. 31 (anti-toxicity test), the activity of 1% PtW6O24/C exhibits negligible changes in the presence of Co2+, Fe2+, Mn2+, and Ni2+ ions, while the performance of 20% Pt/C decreases obviously after three cycles. Such results indicate that 1% PtW6O24/C possesses good anti-toxicity properties.
Besides the aforementioned features, stability is also an important factor for evaluating an excellent electrocatalyst. The accelerated degradation test (ADT) in Fig. 2e was used to estimate the electrocatalytic durability. After 1000 and 3000 cycles, the polarization curves of PtW6O24/C show a slight loss. In addition, the long-term stability test for PtW6O24/C was carried out at an overpotential of 30 mV for 24 h (Fig. 2e, inset). The current density exhibits a small loss, demonstrating the good electrocatalytic stability of PtW6O24/C. The Pt content in PtW6O24/C shows negligible loss before and after the HER, indicating no dissolution of the catalyst during the electrocatalytic process (Supplementary Table 3). The Transmission electron microscopy (TEM) images of PtW6O24/C after long-term electrochemical test demonstrate that its morphology stays the same without aggregation, meaning the good stability of PtW6O24/C (Supplementary Fig. 32). Infrared (IR) spectra of 1% PtW6O24/C after long-term electrocatalytic tests also suggest that 1% PtW6O24/C is stable during the electrochemical reaction process (Supplementary Fig. 33).
Electrocatalytic mechanism of PtW6O24/C
First, a series of control experiments were studied to understand the origins of the excellent HER activities of PtW6O24/C. All the reference catalysts such as LaW10/C, Kenjet black, and Pt(C2H5N4O2)2/C exhibit poor HER activities. These results indicate that PtW6O24/C has excellent HER catalytic properties due to the existence of {PtO6} core (Supplementary Fig. 34).
Furthermore, in situ X-ray adsorption spectroscopy (XAS) was performed under pretreatment and electrocatalytic conditions to gain insight into the electronic state variation of Pt. As shown in Fig. 2f, in initial (before HER) state, the Pt K-edge X-ray absorption near edge structure (XANES) spectrum of PtW6O24/C show similar intensity to the Na2Pt(OH)6 reference, manifesting that the Pt element in PtW6O24/C takes on a similar valence state as in Na2Pt(OH)6. On the other hand, a shift in the edge position to lower energy was observed when moved to HER condition, and the intensity for Pt was significantly decreased. This indicates a reduction in the Pt oxidation state. In addition, the XAS during HER was found closely matching with K2PtCl4 species, strongly suggesting the existence of PtII electronic state during the HER catalytic process. These results demonstrate that the mechanism of the HER process could be attributed to the formation of the PtII intermediate. The valence state of Pt decreased from Pt(IV) to Pt(II), and then the state was maintained between Pt(II) and Pt(I), which is consistent with the results of CV tests. Feature B is due to the interference of W L2-edge, which does not affect the analysis of main peak of PtW6O24/C. During the HER process, the intensity of feature B also decreased, indicating that W might also participate in the HER reaction. This result indicates that no metallic Pt0 is formed during the electrocatalytic HER process. The Pt-O bonds always exist in the PtW6O24/C catalyst, except for the valence state changes of Pt. This result is consistent with the above experimental data, as well as the previous literatures35,36.
DFT calculations were employed at the M06 level to survey the detailed pathways of H2 generation (Fig. 3). [H6PtW6O24] can engage in multiple reduction and protonation reactions under electrocatalytic conditions (Supplementary Table 6). However, H2 evolution from the 2e-reduction state ([H6PtW6O24]2e/2H) is completely restricted due to the high energy demand of 2.41 eV (Supplementary Fig. 35c). Among the considered routes, the most accessible catalytic cycle is proposed to be through the highly reduced [H6PtW6O24]4e/4H state to generate H2 and then regenerate [H6PtW6O24]2e/2H (Fig. 3a). Noticeably, two Pt-O bonds are apparently weakened (or elongated) by the two-electron reduction; therefore, providing an available site for H attack (Supplementary Fig. 14) to form a Pt-H state. The configuration with one H on the Pt site ([H6PtW6O24]4e/4H(Pt)) was revealed to be 0.04 eV energetically more favorable than on the O ([H6PtW6O24]4e/4H(O)) due to a fast intramolecular electronic rearrangement process (Supplementary Figs. 35b and 36). Starting from the [H6PtW6O24]4e/4H(Pt) intermediate, a transition state (TS) search demonstrated that H2 production is kinetically promising with a barrier of only 0.15 eV, and the singlet-state [H6PtW6O24]2e/2H can be spontaneously reconstituted via an exothermic process (Fig. 3b). Our calculations assign a partial charge of QH = 0.12 to the H in the Pt-H moiety (clearly smaller than that on the OH moiety QH = 0.47), supplying the active H and combining with adjacent proton from O to encourage H2 evolution (Fig. 3b, inset). In addition, the catalytic cycle is computed to be catalytically efficient between the Pt(II) and Pt(I)-involved intermediates (Supplementary Fig. 36), and these charge assignments are consistent with the trends obtained in XAS.
Fig. 3. H2 evolution pathways over [PtW6O24].
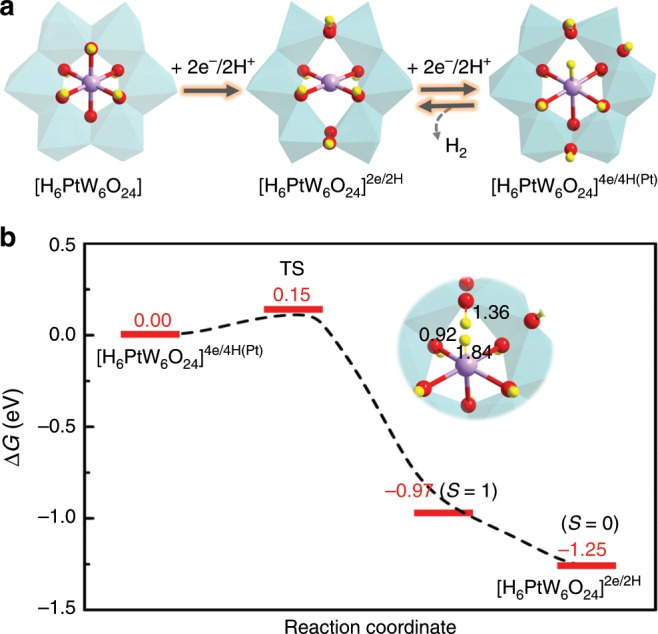
a Mechanistic scheme of the HER catalyzed by PtW6O24/C. [H6PtW6O24] experiences a two-electron/proton-coupled reduction to form [H6PtW6O24]2e/2H (PtII), agreeing well with the intermediate detected by XAS. Two further reductions and intramolecular electronic recombination (Supplementary Fig. 35) were suggested to generate the active [H6PtW6O24]4e/4H(Pt). Catalytic H2 formation is proposed to occur between [H6PtW6O24]2e/2H and [H6PtW6O24]4e/4H. Other pathways are discussed in Supplementary Fig. 35c. b Free energy diagrams for H2 production with an extremely low barrier. H is colored with yellow for clarity.
Discussion
Based on the DFT and XAS results, [H6Pt(II)W6O24]2e/2H may represent an important intermediate for the high HER performance of H6PtW6O24. This result aroused our curiosity to detect the electrocatalytic activity of another POM Na3K5[Pt(II)2(W5O18)2] (Pt2(W5O18)2), since it contains a similar PtII-O moiety in the molecular structure. The overpotential of 1% Pt2(W5O18)2/C is 22 mV at 10 mA cm−2. Its exchange current density and mass activity at 77 mV are 1.65 mA cm−2 and 20.175 A mg−1, respectively, which are quite close to PtW6O24. More detailed data are provided in the Supplementary Figs. 37–57 and Supplementary Tables 4 and 5)38. This result further confirms that Pt-O bond is the active site during the HER process. Herein, it should be also clarified that although commercial Pt/C is widely used as a standard reference for HER research, its performance is already not the best. No matter in size, morphology, and dispersion of metal Pt, there exists enough space to improve its catalytic activity9–14,39. Thus, surpassing commercial Pt/C does not mean that metal Pt-based catalysts are out of date, which exactly suggests an important driving force for deeply developing such state-of-the-art catalysts.
In summary, we selected two structurally well-defined Pt-containing POMs as the platinum oxide model to reveal the role of O atom in Pt-based electrocatalysts towards HER. The electrochemical experiments show that PtW6O24/C and Pt2(W5O18)2/C possess the overpotentials of 22 and 26 mV at a current density of 10 mA cm−2, and their mass activities are 20.175 and 10.976 A mg−1, respectively. In situ XAS experiments and DFT calculations suggest that the Pt-O bond should be the active site for the HER. Specifically, Pt is mainly an electron-obtaining center, while the O acts as the proton-adsorption center. When extra electrons and protons were injected during electrochemical process, the elongated Pt-O site accelerates the coupling of electron and proton and leads to a rapid release of H2 on the Pt-O bond. Therefore, Pt-O can be utilized as a new active site towards HER. This work answers the important role of O atoms in the oxidized platinum-based electrocatalytic HER, which may bring an another enlightenment for the design and preparation of more efficient Pt-based electrocatalysts in the near future.
Methods
Characterization
Single-crystal X-ray diffraction data for Pt-POMs was collected by using a Bruker Smart Apex CCD diffractometer with Mo-Kα radiation (λ = 0.71073 Å) at the temperature of 298(2) K. Powder X-ray diffraction measurements were carried out on a Rigaku D/max-IIB X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å). TEM and high-resolution TEM images were obtained on JEOL-2100F and JEM-F 200 instruments at an accelerating voltage of 200 kV. Scanning transmission electron microscopy (STEM) images were obtained on a Titan Cubed Themis G2 300 equipped with a probe corrector and HF5000. The XPS measurements were performed on a KRATOS Axis ultra DLD X-ray photoelectron spectrometer with a monochromatized Mg Kα X-ray source (hυ = 1283.3 eV). XANES and extended X-ray absorption fine structure (EXAFS) data were collected on the BL14W beamline at the Shanghai Synchrotron Radiation Facility, operated at 3.5 GeV with injection currents of 140–210 mA. The ICP-AES elemental analyses were performed on a Teledyne Leeman Labs ICP-AES spectrometer. Electrochemical measurements and electrocatalytic HER performance were tested by using a CHI760E workstation (CH Instruments, China). The evolved gases during HER decomposition were detected by gas chromatography (Shimadzu, GC-2014C with a thermal conductivity detector). K2PtCl4, Na2Pt(OH)6, Na2WO4·2H2O, La(NO3)3·6H2O, and Ketjen black carbon were purchased from Aladdin Industrial Co., Ltd. Nafion solution (5 wt%) and commercial 40% Pt/C, 20% Pt/C, and 5% Pt/C were purchased from Alfa Aesar China (Tianjin) Co., Ltd. All solution used in experiments were prepared with Millipore water (18.2 MΩ).
Synthesis of PtW6O24/C
Crystal (0.065 g) of Na5[H3Pt(IV)W6O24] was uniformly dispersed in 1 mL H2O, and 5 mg Ketjen black carbon was added to, stirring at room temperature for 2 h. Then, 10 µL melamine-formaldehyde was added to the aqueous and stirred 4 h. The electrocatalyst can be obtained after centrifuged and dried. The obtained sample is denoted as PtW6O24/C.
Synthesis of Pt2(W5O18)2/C
Crystal (0.065 g) of Na3K5[Pt(II)2(W5O18)2] was uniformly dispersed in 1 mL H2O, and 5 mg Ketjen black carbon was added to, stirring at room temperature for 2 h. Then, 10 µL melamine-formaldehyde was added to the aqueous and stirred 4 h. The electrocatalyst can be obtained after centrifuged and dried. The obtained sample is denoted as Pt2(W5O18)2/C.
Electrochemical characterization of Pt-POMs
All reagents are guaranteed reagent and chemicals are of high-purity grade, which were purchased from Aladdin Industrial Co., Ltd. The electrolyte was 0.05 M tetrabutylammonium perchlorate/CH3CN deoxygenated thoroughly for 30 min with pure nitrogen and keep under a positive pressure of this gas during the electrochemical tests. The working electrode was well clean bare glassy carbon. The platinum wire was used as the counter electrode. Non-aqueous Ag/Ag+ electrode served as reference electrode. The filling solution of non-aqueous Ag/Ag+ electrode was 0.01 M AgNO3/CH3CN. All electrochemical tests were carried out by using a CHI760E workstation and performed at room temperature under atmospheric pressure.
Electrochemial measurements of HER
HER tests were carried out a conventional three-electrode electrochemical system in N2-saturated 0.5 M H2SO4 at 300 K. A modified glassy carbon electrode (GCE; d = 3 mm) was used as the working electrode. A saturated calmoel electrode (SCE) and a carbon rod served as reference electrode and counter electrode, respectively. All electrochemical tests were carried out by using a CHI760E workstation. Polarization curves were tested at a scan rate of 5 mV s−1 in 0.5 M H2SO4. The measured potentials vs. SCE were standardized with a reversible hydrogen electrode (RHE) based on E vs. RHE = EθSCE + 0.059 pH (EθSCE = 0.242 V). All data are presented with IR compensation. the electrochemical double layer capacitance (Cdl) was evaluated by cyclic voltammogram (CV) from 0.1 to −0.1 V with different scan rates. The ADT was measured by CV with sweeps at 100 mV at between +0.1 and −0.2 V vs. RHE. The long-term stability was measured at controlled potential.
DFT computational details
All calculations were performed through the facilities provided by the Gaussian09 package (revision D.01)40. Geometry optimizations for all intermediates and TSs were carried out at the M06 level without symmetry restrictions41. The LANL2DZ basis set was employed for the Pt and W, whereas the 6–31G(d, p) basis set was used for the O and H42,43. To confirm the stability of all structures, frequency calculations were performed at the same level as optimization. The TSs were confirmed by the existence of only one imaginary frequency along the reaction coordinate and intrinsic reaction coordinate (IRC) calculations, which indeed connect the right reactants and products (Supplementary Fig. S56)44. The solvation effects of water were introduced by using the PCM model45. Furthermore, the single-point energies of all stationary points were completed at (U)M06/PCM(H2O)/[6–311++G(d,p)/SDD(Pt&W)] level for all energy calculations46. Optimized coordinates (xyz) for all related species are performed Supplementary Table 8. Finally, a data set of computational results is available in the ioChem-BD repository and can be accessed via 10.19061/iochem-bd-6-27 (http://www.iochembd.org/)47.
Supplementary information
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (grant nos. 21671036, 21771033, 51725204, 21771132, 21471106, 51972216), National MCF Energy R&D Program (2018YFE0306105), the Fundamental Research Funds for the Central Universities (grant nos. 2412018BJ001, 2412018ZD007, and 2412018QD005), the Opening Project of Key Laboratory of Polyoxometalate Science of the Ministry of Education (grant no. 130014556), the China Postdoctoral Science Foundation funded project (grant no. 2018M631849), the Foundation of Jilin Educational Committee (grant no. JJKH20190268KJ), and the Scientific Development Project of Jilin Province (grant no. 20190201206JC). The Natural Science Foundation of Jiangsu Province (BK20190041, BK20190828), Guangdong Province Key Area R&D Program (2019B010933001), Collaborative Innovation Center of Suzhou Nano Science and Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the 111 Project. This article is dedicated to the memory of our most beloved supervisor Professor En-Bo Wang, who passed away on 28 September 2019.
Author contributions
Y.-G.L., Z.-H.K., and H.Q.T. conceived the whole experiments. F.Y.Y. performed the main experiments. Z.L.Z. performed all the DFT calculations. L.Y.Y. performed the CE experiments. K.F., Y.J.X. and J.Z. performed the in situ XAS experiments. H.-T.Z. solved the crystals. Y.-G.L. and H.-Q.T. polished the whole paper. All authors discussed the results and implications and commented on the manuscript at all stages.
Data availability
All relevant data are available from the corresponding author on request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Ira Weinstock and other, anonymous, reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Fei-Yang Yu, Zhong-Ling Lang.
Contributor Information
Hua-Qiao Tan, Email: tanhq870@nenu.edu.cn.
Zhen-Hui Kang, Email: zhkang@suda.edu.cn.
Yang-Guang Li, Email: liyg658@nenu.edu.cn.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-019-14274-z.
References
- 1.Tyo EC, Vajda S. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 2015;10:577–588. doi: 10.1038/nnano.2015.140. [DOI] [PubMed] [Google Scholar]
- 2.Stamenkovic VR, et al. Energy and fuels from electrochemical interfaces. Nat. Mater. 2017;16:57–69. doi: 10.1038/nmat4738. [DOI] [PubMed] [Google Scholar]
- 3.She ZW, et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science. 2017;355:eaad4998. doi: 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- 4.Wang, P. et al. Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis. Nat. Commun. 8, 14580 (2017). [DOI] [PMC free article] [PubMed]
- 5.Yin HJ, et al. Ultrathin platinum nanowires grown on single-layered nickel hydroxide with high hydrogen evolution activity. Nat. Commun. 2015;6:6430. doi: 10.1038/ncomms7430. [DOI] [PubMed] [Google Scholar]
- 6.Cabán-Acevedo M, et al. Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide. Nat. Mater. 2015;14:1245–1251. doi: 10.1038/nmat4410. [DOI] [PubMed] [Google Scholar]
- 7.Zou X, et al. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015;44:5148–5180. doi: 10.1039/C4CS00448E. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, et al. Unusual synergistic effect in layered Ruddlesden–Popper oxide enables ultrafast hydrogen evolution. Nat. Commun. 2019;10:149. doi: 10.1038/s41467-018-08117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, et al. Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction. Science. 2016;354:1414–1419. doi: 10.1126/science.aaf9050. [DOI] [PubMed] [Google Scholar]
- 10.Chen G, et al. Interfacial electronic effects control the reaction selectivity of platinum catalysts. Nat. Mater. 2016;15:564–569. doi: 10.1038/nmat4555. [DOI] [PubMed] [Google Scholar]
- 11.Lin L, et al. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature. 2017;544:80–83. doi: 10.1038/nature21672. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, et al. Ultrafine Pt nanoclusters confined in a calixarene-based {Ni24} coordination cage for high-efficient hydrogen evolution reaction. J. Am. Chem. Soc. 2016;138:16236–16239. doi: 10.1021/jacs.6b11218. [DOI] [PubMed] [Google Scholar]
- 13.Bu L, et al. Biaxially strained PtPb/Pt core/shell nanoplate boosts oxygen reduction catalysis. Science. 2016;354:1410–1414. doi: 10.1126/science.aah6133. [DOI] [PubMed] [Google Scholar]
- 14.Wang A, et al. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018;2:65–81. doi: 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- 15.Jones J, et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science. 2016;353:150–154. doi: 10.1126/science.aaf8800. [DOI] [PubMed] [Google Scholar]
- 16.Li H, et al. Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation. Nat. Nanotechnol. 2018;13:411–417. doi: 10.1038/s41565-018-0089-z. [DOI] [PubMed] [Google Scholar]
- 17.Cheng N, et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016;7:13638. doi: 10.1038/ncomms13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, et al. Single-atom Pt as co-catalyst for enhanced photocatalytic H2 evolution. Adv. Mater. 2016;28:2427–2431. doi: 10.1002/adma.201505281. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, et al. Thermally stable single atom Pt/m-Al2O3 for selective hydrogenation and CO oxidation. Nat. Commun. 2017;8:16100. doi: 10.1038/ncomms16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Han L, Liu H, Liu X, Luo J. Potential-cycling synthesis of single platinum atoms for efficient hydrogen evolution in neutral media. Angew. Chem. Int. Ed. 2017;56:13694–13698. doi: 10.1002/anie.201706921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao Linlin, Luo Qiquan, Liu Wei, Lin Yue, Liu Xiaokang, Cao Yuanjie, Zhang Wei, Wu Yuen, Yang Jinlong, Yao Tao, Wei Shiqiang. Identification of single-atom active sites in carbon-based cobalt catalysts during electrocatalytic hydrogen evolution. Nature Catalysis. 2018;2(2):134–141. doi: 10.1038/s41929-018-0203-5. [DOI] [Google Scholar]
- 22.Gao C, Wang J, Xu H, Xiong Y. Coordination chemistry in the design of heterogeneous photocatalysts. Chem. Soc. Rev. 2017;46:2799–2823. doi: 10.1039/C6CS00727A. [DOI] [PubMed] [Google Scholar]
- 23.Zhu C, et al. Single-atom electrocatalysts. Angew. Chem. Int. Ed. 2017;5:13944–13960. doi: 10.1002/anie.201703864. [DOI] [PubMed] [Google Scholar]
- 24.Cheng X, et al. High active, stable oxidized platinum clusters as electrocatalysts for the hydrogen evolution reaction. Energy Environ. Sci. 2017;10:2450–2458. doi: 10.1039/C7EE02537H. [DOI] [Google Scholar]
- 25.Zhan Y, et al. Synthesis of a MoSx-O-PtOx electrocatalyst with high hydrogen evolution activity using a sacrificial counter-electrode. Adv. Sci. 2019;6:1801663. doi: 10.1002/advs.201801663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L, et al. Polyoxometalates in dye-sensitized solar cells. Chem. Soc. Rev. 2019;48:260–284. doi: 10.1039/C8CS00559A. [DOI] [PubMed] [Google Scholar]
- 27.Blasco-Ahicart M, et al. Polyoxometalate electrocatalysts based on earthabundant metals for efficient water oxidation in acidic media. Nat. Chem. 2018;10:2430. doi: 10.1038/nchem.2874. [DOI] [PubMed] [Google Scholar]
- 28.Liu R, et al. Enhanced proton and electron reservoir abilities of polyoxometalate grafted on graphene for high-performance hydrogen evolution. Energy Environ. Sci. 2016;9:1012–1023. doi: 10.1039/C5EE03503A. [DOI] [Google Scholar]
- 29.Chen J, et al. Highly reduced and protonated aqueous solutions of [P2W18O62]6- for on-demand hydrogen generation and energy storage. Nat. Chem. 2018;10:1042–1047. doi: 10.1038/s41557-018-0109-5. [DOI] [PubMed] [Google Scholar]
- 30.Pope MT, et al. Polyoxometalate chemistry: an old field with new dimensions in several disciplines. Angew. Chem. Int. Ed. Engl. 1991;30:34–48. doi: 10.1002/anie.199100341. [DOI] [Google Scholar]
- 31.Zhang B, et al. Atomically dispersed Pt1−polyoxometalate catalysts: how does metal−support interaction affect stability and hydrogenation activity. J. Am. Chem. Soc. 2019;141:8185–8197. doi: 10.1021/jacs.9b00486. [DOI] [PubMed] [Google Scholar]
- 32.Lee U, et al. Structure of pentasodium trihydrogenhexatungstoplatinate(IV) icosahydrate, Na5[H3PtW6O24]·20H2O. Acta Crystallogr. 1983;39:817–819. [Google Scholar]
- 33.Huang X, et al. High-performance transition metal-doped Pt3Ni octahedra for oxygen reduction reaction. Science. 2015;348:1230–1234. doi: 10.1126/science.aaa8765. [DOI] [PubMed] [Google Scholar]
- 34.Li. W, et al. WO3 nanoflakes for enhanced photoelectrochemical conversion. ACS Nano. 2014;8:11770–11777. doi: 10.1021/nn5053684. [DOI] [PubMed] [Google Scholar]
- 35.Rausch B, Symes MD, Chisholm G, Cronin L. Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science. 2014;345:1326–1330. doi: 10.1126/science.1257443. [DOI] [PubMed] [Google Scholar]
- 36.Symes MD, Cronin L. Decoupling hydrogen and oxygen evolution during electrolytic water splitting using an electron-coupled-proton buffer. Nat. Chem. 2013;5:403–409. doi: 10.1038/nchem.1621. [DOI] [PubMed] [Google Scholar]
- 37.Li K, et al. Enhanced electrocatalytic performance for the hydrogen evolution reaction through surface enrichment of platinum nanoclusters alloying with ruthenium in situ embedded in carbon. Energy Environ. Sci. 2018;11:1232–1239. doi: 10.1039/C8EE00402A. [DOI] [Google Scholar]
- 38.Sokolov MN, et al. A Pt(II) isopolytungstate: synthesis and crystal structure. Dalton Trans. 2012;4:11978–11979. doi: 10.1039/c2dt31123b. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, et al. Crystal phase and architecture engineering of lotus-thalamus-shaped Pt-Ni anisotropic superstructures for hydrogen efficent electrochemical hydrogen evolution. Adv. Mater. 2018;30:1801741. doi: 10.1002/adma.201801741. [DOI] [PubMed] [Google Scholar]
- 40.Frisch MJ, et al. Gaussian 09, Revision D.01. Wallingford, CT: Gaussian Inc.; 2013. [Google Scholar]
- 41.Zhao Y, et al. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 42.Hay PJ, et al. Ab initio effective core potentials for molecular calculations—potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985;82:299–310. doi: 10.1063/1.448975. [DOI] [Google Scholar]
- 43.Pople JA, et al. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971;54:724–728. doi: 10.1063/1.1674902. [DOI] [Google Scholar]
- 44.Fukui, K. in Frontier Orbitals and Reaction Paths: Selected Papers of Kenichi Fukui (ed Fukui, K.) 471–476 (World Scientific, Singapore, 1997).
- 45.Tomasi J, et al. Quantum mechanical continuum solvation models. Chem. Rev. 2005;105:2999–3094. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 46.Andrae D, et al. Energy-adjusted ab initio pseudopotentials for the 2nd and 3rd row transition-elements. Theor. Chem. Acc. 1990;77:123–141. doi: 10.1007/BF01114537. [DOI] [Google Scholar]
- 47.Álvarez-Moreno M, et al. Managing the computational chemistry big data problem: the ioChem-BD Platform. J. Chem. Inf. Model. 2015;55:95–103. doi: 10.1021/ci500593j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the corresponding author on request.