

Abstract
Background
Many people with schizophrenia do not achieve a satisfactory treatment response with ordinary anti‐psychotic drug treatment. In these cases, various add‐on medications are used, among them lithium.
Objectives
To assess whether: 1. Lithium alone is an effective treatment for schizophrenia, schizophrenia‐like psychoses and schizoaffective psychoses; and 2. Lithium augmentation of antipsychotic medication is an effective treatment for the same illnesses.
Search methods
In July 2012, we searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials which is based on regular searches of CINAHL, BIOSIS, AMED, EMBASE, PubMed, MEDLINE, PsycINFO, and registries of clinical trials. This search was updated on January 20, 2015. For the first version of the review, we also contacted pharmaceutical companies and authors of relevant studies to identify further trials and obtain original participant data.
Selection criteria
Randomised controlled trials (RCTs) of lithium compared with antipsychotics or placebo (or no intervention), whether as sole treatment or as an adjunct to antipsychotic medication, in the treatment of schizophrenia or schizophrenia‐like psychoses or both.
Data collection and analysis
We extracted data independently. For dichotomous data, we calculated random‐effects meta‐analyses, risk ratios (RRs), and 95% confidence intervals (CI) on an intention‐to‐treat basis. For continuous data, we calculated mean differences (MD) and 95% confidence intervals. We used Grading of Recommendations Assessment, Development and Evaluation (GRADE) to create 'Summary of findings' tables and assessed risk of bias for included studies.
Main results
The update search in 2012 detected two further studies that met our inclusion criteria. We did not find any further studies that met our inclusion criteria in the 2015 search. This review now includes 22 studies, with a total of 763 participants (median mean age: 35 years, range: 26 to 72 years). Most studies were small, of short duration, and incompletely reported. As we detected a high risk of bias in many studies, the overall methodological quality of the included sample was rather low.
Three small studies comparing lithium with placebo as the sole treatment showed no difference in any of the outcomes we analysed.
In eight studies comparing lithium with antipsychotic drugs as the sole treatment, more participants in the lithium group left the studies early (eight RCTs; n = 270, RR 1.77, 95% CI 1.01 to 3.11, low quality evidence).
Thirteen studies examined whether the augmentation of antipsychotic drugs with lithium salts is more effective than antipsychotic drugs alone. More participants who received lithium augmentation had a clinically significant response (10 RCTs; n = 396, RR 1.81, 95% CI 1.10 to 2.97, low quality evidence). However, this effect became non‐significant when we excluded participants with schizoaffective disorders in a sensitivity analysis (seven RCTs; n = 272, RR 1.64, 95% CI 0.95 to 2.81), when we excluded non‐double‐blind studies (seven RCTs; n = 224, RR 1.82, 95% CI 0.84 to 3.96), or when we excluded studies with high attrition (nine RCTs; n = 355, RR 1.67, CI 0.93 to 3.00). The overall acceptability of treatment (measured by the number of participants leaving the studies early) was not significantly different between groups (11 RCTs; n = 320, RR 1.89, CI 0.93 to 3.84, very low quality evidence). Few studies reported on side effects. There were no significant differences, but the database is too limited to make any judgement in this regard. For example, there were no data on thyroid dysfunction and kidney problems ‐ two major and well‐known side effects of lithium.
Authors' conclusions
The evidence base for the use of lithium in schizophrenia is limited to 22 studies of overall low methodological quality. There is no randomised trial‐based evidence that lithium on its own is an effective treatment for people with schizophrenia. There is some GRADE low quality evidence that augmentation of antipsychotics with lithium is effective, but the effects are not significant when more prone‐to‐bias open RCTs are excluded. Nevertheless, further large and well‐designed trials are justified. These should concentrate on two target groups: (1) people with no affective symptoms, so that trialists can determine whether lithium has an effect on the core symptoms of schizophrenia, and (2) people with schizoaffective disorders for whom lithium is widely used in clinical practice, although there is no evidence to support this use.
Plain language summary
Lithium for schizophrenia
Review question
To examine whether the drug lithium alone is an effective treatment for schizophrenia and schizophrenia‐like illnesses. To look at whether lithium is an effective treatment when added on as an extra treatment with antipsychotic drugs.
Background
People with schizophrenia often have two main types of symptoms with their illness, the acute symptoms of hearing voices or seeing things (hallucinations) and strange beliefs (delusions). Examples of chronic symptoms are low mood/depression, social withdrawal, and memory problems. The main treatment for schizophrenia is antipsychotic drugs. However, many people with schizophrenia do not respond fully to these drugs, and certain symptoms of the illness can remain when only an antipsychotic is given. In these cases, various add‐on medications are used, among them lithium. Lithium stabilises a person's mood and is used as an add‐on treatment with antipsychotics for schizophrenia. Lithium can reduce mania and depression.
Study characteristics
The update search in 2012 detected two further studies that met required standards, and no further studies were found in the 2015 search. This review now includes 22 randomised studies, with a total of 763 participants. The studies randomised people with schizophrenia or similar illnesses into groups that received either lithium or placebo (dummy drug), lithium or antipsychotic drugs, lithium plus antipsychotic drugs, or antipsychotic drugs alone.
Key results
The findings in this review show that there is no good quality evidence that lithium on its own is effective for people with schizophrenia or schizoaffective disorder. There is some low quality evidence for the effectiveness of lithium as an add‐on treatment with antipsychotic drugs, but this result is inconclusive. Few studies reported on the side effects of lithium (such as kidney and thyroid problems).
Quality of the evidence
Most of the studies were small, of short duration, and poorly reported. The review authors rated the quality of evidence for the main outcomes to be low or very low quality. Further large and well‐designed trials are needed.
Ben Gray, Senior Peer Researcher, McPin Foundation (http://mcpin.org/), wrote this plain language summary.
Summary of findings
Background
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder; it afflicts approximately one per cent of the population worldwide, with little gender differences. Its typical manifestations are 'positive' symptoms, such as fixed false beliefs (delusions) and perceptions without cause (hallucinations); 'negative' symptoms, such as apathy, lack of drive, and disorganisation of behaviour and thought; and catatonic symptoms, such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable, with 80% to 90% not working (Marvaha 2004) and up to 10% dying (Tsuang 1978).
Description of the intervention
In this review, we examined the role of lithium in the treatment of schizophrenia, schizophrenia‐like psychoses, and schizoaffective psychoses. Lithium is traditionally used for affective psychoses, especially the acute treatment of mania, and for relapse prevention in bipolar affective psychoses. Companion reviews examined carbamazepine (Leucht 2014), valproate (Schwarz 2008), and benzodiazepines (Dold 2012) as sole or adjunctive treatments for schizophrenia.
How the intervention might work
The efficacy of lithium has been identified in a number of psychiatric conditions – it can reduce mania and depression, work as a prophylactic agent in both disorders, and augment the effect of many antidepressants and possibly antipsychotics. Lithium influences virtually every neurotransmitter system, so its efficacy is most probably not due to one specific biochemical mechanism (Jope 1999). Additionally, it has been shown that lithium alters ionic transport and hormonal and circadian regulation as well as mechanisms of gene expression (Lenox 2003).
Why it is important to do this review
Despite the introduction of antipsychotic (neuroleptic) medication in the 1950s, there is still a sizeable minority of people with schizophrenia and related conditions that do not have complete remission of symptoms (Schooler 1993). Over the last 40 years, a variety of adjunctive treatments have been used to treat schizophrenia (Christison 1991). These are often used in addition to antipsychotics in an attempt to alleviate the symptoms of the disease, such as hallucinations and delusional beliefs, although they have also been used instead of antipsychotics. Treatments such as lithium (indicated for bipolar affective disorder), carbamazepine (Leucht 2014), benzodiazepines (Dold 2012), beta‐blockers (Cheine 2001), and electroconvulsive therapy (Tharyan 2005) have all been used for people whose psychoses did not respond to traditional therapy. The situation has improved somewhat with the re‐introduction of clozapine, which has proven efficacy for those that have not responded to traditional medications (Essali 2009). There is evidence that some other 'atypical' antipsychotics are also more effective for the treatment of those with treatment‐resistant schizophrenia, but overall the differences between antipsychotics are small (Leucht 2013). However, many individuals with psychoses have suboptimal responses to treatment, and clinicians are faced with the choice of changing to alternate types of medication or augmenting existing neuroleptics with other drugs or treatments.
Objectives
To assess whether:
1. Lithium alone is an effective treatment for schizophrenia, schizophrenia‐like psychoses and schizoaffective psychoses; and 2. Lithium augmentation of antipsychotic medication is an effective treatment for the same illnesses.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCTs). If a trial was described as 'double blind' but implied randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in statistically significant differences, we did not add the data from these lower quality studies to the results of the better trials, but presented such data within a subcategory. We excluded quasi‐randomised studies, such as those that allocated by alternate days of the week. Where people were given additional treatments as well as lithium, we only included data if the adjunct treatment was evenly distributed between groups and it was only the lithium that was randomised.
Types of participants
Adults, however defined, with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder, and delusional disorder, again, by any means of diagnosis.
We are interested in making sure that information is as relevant to the current care of people with schizophrenia as possible, If studies reported the information we clearly highlighted in the Characteristic of included studies and text, the current clinical state (acute, early postacute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
Lithium alone: any dose.
Placebo (or no intervention).
Lithium in combination with any antipsychotic treatment: any dose.
Placebo (or no intervention) in combination with any antipsychotic treatment.
Antipsychotics alone: any dose.
Types of outcome measures
We grouped all outcomes into short‐term (up to 12 weeks), medium‐term (13 to 26 weeks), and long‐term (over 26 weeks).
Primary outcomes
1. Response to treatment
1.1 Clinically relevant response as defined by the authors 1.2 Relapse ‐ as defined by each of the studies
Secondary outcomes
1. Acceptability of treatment
1.1 Leaving early due to any reason
2. Service utilisation
2.1 Hospital admission 2.2 Days in hospital 2.3 Change in hospital status
3. Clinical response
3.1 Clinically significant improvement of global state 3.2 Average score/change in global state 3.3 Clinically significant improvement in mental state as defined by each of the studies 3.4 Average score/change in mental state 3.5 Clinically significant response on depressive symptoms as defined by each of the studies 3.6 Average score/change in depressive symptoms 3.7 Clinically significant response on manic symptoms as defined by each of the studies 3.8 Average score/change in manic symptoms 3.9 Clinically significant response on negative symptoms as defined by each of the studies 3.10 Average score/change in negative symptoms 3.11 Clinically significant response on positive symptoms as defined by each of the studies 3.12 Average score/change in positive symptoms 3.13 Additional drug use 3.13.1 Antipsychotics 3.13.2 Benzodiazepines
4. Behaviour
4.1 General behaviour 4.2 Specific behaviours 4.2.1 Social functioning 4.2.2 Employment status during trial (employed/unemployed) 4.2.3 Occurrence of violent incidents (to self, others, or property)
5. Adverse events
5.1 General adverse events 5.2 Specific adverse events 5.2.1 Allergic reactions 5.2.2 Blood dyscrasia, such as agranulocytosis 5.2.3 Central nervous system (ataxia, nystagmus, drowsiness, fits, diplopia, tremor) 5.2.4 Death (suicide and non‐suicide deaths) 5.2.5 Gastrointestinal (nausea, vomiting, diarrhoea) 5.2.6 Kidney dysfunction 5.2.7 Movement disorders (extrapyramidal side effects, including neuroleptic malignant syndrome) 5.2.8 Thyroid dysfunction (goitre, thyroid hypofunction)
6. Economic (cost of care)
Search methods for identification of studies
The methods section of reviews of the Cochrane Schizophrenia Group are continuously improved; hence, the methods sections of previous versions of this review can be found in Appendix 1.
Electronic searches
Cochrane Schizophrenia Group’s Trials Register
On January 20, 2015, the Trials Search Co‐ordinator (TSC) searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials using the following search strategy:
*Lithium* in Intervention of STUDY
In such study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, EMBASE, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, hand‐searches, grey literature, and conference proceedings (see Group’s Module). There are no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 2.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
For the original search, we contacted the first author of each included study for information regarding unpublished trials and additional information (however, this was not done in this update).
Data collection and analysis
We present the methods used in data collection and analysis for the current update below; for previous methods, please see Appendix 1.
Selection of studies
Review authors SL and BH independently inspected all citations from the searches to identify relevant abstracts (see Figure 1). Where disputes arose, we acquired the full report for more detailed scrutiny. If citations met inclusion criteria, we obtained full reports of the papers for more detailed inspection. SL and BH independently inspected those articles (with the help of a Chinese translator, if necessary, and in this case, only once). Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
1.

Study flow diagram of trial selection from 2012 and 2015 search.
Data extraction and management
1. Extraction
We initially planned that reviewers MD and BH would extract data from newly included studies, resolving any disagreement by consensus. However, the update search yielded only new studies in the Chinese language, so we had to employ a Chinese translator, Yijun Wang, in order to extract the data. Thus, double extraction was not possible in the case of the two new included studies (Feng 2006 and He 2010).
2. Management
2.1 Forms
We extracted data onto standard, predesigned simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only in the following cases:
a. if the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and
b. if the measuring instrument had not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should have been either i) a self‐report or ii) completed by an independent rater or relative (not the therapist); however, this was often not reported clearly.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided to primarily use endpoint data and only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: a) standard deviations and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the standard deviation when multiplied by two is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996); c) if a scale started from a positive value (such as Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases, skew is present if 2 SD > (S‐S min), where SD is the standard deviation, S is the mean score, and S min is the minimum score. Endpoint scores on scales often have a finite start and endpoint and these rules can be applied. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We entered skewed data from studies of less than 200 participants in additional tables rather than into an analysis. Skewed data pose less of a problem when looking at mean if the sample size is large; we entered such data into syntheses.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week, or per month), to a common metric (e.g., mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that a 50% improvement in a scale‐derived score, such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), can be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 'Summary of findings' table
We used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach to interpret findings (Schünemann 2008) and used GRADE profiler (GRADE Profiler) to import data from RevMan 5 (Review Manager (RevMan)) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes that we rated as critical or important to patient care and decision‐making. We selected the following main outcomes for inclusion in the 'Summary of findings' tables:
Response to treatment ‐ clinically relevant response as defined by the authors
Acceptability of treatment ‐ leaving early due to any reason
Clinically significant response on depressive symptoms (at least 50% Montgomery‐Asberg Depression Rating Scale (MADRS) improvement)
Clinically significant response on manic symptoms (at least 50% Bech‐Rafaelsen improvement)
Clinically significant response on positive symptoms (at least 50% Manchester Scale (MS) improvement)
Adverse events ‐ kidney dysfunction
Adverse events ‐ thyroid dysfunction
Assessment of risk of bias in included studies
BH assessed the risk of bias using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, such as sequence generation, allocation concealment, blinding, incomplete outcome data, and selective reporting. SL supervised BH in this process. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted the authors of the studies in order to obtain further information. We noted the level of risk of bias in both the text of the review (Risk of bias in included studies) and in the tables (Characteristics of included studies).
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that clinicians tend to interpret odds ratios as RRs (Deeks 2000). For statistically significant results, we had originally planned to calculate the number needed to treat to provide benefit/to induce harm statistic (NNTB/H) and its 95% confidence interval (CI) using Visual Rx (www.nntonline.net/visualrx/introduction), taking account of the event rate in the control group. However, 'Summary of findings' tables and calculations therein have superseded this.
2. Continuous data
For continuous outcomes, we estimated the mean difference (MD) between groups. We would have prefered not to calculate effect size measures (standardised mean difference (SMD)). However, if scales of very considerable similarity were used, we would presume there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby, P values are spuriously low, confidence intervals unduly narrow, and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where primary studies did not account for clustering, we presented data in a table with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intraclass correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster‐randomised study, but adjusted for the clustering effect.
We sought statistical advice and were advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation coefficient (ICC) (design effect = 1+(m‐1)*ICC) (Donner 2002). If the ICC was not reported, we assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed, taking into account intraclass correlation coefficients, and relevant data documented in the reports, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g., pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase, the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary, we simply added and combined these within a two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions. Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Intention‐to‐treat
For studies that did not specify the reasons for people leaving the study early, the reviewers assumed that these people had no change in the clinical outcome variables. Wahlbeck 2001 highlighted the problem of high dropout rates in randomised trials of drug treatments for schizophrenia. Since there is no evidence on the degree of attrition, which makes a reasonable analysis of the data possible, we included all trials in the main analysis. However, we used a sensitivity analysis to test whether the exclusion of trials with attrition rates higher than 50% significantly changed the results of the primary outcome parameters. When insufficient data were provided to identify the original group size (prior to attrition), we contacted the authors and allocated the trials to the 'Characteristics of studies awaiting classification' list.
2. Continuous outcomes
If standard deviations were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and confidence intervals were available for group means, and either P value or t value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the standard error (SE) is reported, standard deviations (SDs) are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulas for estimating SDs from P, t, or F values; confidence intervals; ranges; or other statistics. If these formulas did not apply, we calculated the SDs according to a validated imputation method, which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially without seeing comparison data to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise. When such situations or participant groups arose, we discussed them fully.
2. Methodological heterogeneity
We considered all included studies initially without seeing comparison data to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods that we had not predicted would arise. When such methodological outliers arose, we discussed them fully.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I² statistic
We investigated heterogeneity between studies by considering the I² statistic method alongside the Chi² P value. The I² statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of the I² statistic depends on i) magnitude and direction of effects and ii) strength of evidence for heterogeneity (e.g., a P value from a Chi² test, or a confidence interval for an I² statistic). We interpreted I² statistic estimates greater than or equal to around 50% accompanied by a statistically significant Chi² statistic as evidence of substantial levels of heterogeneity (Section 9.5.2, Higgins 2011). When we found substantial levels of heterogeneity in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the nature and direction of results influence the dissemination of research findings (Egger 1997). Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) describes this. We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies or where all studies were of similar sizes. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for the use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us, and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is however a disadvantage to the random‐effects model: it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose random‐effects model for all analyses. The reader is, however, able to choose to inspect the data using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
We reported if inconsistency was high. We first investigated whether data had been entered correctly. Second, if data were correct, we re‐inspected outlier studies to find out whether methodological differences were likely to explain the differences. We also explored such reasons through subgroup analyses, of which we a priori defined the separate examination of studies in first‐episode participants, treatment‐resistant participants, and people with schizoaffective or prominent affective symptoms to be conducted in any case for the primary outcome of comparison three (lithium augmentation). Decisions as to whether we should nevertheless pool the studies, whether we should remove single outlier studies, or whether we would not pool studies at all depended on factors such as whether subgroup effects could explain heterogeneity or whether studies differed in the direction of effects, which is more problematic, or just in the degree of differences between intervention and control (Higgins 2011).
Sensitivity analysis
We only performed sensitivity analyses for the primary outcome, response to treatment in comparison three (lithium augmentation).
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes, we included these studies, and if there was no substantive difference when we added the implied randomised studies to those with better description of randomisation, we employed all data from those studies.
2. Risk of bias
We analysed the effects of excluding trials that we judged to be at a high risk of bias in terms of blinding, i.e., we excluded open RCTs. We also excluded studies with very high attrition (> 50%) in a sensitivity analysis of the primary outcome. We excluded a priori studies with high risk randomisation methods or allocation concealment methods.
3. Cluster‐randomised trials
We also undertook a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
4. Fixed and random effects
We synthesised all data using a random‐effects model; however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates in the fixed‐effect model altered the significance of the results compared with the more evenly distributed weights in the random‐effects model.
Results
Description of studies
For substantive descriptions of the studies, please see the 'Characteristics of included studies' tables and the 'Characteristics of excluded studies' tables.
Results of the search
The initial search in 2002 identified 91 citations, of which 65 appeared relevant and the full studies were inspected. The update search in 2006 yielded 56 further citations, of which four were potentially relevant. We contacted the authors of these studies; we had to exclude three because they were not appropriately randomised (Chen 2001; Gao 2002; Wang 1995). We could obtain no information on the fourth study, Kamisada 1988, and therefore listed it among those studies awaiting assessment. The update search in 2012 identified four further potentially relevant studies. Eventually, we excluded two of them: the first, because of the intervention used (Zhou 2011), and the second, because of a non‐random allocation procedure (Sun 2008). We included the two remaining studies in our analysis (Feng 2006; He 2010). A further search was run in January 2015 and yielded 23 citations; initial scanning of this search found no new included studies, all were either studies to be excluded or additional references to studies already included in the review (see Figure 1).
Included studies
We identified 22 studies for inclusion in this review.
1. Study design
Most studies used a parallel group design, but Garver 1983; Simpson 1976; Small 1975; Small 2001; and Terao 1995 were cross‐over studies. Of the latter, we used only the results of the first phase, as described in the methods section.
2. Length of trials
Mattes 1984 was the longest study, with a duration of one year. He 2010; Small 1975; Small 2001; and Terao 1995 were medium‐term studies, with a duration of 13 to 19 weeks. All of the others were in the 'short‐term' category, lasting between three and eight weeks within a single treatment phase.
3. Participants
The 22 studies included a total of 763 people. The number of people in each study was rather low and ranged between 10 and 92. Fourteen out of 22 studies provided the mean age of the participants (median: 35 years, range: 26 to 72 years). Fifteen studies provided information about severity, out of which 11 described their participants as chronic or non‐responsive to previous treatment. Only two studies, Mattes 1984 and Schulz 1999, were undertaken in the community; all others were carried out with people who were in hospital at the beginning of the trial.
Most participants had schizophrenia, but there were also some people with schizoaffective disorder (n = 196 ‐ of these, 155 had schizomania), schizophreniform disorder (n = 22), atypical psychoses (n = 7), and delusional disorder (n = 5), and there were 29 participants where the diagnosis was not clearly indicated. Diagnostic criteria varied to a considerable extent, because the studies were carried out over a long period of time, but most of the studies used some sort of standard diagnostic criteria. Several studies examined specific groups of participants. Biederman 1979 studied participants with "motor hyperactivity" or elevated mood, Hogarty 1995 examined participants with "persistent distress and/or anxiety", Simpson 1976 analysed a chronic group with tardive dyskinesia, and Brockington 1978; Collins 1991; Feng 2006; He 2010; Hogarty 1995; Small 1975; Small 2001; Schulz 1999; Simhandl 1996; and Wilson 1993 included only treatment‐resistant participants according to a variety of criteria.
4. Interventions
Three studies compared lithium as a sole agent with placebo as a sole agent (Garver 1983; Johnstone 1988; Simpson 1976); eight studies compared lithium as a sole agent with antipsychotics (Braden 1982; Brockington 1978; Dube 1981; Johnson 1971; Johnstone 1988; Mattes 1984; Prien 1972; Shopsin 1971); and 13 studies compared lithium added to antipsychotic drugs versus placebo added to antipsychotic drugs (Biederman 1979; Collins 1991; Feng 2006; He 2010; Hogarty 1995; Huang 1984; Johnstone 1988; Schulz 1999; Simhandl 1996; Small 1975; Small 2001;Terao 1995; Wilson 1993). Johnstone 1988 provided data for all three comparisons.
The lithium dose was commonly adjusted to be within a therapeutic blood‐level range. All but one study that compared lithium as a sole agent with antipsychotics used chlorpromazine as a comparator. The exception was the study by Mattes 1984, which used fluphenazine. The range of chlorpromazine or its equivalent daily doses in the other studies was 300 to 2000 mg.
Of the 13 studies that examined lithium as an adjunct to antipsychotics, three used haloperidol as an antipsychotic (Biederman 1979; Huang 1984; Wilson 1993); two, clozapine (Feng 2006; Small 2001); two, fluphenazine (Hogarty 1995; Schulz 1999); one, pimozide (Johnstone 1988); and one, risperidone (He 2010). In the other four studies, the therapists could choose any antipsychotic drug that they preferred (Collins 1991; Simhandl 1996; Small 1975; Terao 1995).
5. Outcomes
A variety of scales were used to assess clinical response and adverse events. The reporting on efficacy and on side effects was incomplete in the original publications. However, we were able to substantially improve this situation by contacting the authors, many of whom agreed to share their data with us.
5.1 Outcome scales
We present details of scales that provided usable data below. We have given reasons for exclusion of data provided by other instruments under 'outcomes' in the 'Characteristics of included studies' tables.
5.1.1 Global functioning
Clinical Global Impression (CGI) Scale (Guy 1976)
A rating instrument commonly used in studies of schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used, with low scores indicating decreased severity or greater recovery or both.
5.1.2 Mental state
Brief Psychiatric Rating Scale (BPRS) (Overall 1962)
A brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The scale has 18 items, and each item can be defined on a seven‐point scale varying from 'not present' (1) to 'extremely severe' (7). Scoring goes from 18 to 126.
Positive and Negative Syndrome Scale (PANSS) (Kay 1987)
This scale was developed to evaluate the positive, negative, and general symptoms in schizophrenia. The scale has 30 items, and each item can be defined on a seven‐point scoring system varying from one (absent) to seven (extreme). This scale can be divided into subscales for measuring the severity of general psychopathology, positive symptoms, negative symptoms, mania (excited component), and aggression (Supplemental Aggression Risk Profile). Higher scores indicate more pronounced symptomatology.
Bech‐Rafaelsen Mania Scale (BRMS) (Bech 1978)
This rating scale has 11 items addressing symptoms of mania. The single items can all be defined on a five‐point scale from zero (normal) to four (extreme). A total score is calculated, which goes from zero to 44.
New Haven Schizophrenia Index (NHSI) (Astrachan 1972)
A symptom checklist for the evaluation of schizophrenic pathology.
Manchester Scale (MS) (Krawiecka 1977)
A brief rating scale used to assess the severity of symptoms associated with schizophrenia and comprising positive, negative, and depressive symptoms. Each item can be defined on a five‐point scale varying from 'not present' (zero) to 'severe' (four).
Structured Clinical Interview (SCI) (Burdock 1968)
A scale to assess the mental state of those with psychiatric disorders. Higher scores indicate more symptoms.
Hamilton Rating Scale for Depression (HRSD) (Hamilton 1960)
The instrument is designed to be used only on patients already diagnosed as suffering from affective disorder of depressive type. It is used for quantifying the results of an interview, and its value depends entirely on the skill of the interviewer in eliciting the necessary information. The scale contains 17 variables measured on either a five‐point or a three‐point rating scale, the latter being used where quantification of the variable is either difficult or impossible. Among the variables are depressed mood, suicide, work and loss of interest, retardation, agitation, gastrointestinal symptoms, general somatic symptoms, hypochondriasis, loss of insight, and loss of weight. It is useful to have two raters independently scoring a patient at the same interview. The scores of the patient are obtained by summing the scores of the two physicians.
Montgomery‐Asberg Depression Rating Scale (MADRS) (Montgomery 1979)
A 10‐item checklist to measure the overall severity of depression symptoms. Items are rated on a scale of zero to six with anchors at two‐point intervals. Higher scores indicate more symptoms. Scoring ranges from zero to 60.
Scale for the Assessment of Negative Symptoms (SANS) (Andreasen 1989)
This six‐point scale gives a global rating of the following negative symptoms: alogia, affective blunting, avolition‐apathy, anhedonia‐asociality, and attention impairment. Higher scores indicate more symptoms.
5.1.3 Adverse events
Abnormal Involuntary Movement Scale (AIMS) (NIMH 1970)
The Abnormal Involuntary Movement Scale has been used to assess tardive dyskinesia, a long‐term, drug‐induced movement disorder. However, using this scale in short‐term trials may also be helpful to assess some rapidly occurring abnormal movement disorders, such as tremor.
Simpson‐Angus Scale (SAS) (Simpson 1970)
This 10‐item scale, with a scoring system of zero to four for each item, measures drug‐induced parkinsonism, a short‐term, drug‐induced movement disorder. A low score indicates low levels of parkinsonism.
Barnes Akathisia Scale (BAS) (Barnes 1989)
Akathisia is a drug‐induced movement disorder. The scale comprises items rating the observable, restless movements that characterise akathisia, the subjective awareness of restlessness, and any distress associated with the condition. These items are rated from zero (normal) to three (severe). In addition, there is an item for rating the global severity, which starts from zero (absent) to five (severe). A low score indicates low levels of akathisia.
5.1.4 Missing outcomes
No data were available for many important outcomes, such as length of hospital stay, ability to work, or quality of life.
Excluded studies
We excluded 64 studies. The main reasons for exclusion were that studies did not (or did not adequately) randomise (n = 38), did not have a majority of participants with schizophrenia (n = 13), did not include a placebo or no‐intervention group (n = six), or did not present any data that we could meta‐analyse (n = seven). We contacted the authors of four studies, Carman 1981; Gerlach 1975; Growe 1979; Jus 1978, but were unable to obtain further data. After translation, we excluded Liu 2006 as participants were people with periodic psychosis (according to the Chinese Classification of Mental Disorders (CCMD‐3)), not people with schizophrenia.
Awaiting assessment
Three studies are in awaiting assessment: one needs translation (Mosolov 1998), and we are seeking more information from the other studies (Kamisada 1988; McGorry 2002).
Ongoing Studies
There are currently no ongoing studies that we are aware of.
Risk of bias in included studies
For graphical representations of our judgements of risk of bias, please refer to Figure 2 and Figure 3. Full details of judgements can be seen in the 'Risk of bias' tables.
2.
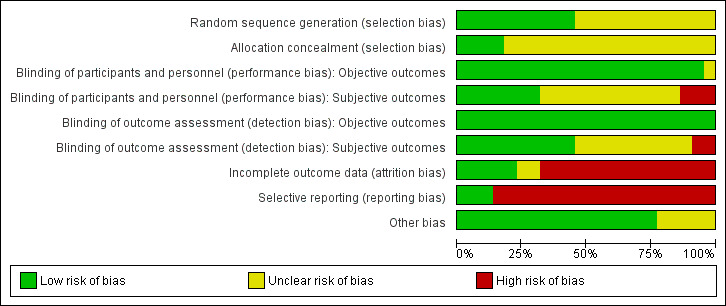
'Risk of bias' graph: review authors' judgements about each 'Risk of bias' item presented as percentages across all included studies.
3.
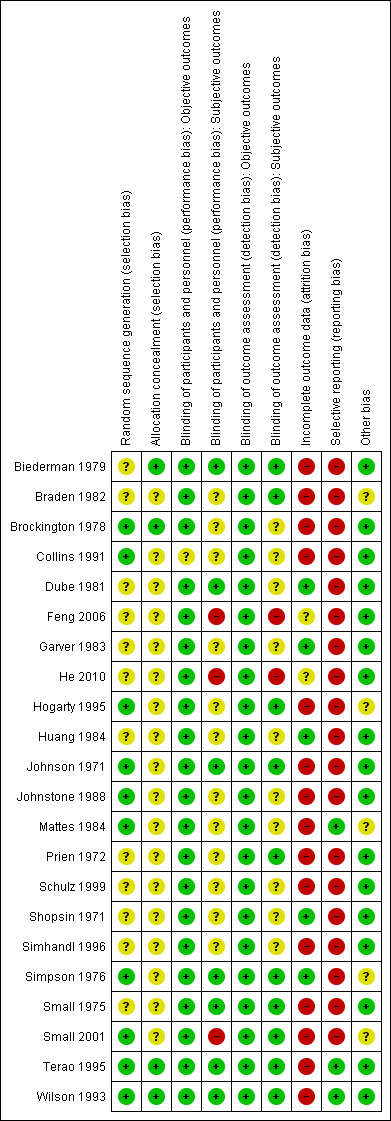
'Risk of bias' summary: review authors' judgements about each 'Risk of bias' item for each included study.
Allocation
All included studies were stated to be randomised. Brockington 1978; Collins 1991; Hogarty 1995; Johnson 1971; Johnstone 1988; Mattes 1984; Small 2001; Terao 1995; and Wilson 1993 used lists of random numbers for allocating the participants. Simpson 1976 used a coin‐toss method. We classified these studies as having a low risk of selection bias. The other studies did not provide a specific method used in the randomisation procedure, and we thus classified these as having an unclear risk of selection bias.
Regarding the allocation concealment, we classified all but four studies as unclear in this respect.
Blinding
We rated separately objective and subjective outcomes. We also rated performance and detection bias separately, resulting in four independent assessments of blinding.
All but five studies were double‐blind. Collins 1991 was randomised but open. Simpson 1976 only blinded the raters. Feng 2006 and He 2010 did not provide any information about blinding so we assumed an open‐label design. Prien 1972; Terao 1995; and Wilson 1993 used identical capsules for blinding. In Shopsin 1971 and Schulz 1999, the raters and the participants, but not the therapists, were blind to medication. The other studies did not describe their blinding method; they were just said to be blind.
As for objective outcomes, for both performance and detection bias, we rated all studies as having a low risk of bias because we considered blinding to be less important for objective outcomes as it was for subjective outcomes.
Incomplete outcome data
Although all but two studies, Feng 2006; He 2010, indicated the numbers of participants who left the study before its completion, the reasons for leaving the studies early were not consistently indicated. Thus, we could only analyse the specific reasons for leaving the studies early in only one study (Biederman 1979).
We assessed only five studies as having a low risk of attrition bias, but only because there were no participants leaving early in these studies. We judged two studies as unclear and 15 studies as having a high risk of attrition bias. Many of these 15 studies often had a high attrition rate (more than 25%) that was not evenly distributed between groups. Moreover, none of these studies gave specific reasons for discontinuation.
Selective reporting
Selective reporting was a major problem within the included studies. We judged 19 out of 22 studies as having a high risk of reporting bias due to lack of information about predefined outcomes. Very often the studies did not report neither standard deviations nor means. The remaining three studies did not selectively report on any outcomes; thus, we judged these as having a low risk of bias in this respect.
Other potential sources of bias
We did not identify any other potential sources of bias in 17 out of 22 of the included studies. We judged five studies as unclear in this respect (for various reasons).
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. Lithium as sole treatment compared with placebo as sole treatment for schizophrenia.
| Lithium as sole treatment compared with placebo as sole treatment for schizophrenia | ||||||
| Patient or population: participants with schizophrenia Setting: hospital Intervention: lithium as sole treatment Comparison: placebo as sole treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo as sole treatment | Lithium as sole treatment | |||||
| Response to treatment clinically relevant response as defined by the authors Follow‐up: mean 3 weeks | 458 per 1000 | 380 per 1000 (202 to 710) | RR 0.83 (0.44 to 1.55) | 54 (2 studies) | ⊕⊕⊝⊝ low¹, ² | ‐ |
| Acceptability of treatment leaving early due to any reason Follow‐up: mean 3 weeks | 100 per 1000 | 114 per 1000 (29 to 444) | RR 1.14 (0.29 to 4.44) | 65 (3 studies) | ⊕⊕⊝⊝ low², ³ | ‐ |
| Clinically significant response on depressive symptoms at least 50% MADRS improvement Follow‐up: mean 8 weeks | 333 per 1000 | 477 per 1000 (217 to 1000) | RR 1.43 (0.65 to 3.16) | 39 (1 study) | ⊕⊕⊝⊝ low², ⁴ | ‐ |
| Clinically significant response on manic symptoms at least 50% Bech‐Rafaelsen improvement Follow‐up: mean 8 weeks | 278 per 1000 | 286 per 1000 (106 to 781) | RR 1.03 (0.38 to 2.81) | 39 (1 study) | ⊕⊕⊝⊝ low², ⁴ | ‐ |
| Clinically significant response on positive symptoms at least 50% MS improvement Follow‐up: mean 8 weeks | 500 per 1000 | 430 per 1000 (220 to 845) | RR 0.86 (0.44 to 1.69) | 39 (1 study) | ⊕⊕⊝⊝ low², ⁴ | ‐ |
|
Kidney dysfunction outcome not available in any study |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Thyroid dysfunction outcome not available in any study |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The basis for the assumed risk (e.g., the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MADRS: Montgomery‐Asberg Depression Rating Scale; MS: Manchester Scale; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Risk of bias: there was a high risk of selective reporting in both studies. ²Imprecision: the 95% confidence interval includes both benefit and harm. Moreover, the number of events was small. ³Indirectness: acceptability was measured by the number of participants leaving the studies for any reason, which is an indirect measure of acceptability. ⁴Risk of bias: there was an unclear risk of performance and detection bias. Selective reporting was present.
Summary of findings 2. Lithium compared with antipsychotics for schizophrenia.
| Lithium compared with antipsychotics for schizophrenia | ||||||
| Patient or population: participants with schizophrenia Setting: hospital Intervention: lithium Comparison: antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Antipsychotics | Lithium | |||||
| Response to treatment clinically relevant response as defined by the authors Follow‐up: mean 5 weeks | 500 per 1000 | 360 per 1000 (140 to 920) | RR 0.72 (0.28 to 1.84) | 80 (3 studies) | ⊕⊕⊝⊝ low¹, ² | ‐ |
| Acceptability of treatment leaving early due to any reason Follow‐up: mean 10.6 weeks | 139 per 1000 | 246 per 1000 (140 to 432) | RR 1.77 (1.01 to 3.11) | 270 (8 studies) | ⊕⊝⊝⊝ very low², ³, ⁴, ⁵ | ‐ |
| Clinically significant response on depressive symptoms at least 50% MADRS improvement Follow‐up: mean 8 weeks | 652 per 1000 | 476 per 1000 (280 to 815) | RR 0.73 (0.43 to 1.25) | 44 (1 study) | ⊕⊕⊝⊝ low², ⁶ | ‐ |
| Clinically significant response on manic symptoms at least 50% Bech‐Rafaelsen improvement Follow‐up: mean 8 weeks | 565 per 1000 | 288 per 1000 (136 to 616) | RR 0.51 (0.24 to 1.09) | 44 (1 study) | ⊕⊕⊝⊝ low², ⁶ | ‐ |
| Clinically significant response on positive symptoms at least 50% MS improvement Follow‐up: mean 8 weeks | 870 per 1000 | 426 per 1000 (252 to 722) | RR 0.49 (0.29 to 0.83) | 44 (1 study) | ⊕⊕⊝⊝ low⁵, ⁶ | ‐ |
|
Kidney dysfunction outcome not available in any study |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Thyroid dysfunction outcome not available in any study |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The basis for the assumed risk (e.g., the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MADRS: Montgomery‐Asberg Depression Rating Scale; MS: Manchester Scale; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Risk of bias: there was a high risk of selection bias. ²Imprecision: the 95% confidence interval includes both benefit and harm. Moreover, the number of events was small. ³Risk of bias: there was a high risk of selection bias in the majority of studies. Moreover, a high risk of attrition bias was also prevalent. ⁴Indirectness: acceptability was measured by the number of participants leaving the studies for any reason, which is an indirect measure of acceptability. ⁵Imprecision: the number of events was small. ⁶Risk of bias: there was an unclear risk of performance and detection bias. Selective reporting was present.
Summary of findings 3. Adjunctive lithium + antipsychotics compared with placebo/no adjunctive treatment + antipsychotics for schizophrenia.
| Adjunctive lithium + antipsychotics compared with placebo/no adjunctive treatment + antipsychotics for schizophrenia | ||||||
| Patient or population: participants with schizophrenia Setting: hospital Intervention: adjunctive lithium + antipsychotics Comparison: placebo/no adjunctive treatment + antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo/no adjunctive treatment+ antipsychotics | Adjunctive lithium + antipsychotics | |||||
| Response to treatment clinically relevant response as defined by the authors Follow‐up: mean 10 weeks | 228 per 1000 | 413 per 1000 (251 to 677) | RR 1.81 (1.1 to 2.97) | 396 (10 studies) | ⊕⊕⊝⊝ low¹, ² | ‐ |
| Acceptability of treatment leaving early due to any reason Follow‐up: mean 10.2 weeks | 130 per 1000 | 245 per 1000 (121 to 499) | RR 1.89 (0.93 to 3.84) | 320 (11 studies) | ⊕⊝⊝⊝ very low², ³, ⁴, ⁵ | ‐ |
| Clinically significant response on depressive symptoms at least 50% MADRS improvement Follow‐up: mean 8 weeks | 652 per 1000 | 685 per 1000 (450 to 1000) | RR 1.05 (0.69 to 1.58) | 45 (1 study) | ⊕⊕⊝⊝ low¹, ⁶ | ‐ |
| Clinically significant response on manic symptoms at least 50% Bech‐Rafaelsen improvement Follow‐up: mean 8 weeks | 565 per 1000 | 639 per 1000 (396 to 1000) | RR 1.13 (0.7 to 1.82) | 45 (1 study) | ⊕⊕⊝⊝ low¹, ⁶ | ‐ |
| Clinically significant response on positive symptoms at least 50% MS improvement Follow‐up: mean 8 weeks | 870 per 1000 | 730 per 1000 (539 to 983) | RR 0.84 (0.62 to 1.13) | 45 (1 study) | ⊕⊕⊝⊝ low¹, ⁶ | ‐ |
|
Kidney dysfunction outcome not available in any study |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
|
Thyroid dysfunction outcome not available in any study |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MADRS: Montgomery‐Asberg Depression Rating Scale; MS: Manchester Scale; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Risk of bias: there was a high risk of reporting bias. ²Imprecision: there was a small number of events. ³Risk of bias: there was high reporting and attrition bias and some evidence of performance bias. ⁴Inconsistency: there were differences between studies (number of events, effects sizes). ⁵Indirectness: acceptability was measured by the number of participants leaving the studies for any reason, which is an indirect measure of acceptability. ⁶Imprecision: the 95% confidence interval includes both benefit and harm. Moreover, the number of events was small.
1. Comparison 1: lithium versus placebo as sole treatment
Three studies fell into this category (Garver 1983; Johnstone 1988; Simpson 1976).
1.1 Clinically significant response as defined by the authors
The studies by Garver 1983 and Johnstone 1988 both provided data for the calculation of the clinically significant response defined as at least 50% improvement of the baseline score on the New Haven Schizophrenia Index or on the Manchester Scale. No significant difference between lithium and placebo as sole treatments was found (2 RCTs; n = 54, RR 0.83; 95% CI 0.44 to 1.55). No significant heterogeneity was found (Chi² = 0.08, P = 0.78; I² = 0%).
1.2 Leaving the study early
In the studies by Simpson 1976 and Garver 1983 no participants left the studies before the end so that only the data from Johnstone 1988 had an impact on the statistical analysis. There was no significant difference between groups (3 RCTs; n = 65, RR 1.14; 95% CI 0.29 to 4.44)
1.3 Mental state
1.3.1 General response/non‐response according to two scores
In Garver 1983 no significant differences were found in outcomes in terms of improvement on the New Haven Schizophrenia Index (at least 20% improvement, 1 RTC; n = 15, RR 0.67, CI 0.13 to 3.53; at least 35% improvement, 1 RCT; n = 15, RR 0.67, CI 0.13 to 3.53; at least 50% improvement, 1 RCT; n = 15, RR 0.67, CI 0.13 to 3.53). The same applies to Johnstone 1988 for improvements in the Manchester Scale (at least 20% improvement, 1 RCT; n = 39, RR 0.92, CI 0.61 to 1.40; at least 35% improvement, 1 RCT; n = 39, RR 0.86, CI 0.47 to 1.58; at least 50% improvement, 1 RCT; n = 39, RR 0.86, CI 0.44 to 1.69).
1.3.2 General ‐ average global scores at endpoint
In Johnstone 1988, the mean Manchester Scale scores at endpoint were similar in both groups (1 RCT; n = 39, WMD 0.0, CI ‐2.9 to 2.9). The mean New Haven Schizophrenia Index scores in Garver 1983 were skewed (see 'other data table').
1.3.3 Specific ‐ depression, mania, negative symptoms, positive symptoms
Only Johnstone 1988 provided usable data on depression, manic symptoms, negative symptoms, and positive symptoms. No significant differences in terms of any level of improvement of these symptoms were found.
1.4 Adverse events ‐ movement disorders
Johnstone 1988 presented scale‐derived data on extrapyramidal side effects in general and on tardive dyskinesia that were skewed; therefore, we only presented these in the 'other data table'. Johnstone 1988 reported no significant differences between groups.
1.5 Missing outcomes
We could not extract information about any of the other outcomes that were listed in the methods section.
1.6 Publication bias
We could not assess publication bias because there were fewer than 10 included studies.
2. Comparison 2: lithium versus antipsychotics as sole treatment
Eight studies fell into this category (Braden 1982; Brockington 1978; Dube 1981; Johnson 1971; Johnstone 1988; Mattes 1984; Prien 1972; Shopsin 1971).
2.1 Clinically significant response as defined by the authors
Three studies provided dichotomised data on clinically significant response. Brockington 1978 and Johnson 1971 indicated the numbers of participants who were improved, and Johnstone 1988, the number of participants with at least 50% Manchester Scale improvement. Data showed no difference between lithium and antipsychotic drugs (3 RTCs; n = 80, RR 0.72; 95% CI 0.28 to 1.84). No significant heterogeneity was found (Chi² = 3.02, P = 0.22; I² = 34%).
2.2 Leaving the study early
Significantly more participants who received lithium as a sole treatment left the studies early (8 RTCs; n = 270, RR 1.77, CI 1.01 to 3.11). Only four studies however reported the reasons (Dube 1981; Mattes 1984; Prien 1972; Shopsin 1971). Combining the results of these studies, it appears that lack of efficacy (4 RCTs; n = 178, RR 2.89, CI 1.09 to 7.67; P = 0.03) rather than poor tolerability (n = 178, RR 1.24, CI 0.08 to 19.21; P = 0.88) led to the attrition.
2.3 Global state
2.3.1 Improved according to the Clinical Global Impression (CGI) Scale
Brockington 1978 and Johnson 1971 presented data on the number of participants with improvement of their global state according to the Global Clinical Impression (CGI) Scale. There was no significant difference between lithium and antipsychotic drugs (2 RCTs; n = 36, RR 0.62, CI 0.06 to 5.98).
2.3.2 Relapse
Only Mattes 1984 reported relapse at one year. More participants who were treated with lithium relapsed (six out of seven) compared with one out of seven participants taking an antipsychotic. This result did not reach statistical significance (1 RTC; n = 14, RR 6.0, CI 0.95 to 37.8; P = 0.06).
2.4 Mental state ‐ general
2.4.1 At least 20%, 35%, or 50% improvement on the Manchester Scale
Johnstone 1988 was the only study that allowed an analysis of dichotomised data derived from the Manchester Scale. More participants in the lithium group than in the pimozide group achieved at least 20% (1 RCT; n = 44, RR 0.73, CI 0.53 to 1.01; P = 0.06), 35% (1 RCT; n = 44, RR 0.61, CI 0.37 to 1.00; P = 0.05), or 50% improvement (1 RCT; n = 44, RR 0.58, CI 0.33 to 1.01; P = 0.05) on their initial score in this scale.
2.4.2 Mean BPRS at endpoint
Homogeneous results of three trials, Dube 1981; Johnson 1971; Shopsin 1971, showed a significant superiority of antipsychotic drugs in this regard (3 RCTs; n = 92, WMD 10.23, CI 6.32 to 14.15). We calculated the standard deviations from the study by Dube 1981 and Johnson 1971 from exact P values in these publications. Although the data might be skewed according to the criteria stipulated in the methods section, our means of analysing it might justify us deviating from this. Furthermore, both studies used parametric tests in their analyses, and Dube 1981 confirmed by e‐mail that the data were normally distributed.
2.4.3 Mean MS at endpoint
Johnstone 1988 used the Manchester Scale and also found a significant superiority of antipsychotic drugs (1 RCT; n = 44, WMD 3.00, CI 0.38 to 5.62). Original participant data were available for this analysis, and these appeared to be normally distributed.
2.4.4 Mean SCI at endpoint
Johnson 1971 also used the SCI for the evaluation of the mental state and found no significant difference between groups (1 RCT; n = 11, WMD ‐2.0, CI ‐81.29 to 77.29).
2.5 Mental state ‐ specific
Only Johnstone 1988 provided data for the analysis of specific aspects of the mental state, reporting on depressive, manic, negative, and positive symptoms.
2.5.1 Depressive symptoms
No significant differences were found between lithium and pimozide in terms of an improvement on the Montgomery‐Asberg Depression Rating Scale of at least 20% (1 RCT; n = 44, RR 0.79, CI 0.53 to 1.18), 35% (1 RCT; n = 44, RR 0.75, CI 0.44 to 1.14), and 50% (1 RCT; n = 44, RR 0.73, CI 0.43 to 1.25).
2.5.2 Manic symptoms
No significant differences were found between lithium and pimozide in terms of an improvement on the Bech‐Rafaelsen Mania Scale of at least 20% (1 RCT; n = 44, RR 0.89, CI 0.58 to 1.37), 35% (1 RCT; n = 44, RR 0.67, CI 0.35 to 1.29) and 50% (1 RCT; n = 44, RR 0.51, 0.24 to 1.09; P = 0.08), although in this last comparison, statistical significance became borderline.
2.5.3 Negative symptoms
Lithium was significantly less effective than pimozide in terms of an improvement in the negative symptoms subscore of the Manchester Scale of at least 20% (1 RCT; n = 44, RR 0.40, CI 0.23 to 0.69), 35% (1 RCT; n = 44, RR 0.37, CI 0.20 to 0.68) and 50% (1 RCT; n = 44, RR 0.37, CI 0.20 to 0.68).
2.5.4 Positive symptoms
Lithium was significantly less effective than pimozide in terms of an improvement in the positive subscore of the Manchester Scale of at least 20% (1 RCT; n = 44, RR 0.55, CI 0.36 to 0.83), 35% (1 RCT; n = 44, RR 0.49, CI 0.29 to 0.83) and 50% (1 RCT; n = 44, RR 0.49, CI 0.29 to 0.83).
2.6 Adverse events
2.6.1 Anticholinergic adverse events
Prien 1972 presented data on blurred vision, dry mouth, and constipation. No significant differences between groups were found.
2.6.2 Central nervous system
Only two studies, Prien 1972; Shopsin 1971, reported data on various adverse events relating to the central nervous system (ataxia, dizziness, hyperactive reflexes, muscle weakness, slurred speech, somnolence, and toxic confusion). Somnolence was significantly more frequent in the antipsychotics group (1 RCT; n = 83, RR 0.2, CI 0.04 to 0.73) and toxic confusion was more frequent in the lithium group (2 RCTs; n = 104, RR 8.83, CI 1.16 to 67.38). The other adverse events occurred with similar frequency in both groups.
2.6.3 Dermatologic
In the study by Prien 1972, no significant difference in the occurrence of pruritus was found (1 RCT; n = 83, RR 0.41, CI 0.02 to 9.83).
2.6.4 Gastrointestinal adverse events
Again, only Prien 1972 presented data. There were no significant differences between groups in terms of dehydration (1 RCT; n = 83, RR 3.71, CI 0.16 to 88.51), nausea (1 RCT; n = 83, RR 0.62, CI 0.12 to 3.21) and vomiting (1 RCT; n = 83, RR 0.83, CI 0.15 to 4.70).
2.6.5 Movement disorder
In the two trials that presented dichotomous data on movement disorder, Prien 1972; Shopsin 1971, no significant differences were found in terms of parkinsonism (1 RCT; n = 83, RR 0.25, CI 0.01 to 5.00), tremor (1 RCT; n = 83, RR 2.18, CI 0.69 to 6.87)or use of antiparkinson medication (1 RCT; n = 21, RR 0.1, CI 0.01 to 1.68). Johnstone 1988 presented scale‐derived data about tardive dyskinesia, which were skewed (see Analysis 1.12).
1.12. Analysis.
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 12 Adverse events: 1. Movement disorder ‐ unable to use (skewed data).
| Adverse events: 1. Movement disorder ‐ unable to use (skewed data) | ||
|---|---|---|
| Study | Mean + SD | Mean + SD |
| Johnstone 1988 | AIMS global score at endpoint ‐ high = poor. Placebo: mean = 2.1, SD = 2.2. N = 18. Lithium: mean = 3.1, SD = 2.7. N = 21. | TAKE global score at endpoint ‐ high = poor. Placebo: mean = 2.5, SD = 1.9. N = 18. Lithium: mean = 3.2, SD = 2.4. N = 21. |
2.7 Laboratory abnormalities
Only Shopsin 1971 reported data on laboratory abnormalities. More participants in the antipsychotic group had a decreased white blood cell count, which was of borderline statistical significance (1 RCT; n = 21, RR 0.10, CI 0.00 to 1.11; P = 0.06), whereas more participants in the lithium group had an increased white blood cell count (one RCT; n = 21, RR 17.42, CI 1.14 to 265.34; P = 0.04). No significant differences in terms of increased uric acid blood level (1 RCT; n = 21, RR 6.42, CI 0.37 to 110.71) or proteinuria (1 RCT; n = 21, RR 4.58, CI 0.25 to 85.33) were found.
2.8 Missing outcomes
No data were found for 'service' outcomes, such as 'duration of hospital stay'. There were neither data on satisfaction with treatment or costs nor usable information about specific aspects of mental state, such as aggression, positive symptoms, negative symptoms, or mood.
2.9 Publication bias
We could not assess publication bias because there were fewer than 10 included studies.
3. Comparison 3: lithium versus placebo as an adjunct to antipsychotic drugs
Twelve studies fell under this category (Biederman 1979; Collins 1991; Feng 2006; He 2010; Hogarty 1995; Huang 1984; Johnstone 1988; Schulz 1999; Simhandl 1996; Small 1975; Small 2001; Terao 1995).
3.1 Clinically significant response as defined by the authors
According to the data from 10 studies, there were more responders in the lithium augmentation group than in the group that received antipsychotic drugs alone (10 RCTs; n = 396, RR 1.81, CI 1.10 to 2.97; P = 0.02). As significant heterogeneity was found (Chi² = 17.71, P = 0.04; I² = 49%), we explored it in a subgroup analysis (see Analysis 4.1 and Analysis 4.2).
4.1. Analysis.
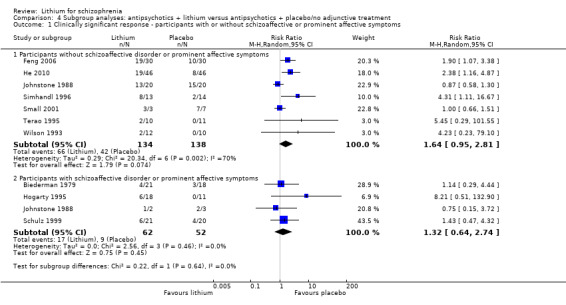
Comparison 4 Subgroup analyses: antipsychotics + lithium versus antipsychotics + placebo/no adjunctive treatment, Outcome 1 Clinically significant response ‐ participants with or without schizoaffective or prominent affective symptoms.
4.2. Analysis.
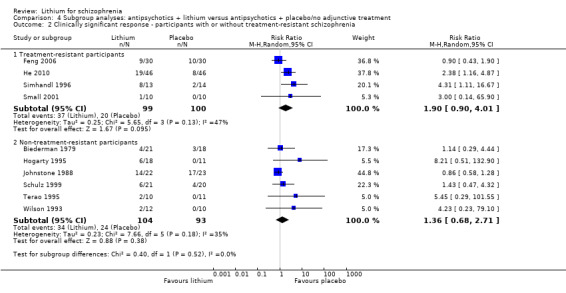
Comparison 4 Subgroup analyses: antipsychotics + lithium versus antipsychotics + placebo/no adjunctive treatment, Outcome 2 Clinically significant response ‐ participants with or without treatment‐resistant schizophrenia.
3.2 Leaving the study early
Although there was a trend showing that more participants treated with lithium augmentation left the studies early than participants who received antipsychotics alone, it did not reach conventional levels of significance (11 RCTs; n = 320, RR 1.89, CI 0.93 to 3.84; P = 0.08). Only Biederman 1979 reported on 'leaving early due to adverse events'. There was no significant difference between groups (1 RCT; n = 39, RR 4.32, CI 0.22 to 84.48).
3.3 Global state
3.3.1 Improved according to the Clinical Global Impression (CGI) Scale
There was no statistically significant difference between participants who received lithium augmentation and those who received placebo in addition to antipsychotics in terms of improvement of their global state according to the Clinical Global Impression Scale (4 RCTs; n = 115, RR 1.52, CI 0.57 to 4.02).
3.3.2 Relapse
Only Hogarty 1995 reported relapse rates; no significant differences between groups were found (1 RCT; n = 29, RR 0.21, CI 0.01 to 4.76).
3.4 Mental state ‐ general
3.4.1 At least 20%, 35%, or 50% improvement inf the BPRS total score
No significant differences in terms of at least 20% BPRS improvement were found (5 RCTs; n = 131, RR 1.21, CI 0.83 to 1.76), but significantly more participants who received lithium augmentation had at least 35% (5 RCTs; n = 131, RR 1.77, CI 1.02 to 3.10) and at least 50% (5 RCTs; n = 131, RR 2.52, CI 1.17 to 5.42) improvement of this score.
3.4.2 At least 20%, 35%, or 50% improvement in the MS total score
Johnstone 1988 used the Manchester Scale for the evaluation of the general mental state. No significant differences between groups were found in terms of an improvement of the baseline MS score of at least 20% (1 RCT; n = 45, RR 0.90, CI 0.71 to 1.13), 35% (1 RCT; n = 45, RR 0.93, CI 0.67 to 1.30) or 50% (1 RCT; n = 45, RR 0.98, CI 0.69 to 1.40).
3.4.3 At least 50% improvement in the PANSS total score
Feng 2006 and He 2010 used the PANSS total score to evaluate general mental state. Significantly more participants who received lithium augmentation than those who received placebo in addition to antipsychotics had an improvement of at least 50% (2 RCTs; n = 152, RR 1.49, CI 1.16 to 1.90).
3.4.4 Mean BPRS total score at endpoint
Four studies presented usable data on the mean BPRS at endpoint. Although all studies showed a trend in favour of lithium augmentation, the pooled mean difference did not reveal a statistically significant superiority (4 RCTs; n = 102, WMD ‐3.26, CI ‐7.10 to 0.57). Individual participant data were available for Terao 1995 and appeared to be normally distributed.
3.4.5 Mean MS total score at endpoint
In Johnstone 1988, the mean MS total score at endpoint was similar in both groups (1 RCT; n = 45, WMD 0.70, CI ‐1.53 to 2.93). Again individual participant data were available that seemed to be normally distributed.
3.4.5 Mean PANSS total score at endpoint
Feng 2006 and He 2010 showed a significant difference in mean PANSS total score at endpoint in favour of lithium (2 RCTs; n = 152, WMD ‐8.53, CI ‐11.44 to ‐5.62). No individual participant data were available.
3.5 Mental state ‐ specific
3.5.1 Depressive symptoms
Johnstone 1988 used the Montgomery‐Asberg Depression Scale to monitor depression and found no significant differences in terms of participants who had an improvement of at least 20% (1 RCT; n = 45, RR 0.99, CI 0.72 to 1.35), 35% (1 RCT; n = 45, RR 0.98, CI 0.66 to 1.45) or 50% (1 RCT; n = 45, RR 1.05, CI 0.69 to 1.58) in their baseline score. Wilson 1993 used the depression/anxiety subscore of the BPRS and found no significant differences in terms of participants who had an improvement of at least 20% (1 RCT; n = 22, RR 0.97, CI 0.48 to 1.95), 35% (1RCT; n = 22, RR 1.17, CI 0.53 to 2.55) or 50% (1 RCT; n = 22, RR 1.17, CI 0.53 to 2.55) in their baseline score. For Johnstone 1988, it was possible to analyse the mean Montgomery‐Asberg Depression Scale score at endpoint as well. There was no significant difference between groups (1 RCT; n = 45, WMD ‐0.34, CI ‐1.64 to 0.96). Using the Hamilton Depression Scale, Schulz 1999 found no significant differences in mean endpoint scores between groups (1 RTC; n = 16, WMD 3.40, CI ‐7.66 to 14.46).
3.5.2 Manic symptoms
Johnstone 1988 found no significant differences in terms of improvement of the Bech‐Rafaelsen Mania Scale of at least 20% (1 RCT; n = 45, RR 1.05, CI 0.72 to 1.52), 35% (1 RCT; n = 45, RR 1.13, CI 0.70 to 1.82) or 50% (1 RCT; n = 45, RR 1.13, CI 0.70 to 1.82).
Feng 2006 provided endpoint data on the Excited Component of PANSS. A significant difference in favour of lithium was found (1 RCT; n = 60, WMD ‐2.47, CI ‐3.62 to ‐1.32).
3.5.3 Negative symptoms
Three studies, Simhandl 1996; Terao 1995; Wilson 1993, used the SANS and showed no significant differences in terms of an improvement in this scale of at least 20% (3 RCTs; n = 70, RR 1.43, CI 0.66 to 3.10), 35% (3 RCTs; n = 70, RR 1.14, CI 0.66 to 2.04) or 50% (3 RCTs; n = 70, RR 1.04, CI 0.50 to 2.17). Johnstone 1988 used the negative symptoms score of the Manchester Scale. Again, no significant differences were found in terms of an improvement in this scale of at least 20% (1 RCT; n = 45, RR 0.95, CI 0.81 to 1.11), 35% (1 RCT; n = 45, RR 1.00, CI 0.83 to 1.20) or 50% (1 RCT; n = 45, RR 1.00, CI 0.83 to 1.20). Finally, Small 2001 used the negative symptoms score of the PANSS and revealed no significant differences in terms of an improvement in this scale of at least 20% (1 RCT; n = 20, RR 0.50, CI 0.12 to 2.14), 35% (1 RCT; n = 20, RR 1.00, CI 0.07 to 13.87) or 50% (1 RCT; n = 20, RR 3.00, 0.14 to 65.90).
For several studies, it was possible to analyse mean scores of rating scales for negative symptoms as well. Two studies, Schulz 1999; Wilson 1993, used the negative symptom subscore of the BPRS and found no significant difference between groups (n = 61, WMD 0.6, CI ‐1.1 to 2.3). Johnstone 1988 provided data about the negative symptom subscore of the MS and found no significant difference (n = 45, RR 0.3, CI ‐0.3 to 0.9). He 2010 and Small 2001 used the negative symptoms subscore of the PANSS and found a similar result (n = 112, WMD ‐0.07, CI ‐2.85 to 2.72). Finally, Simhandl 1996 and Terao 1995 used the SANS. On combining the results of both trials, we found a statistically significant superiority of lithium augmentation (n = 41, WMD ‐4.5, CI ‐8.8 to ‐0.2; P = 0.04).
3.5.4 Positive symptoms
Again, the trials used a variety of scales, and no significant differences between groups were found in almost all of them. Simhandl 1996 and Wilson 1993 used the positive symptoms subscore of the BPRS and showed no significant differences in terms of an improvement in this scale of at least 20% (2 RCTs; n = 49, RR 1.26, CI 0.87 to 1.82), 35% (2 RCTs; n = 49, RR 1.14, CI 0.66 to, 1.89) or 50% (2 RCTs; n = 49, RR 1.27, CI 0.71 to 2.28). Johnstone 1988 used the positive symptoms score of the Manchester Scale. Again, no significant differences were found in terms of an improvement of at least 20% (1 RCT; n = 45, RR 1.04, CI 0.93 to 1.18), 35% (1 RCT; n = 45, RR 0.94, CI 0.73 to 1.21) or 50% (1 RCT; n = 45, RR 0.84, CI 0.62 to 1.13). Finally, Small 2001 used the positive symptoms score of the PANSS and found no significant differences in terms of an improvement of at least 20% (1 RCT; n = 20, RR 1.00, CI 0.42 to 2.40), 35% (1 RCT; n = 20, RR 1.00, CI 0.34 to 2.93) or 50% (1 RCT; n = 20, RR 1.33, CI 0.40 to 4.49).
Concerning mean endpoint scores of scales, Schulz 1999 analysed the BPRS positive subscore at endpoint and found no significant difference between groups (1 RCT; n = 41, WMD 0.14, CI ‐3.45 to 3.73). Johnstone 1988 used the MS positive subscore and found no significant difference between groups (1 RCT; n = 45, WMD 0.23, CI ‐1.34 to 1.80). He 2010 and Small 2001 monitored the participants using the positive subscore of the PANSS and found a significant difference between groups in favour of lithium augmentation (2 RCTs; n = 112, WMD ‐3.15, CI ‐4.91 to ‐1.39).
3.5.5 Aggressive behaviour
Feng 2006 showed a statistical trend (P = 0.06) in favour of lithium in mean PANSS aggression scores at endpoint between groups (1 RCT; n = 60, WMD ‐1.34, CI ‐2.76 to 0.08).
3.6 Medication use
3.6.1 Mean haloperidol dose
The lithium augmentation group received a significantly lower mean haloperidol dose than the group that received haloperidol alone in the study by Wilson 1993 (1 RCT; n = 21, WMD ‐6.90, CI ‐13.62 to ‐0.18). The data from other studies were skewed.
3.6.2 Number of participants using benzodiazepines
In the study by Wilson 1993, a similar number of participants in both groups received benzodiazepines during the study (1 RCT; n = 22, RR 0.83, CI 0.50 to 1.38).
3.7 Adverse events
3.7.1 Central nervous system
Two participants in the lithium augmentation groups of Biederman 1979 and Wilson 1993 developed delirium, but this was not statistically significant (2 RCTs; n = 61, RR 2.56, CI 0.28 to 23.28).
3.7.2 Movement disorder
3.7.2.1 At least one extrapyramidal adverse event
He 2010 and Wilson 1993 found no significant difference between groups (2 RCTs; n = 113, RR 1.44, CI 0.22 to 9.40).
3.7.2.2 Use of antiparkinson medication
The pooled data of Simhandl 1996 and Wilson 1993 showed no significant differences between groups (2 RCTs; n = 49, RR 0.98, CI 0.58 to 1.63).
3.7.2.3. Atkathisia
He 2010 found no significant difference between groups (1 RCT; n = 92, RR 0.75, CI 0.28 to 1.99).
3.7.2.4 Tremor
He 2010 found no significant difference between groups (1 RCT; n = 92, RR 0.88, CI 0.35 to 2.21).
3.7.2.5 Mean Simpson‐Angus Scale score at endpoint
The data from Wilson 1993 showed no significant differences between groups (1 RCT; n = 21, WMD 2.7, CI ‐0.53 to 5.93).
3.7.2.6 Movement disorder ‐ unable to use
Some studies indicated other scale‐derived data about movement disorder, but these were skewed (see Analysis 3.32).
3.32. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 32 Adverse events: 4. Movement disorder ‐ unable to use (skewed data).
| Adverse events: 4. Movement disorder ‐ unable to use (skewed data) | |||
|---|---|---|---|
| Study | Global EPS | Tardive dyskinesia | Akathisia |
| Johnstone 1988 | TAKE global score at endpoint (high = poor). Pimozide + placebo: mean = 5.5, SD = 4.0. N = 23. Pimozide + lithium: mean = 6.9, SD = 3.2. N = 21. | AIMS global score at endpoint (high = poor). Pimozide + placebo: mean = 3.6, SD = 3.2. N = 23. Pimozide + lithium: mean = 4.3, SD = 4.0. N = 21. | ‐ |
| Simhandl 1996 | Mean SAS at endpoint (high = poor). Lithium + antipsychotics: mean = 0.24, SD = 0.16. N = 11. Placebo + antipsychotics: mean = 0.31, SD = 0.35. N = 12. | Mean AIMS at endpoint (high = poor). Lithium + antipsychotics: mean = 8.1, SD = 7.3. N = 11. Placebo + antipsychotics: mean = 13.08, SD = 14.2. N = 12. | ‐ |
| Wilson 1993 | ‐ | Mean AIMS at endpoint (high = poor). Lithium + antipsychotics: mean = 1.7, SD = 2.2. N = 11. Placebo + antipsychotics: mean = 1.2, SD = 0.9. N= 10. | Mean BAS at endpoint (high = poor). Lithium + antipsychotics: mean = 1.3, SD = 2.0. N = 11. Placebo + antipsychotics: mean = 2.1, SD = 1.7. N = 10. |
3.7.3 Non‐specific discomfort
In the study by Wilson 1993, one participant in the lithium group had 'non specific discomfort', but this did not lead to a statistically significant difference between groups (1 RCT; n = 21, RR 2.54, CI 0.11 to 56.25).
3.7.4 UKU Side Effect Rating Scale ‐ unable to use
Terao 1995 provided data about the UKU global score at endpoint, but these data were skewed (see Analysis 3.34).
3.34. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 34 Adverse events: 6. UKU at endpoint ‐ unable to use (skewed data).
| Adverse events: 6. UKU at endpoint ‐ unable to use (skewed data) | |
|---|---|
| Study | |
| Terao 1995 | Antipsychotics + placebo: mean = 8.2, SD = 5.4. N = 9. Antipsychotics + lithium: mean = 8.3, SD = 4.6. N = 9. |
3.7.5 Specific adverse events
Only two studies reported on specific adverse events. Feng 2006 analysed abnormalities in electroencephalogram (EEG), liver functions, and QTC (corrected QT interval)/electrocardiogram (ECG). He 2010 provided data on constipation, dizziness, dry mouth, insomnia, lactation, nausea, somnolence, tachycardia, and weight gain. For each of these adverse events, no significant difference between groups was found.
3.8 Missing outcomes
No data were found for 'service' outcomes, such as 'duration of hospital stay'. Again, there were no data on satisfaction with treatment or costs.
3.9 Publication bias
Funnel plots revealed no obvious publication bias in the primary outcome, but the small number of trials available for the plot hampered this.
4. Subgroup analyses
We performed all subgroup analyses only on the primary outcome response to treatment in comparison three (adding lithium to antipsychotics versus monotherapy with antipsychotics). The preplanned subgroup analysis on participants with a first episode of schizophrenia did not apply, because we did not find relevant studies.
4.1. Participants with or without schizoaffective or prominent affective symptoms
Lithium augmentation was not associated with statistically significantly more responders, neither when studies with an inclusion criterion of schizoaffective disorder or prominent affective symptoms were analysed separately (4 RCTs, n = 114, RR 1.32, CI 0.64 to 2.74), nor when the studies which did not have such a criterion were analysed separately (7 RCTs, n = 272, RR 1.64, CI 0.95 to 2.81; P = 0.07). The direction of the effect was the same, confidence intervals overlapped broadly, and the test for subgroup differences was not statistically significant (Chi² statistic = 0.22, df = 1 (P = 0.64); I² statistic = 0%).
4.2. Participants with or without treatment‐resistant schizophrenia
The effect of lithium augmentation for the primary outcome 'clinically significant response' was not statistically significant in neither studies on treatment‐resistant participants (4 RCTs, n = 199, RR 1.90, CI 0.90 to 4.01) nor in studies without such an inclusion criterion (6 RCTs, n = 197, RR 1.36, CI 0.68, 2.71). The test for subgroup differences was not statistically significant (Chi² statistic = 0.40, df = one (P = 0.52); I² statistic = 0%).
5. Sensitivity analyses
We performed all sensitivity analyses only on the primary outcome response to treatment in comparison three (adding lithium to antipsychotics versus monotherapy with antipsychotics). The preplanned sensitivity analyses excluding studies with 'implied randomisation' and excluding 'cluster randomised trials' did not apply.
5.1 Exclusion of participants with schizoaffective disorder
We examined whether the exclusion of studies with an inclusion criterion of schizoaffective disorder or prominent affective symptoms led to any important change in terms of the primary outcome 'clinically significant response', and the superiority of lithium augmentation in terms of a clinically significant response no longer met conventional thresholds of statistical significance (7 RCTs, n = 272, RR 1.64, CI 0.95 to 2.81, p = 0.07).
5.2 Exclusion of trials with attrition > 50%
In comparison three, only Schulz 1999 had an attrition rate of higher than 50%. When we excluded this trial from the analysis of the primary outcome 'clinically significant response', the overall result no longer met conventional thresholds of statistical significance (9 RTCs; n = 355, RR 1.67, CI 0.93 to 3.00; P = 0.08).
5.3 Exclusion of non‐double‐blind studies
When we excluded three non‐double‐blind studies from the analysis of the primary outcome 'clinically significant response' (He 2010; Feng 2006; Small 2001), the effect of lithium augmentation became statistically non‐significant (7 RCTs; n = 224, RR 1.82, CI 0.84 to 3.96; P = 0.13).
5.4 Fixed‐effect model
Where we changed the method of data synthesis from the random‐effects model to the fixed‐effect model, the results for our primary outcome remained statistically significant (10 RCTs; n = 396, RR 1.77 CI 1.33 to 2.37).
Discussion
Summary of main results
General
The use of lithium for schizophrenia and schizoaffective disorders has been of interest to researchers for a long period of time. The first randomised controlled trials were published in the early 1970s, with the most recent in 2010. The available studies are well distributed throughout the decades, with seven studies published in the 1970s, six in the 1980s, six in the 1990s, and three in the 21st century. Although we were able to include 22 studies in this review, they had rather small sample sizes, which lacked sufficient power to detect a small to moderate effect, and were incompletely reported in the original publications. However, we are indebted to a number of authors who shared their data with us and made a much better assessment of the available evidence possible (see Acknowledgements).
1. Comparison 1: lithium versus placebo as the sole treatment for schizophrenia
We found no difference between lithium and placebo as the sole treatments for schizophrenia in terms of any outcome parameter analysed. Although it should be noted that only three very small trials were relevant for this comparison, if lithium had a strong effect on the symptoms of schizophrenia, at least some difference compared with placebo might have been found. As lithium has a number of well‐known side effects, its use as a treatment for those with schizophrenia is not justified.
2. Comparison 2: lithium versus antipsychotics as the sole treatment for schizophrenia
2.1 Clinically significant response as defined in the studies
Only three trials reported on this simple measure, and no significant differences were found. Little can be concluded from this analysis including 80 participants. It is disappointing that more trials did not report this simple outcome.
2.2 Leaving the study early
More participants who received lithium salts than those who were on antipsychotics left the studies early. According to a subset of trials, this might reflect a lower efficacy of lithium when compared with antipsychotic agents.
2.3 Global state
2.3.1 Improved according to the Clinical Global Impression Scale
The data revealed no differences between groups, but again only two very small studies provided usable data.
2.3.2 Relapse
It is surprising that there was only one long‐term study (Mattes 1984), because the main indication for lithium salts in affective disorders is the prevention of relapse. A statistically borderline superiority of antipsychotics was found (P = 0.06), but given the very small number of participants included (n = 14), this cannot be considered robust.
2.4 Mental state ‐ general
Pimozide was superior to lithium salts according to several levels of response derived from the Manchester Scale (MS) global score, but only one study, Johnstone 1988, provided data. However, the mean MS global score at endpoint in the same study and the mean Brief Psychiatric Rating Scale (BPRS) at endpoint in three other studies also showed a significant superiority of antipsychotic drugs. Thus, there is some evidence that lithium alone is not as effective as antipsychotic drugs alone for the treatment of schizophrenia, as was expected.
2.5 Mental state ‐ specific
Only one study, Johnstone 1988, provided data for the analysis of specific aspects of the mental state. No significant differences between groups in terms of depressive or manic symptoms were found, but lithium was less effective than pimozide in improving the negative and positive symptoms of schizophrenia. Thus, lithium does not seem to be a viable alternative to antipsychotic drugs for the treatment of the core symptoms of the disorder.
2.6 Adverse events
Again, the fact that only one of two trials reported usable data on adverse events clearly hampered the analysis. There were no significant differences in terms of anticholinergic, dermatologic, gastrointestinal, or extrapyramidal adverse events. Lithium can lead to toxic confusion, especially when its plasma level is outside the therapeutic range, but it is also possible that it is less sedating than antipsychotic drugs. One trial reported a significant difference in terms of an increase of the white blood cell count, but the clinical meaning of this was unclear.
3. Comparison 3: lithium as an adjunct to antipsychotics for schizophrenia
3.1 Clinically significant response
Overall, more participants who received lithium in addition to antipsychotic drugs were classified as having had a clinically significant response. Thus, it is possible that lithium augmentation moderately improves the outcome compared with treatment with antipsychotic drugs alone. Importantly, however, this finding became non‐significant when we excluded those with schizoaffective disorders, non‐double‐blind studies, and studies with a high attrition rate (see Analysis 5.1, Analysis 6.1, and Analysis 7.1). The confidence interval was also quite large, meaning it was possible that the lithium effect was very small. Furthermore, it was not possible to show any effects of specific symptoms of schizophrenia, so lithium may act rather on general symptoms of the disorder. All in all, we do not think that lithium augmentation can be recommended on this basis.
5.1. Analysis.
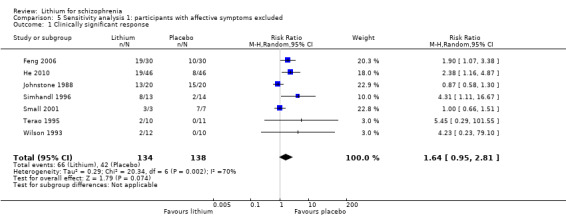
Comparison 5 Sensitivity analysis 1: participants with affective symptoms excluded, Outcome 1 Clinically significant response.
6.1. Analysis.
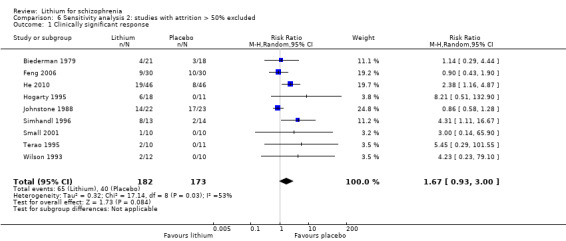
Comparison 6 Sensitivity analysis 2: studies with attrition > 50% excluded, Outcome 1 Clinically significant response.
7.1. Analysis.
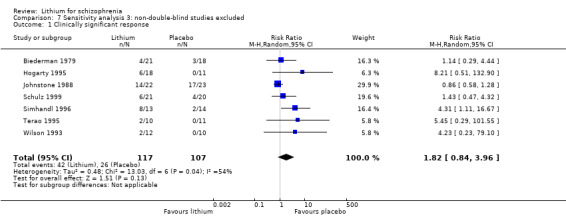
Comparison 7 Sensitivity analysis 3: non‐double‐blind studies excluded, Outcome 1 Clinically significant response.
3.2 Leaving the study early
The difference in the outcome 'leaving the studies early due to any reason' did not reach conventional levels of significance; thus, no difference between groups was identified. The reasons for discontinuing the studies were rarely specified; thus, we judged the vast majority (n = 15) of studies as having a high risk in terms of attrition bias. Based on these low quality results, one should not speculate about the tolerability of lithium augmentation for schizophrenia.
3.3 Global state
There were no significant differences between groups in terms of improvement of global state measured by Clinical Global Impression (CGI) of relapse rates, so any conclusions about the efficacy of lithium augmentation cannot be drawn based on these parameters.
3.4 Mental state ‐ general
It is possible that augmentation with lithium somewhat improves the general mental state of those with schizophrenia. However, this effect was shown for an at least 50% or 35% improvement of the BPRS, but not for an at least 20% improvement. This effect is thus not consistent. Furthermore, one trial that used the Manchester Scale instead of the BPRS as an outcome measure did not find a superiority of lithium augmentation (Johnstone 1988). The reasons for this difference in effect are unclear. Possible explanations are the fact that Johnstone 1988 used a different scale, that it studied acutely ill participants whereas many of the other studies examined treatment‐resistant participants, or that the study used pimozide as an antipsychotic in the study whereas most of the other studies used haloperidol. On the other hand, Feng 2006 and He 2010 found a significant effect of lithium augmentation, measured as at least 50% improvement on the Positive and Negative Syndrome Scale (PANSS) total score. However, it has to be noted that both these studies were not double‐blind, which might have biased this result. To conclude, only limited evidence exists that lithium improves general mental state in schizophrenia.
3.5 Mental state ‐ specific
Few studies reported responses to specific symptoms of the mental state (e.g., depressive, manic, negative, or positive symptoms). Feng 2006 found a significant difference in favour of lithium in the Excited Component of PANSS. He 2010 and Small 2001 showed the same effect for the PANSS positive symptoms scale. No other studies found any apparent differences for the remaining outcomes. The fact that the trials used a number of different scales hampered analysis. In addition, it should again be noted that Feng 2006 and He 2010 were not double‐blind studies.
3.6 Medication use
Less haloperidol was given in the lithium augmentation group. As only one study provided usable data (Wilson 1993), the meaning of this result is unclear. In the same study, a similar number of participants in both groups used benzodiazepines at least once. Again, due to the small sample size, we cannot draw conclusions.
3.7 Adverse events
3.7.1 Delirium
Based on data from two studies (Biederman 1979 and Wilson 1993), two participants in the lithium augmentation group developed delirium, but this was not statistically significant (2 RCTs; n = 61, RR 2.56, CI 0.28 to 23.28).
3.7.2 Movement disorder
No significant differences between groups were found. This is not surprising, because lithium is not known to be frequently associated with movement disorders.
3.7.3 Specific adverse events
Only two studies reported on specific adverse events and found no differences between groups in terms of constipation, dizziness, dry mouth, electroencephalogram (EEG) abnormalities, insomnia, lactation, liver function abnormalities, nausea, QTC (corrected QT interval)/ECG (electrocardiogram) abnormalities, somnolence, tachycardia, and weight gain. Based on this scarce information, we cannot draw firm conclusions about the adverse events. The studies did not record thyroid dysfunction and kidney problems ‐ two major side effects of lithium.
4. Missing outcomes
It is surprising that although the main indication of lithium for affective disorders is relapse prevention, only one small study provided relevant data (Mattes 1984). It is hoped that more data on long‐term effects and on important 'service' outcomes, such as duration of hospital stay, satisfaction with treatment, or costs, will be available for future updates of this review.
5. Subgroup analysis
We performed all subgroup analyses for the primary outcome (clinically significant response) of lithium augmentation. We did not perform any separate subgroup analysis for people with a first episode of schizophrenia, because there were no studies on this subgroup.
5.1 Studies on participants with versus without schizoaffective disorder or prominent affective symptoms
When we analysed separately the studies with an inclusion criterion of prominent affective symptoms/schizoaffective disorder and the studies without such inclusion criteria, there was no significant effect in either subgroup. The direction of effect was the same in both subgroups. Although the subgroup test was far from ideal to address this question, there is no evidence that lithium works better for patients with affective symptoms based on this analysis.
5.2 Participants with treatment‐resistant schizophrenia
We found a statistically significant effect of lithium augmentation for participants with treatment‐resistant schizophrenia. This effect was not significant for non‐treatment‐resistant participants. However, as we identified no significant difference between subgroups, one should not assume that lithium might be more helpful for patients that did not respond to previous antipsychotic treatment. It could be a spurious finding.
6. Sensitivity analysis
We performed all sensitivity analyses for the primary outcome (clinically significant response) of lithium augmentation (comparison three).
6.1 Exclusion of participants with schizoaffective disorder
Excluding people with schizoaffective disorder from the overall analysis resulted in loss of statistical superiority of lithium augmentation over placebo or usual treatment. Therefore, the overall effect might have been in part driven by these participants.
6.2 Exclusion of trials with attrition higher than 50%
Only two studies had an attrition rate that was higher than 50%. Nevertheless, exclusion of these two studies led to a reduction in statistical significance of the primary outcome, meaning it then did not meet conventional criteria of statistical significance anymore (P = 0.08). This finding shows that the superiority of lithium augmentation is not robust and may have been in part based on studies that are more prone to bias.
6.3 Exclusion of non‐double‐blind trials
Excluding non‐double‐blind trials from the overall analysis resulted in a loss of statistical superiority of lithium augmentation over placebo. This result indicates that the lower quality non‐double‐blind studies in part drove the effect. The more carefully controlled double‐blind studies did not show statistically significant effects, which needs to be interpreted as lithium augmentation is not proven as an efficacious treatment for schizophrenia.
6.4 Application of random‐effects model
Application of the fixed‐effect model instead of the random‐effects model did not result in a loss of statistical superiority of lithium augmentation over placebo or usual treatment. As expected, the confidence interval got smaller, but the point estimate remained similar.
Overall completeness and applicability of evidence
Strengths in terms of completeness are that several studies were conducted in participants with treatment‐resistant schizophrenia for which effective adjuncts are most needed. On the other hand, there are open questions. For example, as lithium is effective for bipolar disorder, more studies on participants with schizoaffective disorder or prominent affective symptoms would be useful. The data are also clearly not complete in terms of side effects and long‐term effects. Moreover, the small number of included studies, together with a poor quality of all available data, makes any conclusive judgement about the potential applicability of the obtained results doubtful. Trials with small sample sizes lack sufficient power to detect a small to moderate effect; thus, results from such trials are often inconclusive, even when a real effect does exist. Moreover, it has been suggested that meta‐analyses based on summation of small trials should be interpreted as inconclusive regardless of whether the combined estimate was significant (Davey Smith 1998).
Quality of the evidence
First, as many as 31 out of 50 studies that we eventually excluded form the final analysis failed to randomise the trial sequence. Due to this low quality, we included only 22 studies in the meta‐analysis.
Almost all studies were randomised and double‐blind, but many of them did not present detailed descriptions. Therefore, it is unclear whether some these studies were really adequately randomised and whether treatment allocation was concealed and blinding assured throughout the whole procedure (Feng 2006 and He 2010 were especially problematic in that respect, as they did not provide any information about blinding).
Reporting bias is especially prevalent in this study pool, with high risk being a problem in a majority of reports often but not restricted to missing standard deviations. Without original study protocols and raw data sets, one cannot really use those trials to the full extent.
The threat of other biases was typically absent, although we emphasise that we focused on clear 'other biases' in this regard, because any study can be criticised for one aspect or another.
Overall, using the GRADE approach to interpret findings (Schünemann 2008) and GRADE profiler to analyse the data (GRADE Profiler), we found that the overall quality of the recommendations that we could derive from the preselected outcomes was low.
Potential biases in the review process
We went to considerable lengths to obtain as many relevant studies as possible. However, one can never be certain whether some additional (unpublished) material exist that was not available for the analysis. Moreover, the change from RevMan 4 to the new requirements of RevMan 5 (Review Manager (RevMan)) was not without problems, because we had to re‐examine the old studies.
Agreements and disagreements with other studies or reviews
Some previous unsystematic reviews yielded conflicting results about the applicability and effectiveness of lithium for schizophrenia. One early review stated that there was a beneficial effect of lithium in schizophrenia (Delva 1982), whereas another from approximately the same period of time did not find any evidence to support such a claim (Atre‐Vaidya 1989). A recent review of adjunctive lithium also emphasised this confusion by stressing that many positive early findings are denied by newer contradictory evidence and concluded that any firm conclusion could not be reached (Citrome 2009).
Authors' conclusions
Implications for practice.
For clinicians
There is currently no evidence from randomised controlled trials to support the use of lithium alone as treatment for people with schizophrenia. Lithium is also associated with a wide range of side effects. Although there is some limited evidence to support the use of lithium as an augmentation of antipsychotics for people with schizophrenia, the data is of low quality and cannot be considered conclusive. While we lack evidence to predict which people with schizophrenia (if any) might most benefit from lithium augmentation, issues such as past response to lithium or prominent affective symptoms may warrant consideration by clinicians. For those currently caring for patients who have been receiving lithium as a treatment for schizophrenia, clinicians need to consider whether this treatment should continue to be used.
Those with schizophrenia and schizoaffective disorder
People with schizophrenia should be made aware of the lack of any empirical basis for the use of lithium as a sole agent for their illness. They should know that there is only very limited evidence that lithium is effective as an adjunct to antipsychotics. It should be kept in mind that the underlying evidence base was small and of low quality.
For managers and policymakers
The data cannot support the use of lithium augmentation as a routine measure. Nevertheless, since some superiority was shown in certain efficacy outcomes, managers and policymakers should support further trials on the question.
Implications for research.
General (seeTable 4)
1. Suggested study design.
| Methods | Allocation: random (with details of the specific randomisation method). Blinding: double‐blind (blind or independent raters). Duration: over 26 weeks. |
| Participants |
Diagnosis: people with schizophrenia without prominent affective symptoms; people with schizoaffective disorders; and people with treatment‐resistant schizophrenia.
Age: adults.
Sex: male and female. N = 750*. |
| Interventions |
|
| Outcomes | Clinically significant response** (including relapse), leaving the study early**, service utilisation, behaviour, adverse events, and economic outcomes. All outcomes grouped by time ‐ short‐term (up to 12 weeks), medium‐term (13 to 26 weeks), and long‐term (over 26 weeks). |
| Notes | *150 in each arm. **primary outcomes of interest. |
Any future studies should respect standards of measuring outcomes and of reporting data in order to enhance the comparability of study results (Schulz 2010).
Specific
This review revealed some evidence that lithium augmentation for people with schizophrenia may be effective, but the results were not conclusive. Therefore, further studies are warranted. The following populations are of special research interest:
People with schizoaffective disorders, because lithium is frequently used in addition to antipsychotic drugs for this disorder in the clinical routine.
People with treatment‐resistant schizophrenia. Here, drugs with additional effects on schizophrenic symptoms are most needed. Even small differences in outcome may be of great importance in this subgroup, and therefore, a large simple trial is justified.
People with aggressive behaviour. Lithium is used for those with aggressive or violent episodes, and its evaluation within trials in this subgroup of people with schizophrenia would be valuable.
Relapse prevention. Lithium is frequently used as a 'mood stabiliser' in bipolar disorder. It would be useful to assess its efficacy in relapse prevention for schizophrenia.
What's new
| Date | Event | Description |
|---|---|---|
| 27 August 2015 | New citation required but conclusions have not changed | The results of 2 update searches (2012, 2015) added to the review; new data did not change the overall conclusions of the review. Substantial changes have been made to the structure of the review, with the addition of a 'Summary of findings' table and the updating of the risk of bias of the trials. |
| 20 January 2015 | New search has been performed | 2012 search updated, no new studies found. Results from 2012 search added to review; 2 new studies included. |
History
Protocol first published: Issue 4, 2002 Review first published: Issue 3, 2003
| Date | Event | Description |
|---|---|---|
| 31 October 2008 | Amended | New review format. 2002 update: a number of individual patient data were received from the original authors. This enabled us to examine whether the data were normally distributed. An amendment was added to the methods section. 2006 update: of 4 potentially relevant studies, 3 were not appropriately randomised and one had to be classified as 'awaiting assessment'. |
Acknowledgements
The Cochrane Schizophrenia Group (CSG) maintain a template for the methods section of their reviews; we have used and adapted this.
We would like to thank Profs. and Drs. Braden, Carman, Dube, Gershon, Hogarty, Johnstone, Kendell, Larkins, Lerner, Mattes, Prien, Schulz, Simhandl, Small, Wilson, and Terao for their replies to our letters or for sending us their original participant data. Without the contributions of these colleagues, this review would not have been possible. We thank Chunbo Li for contacting Chinese authors and Yijun Wang for translating Chinese studies. We also wish to thank Mark Fenton, Judy Wright, Samantha Roberts, and Farhad Shokraneh for the trial searches and the editorial base of the CSG for their continuous help.
Hong‐ling Yang kindly peer‐reviewed this version of the review.
Appendices
Appendix 1. Previous methods sections
1. Selection of trials
Material downloaded from electronic sources included details of author, institution, or journal of publication.
The principal reviewer (SL) inspected all reports. JM then re‐inspected thesein order to ensure reliable selection. We resolved any disagreement by discussion, and where there was still doubt, we acquired the full article for further inspection. Upon obtaining the full articles, we decided whether the studies met the review criteria. If we could not resolve disagreement by discussion, we sought further information and added these trials to the list of those awaiting assessment.
2. Quality assessment
We assessed the methodological quality of the trials included in this review using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). These criteria are based on the evidence of a strong relationship among the potential for bias in the results and the allocation concealment (Schulz 1995) and is defined below:
A. Low risk of bias (adequate allocation concealment).
B. Moderate risk of bias (some doubt about the results).
C. High risk of bias (inadequate allocation concealment).
For the purpose of the analysis in this review, we included trials if they met the criteria A or B in the Cochrane Handbook for Systematic Reviews of Interventions.
3. Data extraction
We (SL and JM) independently extracted the data from included studies. We discussed any disagreement, documented the decisions, and, if necessary, contacted the authors of the studies for clarification. We documented justification for excluding references from the review.
4. Data management
4.1 Intention‐to‐treat
For studies that did not specify the reasons for people leaving the study early (dropped out), we assumed that these people had no change in the clinical outcome variables. Wahlbeck 2001 highlighted the problem of high dropout rates in randomised trials of drug treatments for schizophrenia. Since there is no evidence on the degree of attrition that makes a reasonable analysis of the data possible, we included all trials in the main analysis. However, we used a sensitivity analysis to test whether the exclusion of trials with dropout rates higher than 50% significantly changed the results of the primary outcome parameters. When insufficient data were provided to identify the original group size (prior to dropouts), we contacted the authors and allocated the trials to the 'awaiting assessment' list.
4.2 Cross‐over design
We expected that some trials would use a cross‐over design. In order to exclude the potential additive effect in the second or more stages of these trials, we only analysed data from the first stage.
4.3 Data types
We assessed outcomes using continuous (for example, changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change', or 'much change'), or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Currently, RevMan does not support categorical data, so they could not be analysed as such.
4.3.1 Dichotomous data
Where possible, we converted outcome measures to dichotomous data. This may be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. If the authors of a study had used a predefined cut‐off point for determining clinical effectiveness, we used this where appropriate. Otherwise, we generally assumed that we could consider a 50% reduction of a scale (e.g., the Brief Psychiatric Rating Scale ‐ Overall 1962) or a rating of 'at least much improved' according to the Clinical Global Impression Scale (Guy 1976) as a clinically significant response.
For dichotomous outcomes, we estimated a relative risk (RR) with the 95% confidence interval (CI) based on a fixed‐effect model in case of homogeneous outcomes and based on a random‐effects model in the case of heterogeneous outcomes. When overall results were significant, we calculated the number needed to treat (NNT) or the number needed to harm (NNH) or both as the inverse of the risk reduction. It has been shown that RRs are more intuitive (Boissel 1999) than odds ratios and that clinicians tend to interpret odds ratios as relative risks (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. However, we inspected data to see if an analysis using a Mantel‐Haenszel odds ratio and a random‐effects model made a substantive difference.
4.3.2 Continuous data
4.3.2.1 Normal distribution
Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to data derived from continuous measures of endpoint ('state' data). The criteria were used before inclusion: When a scale started from zero, the standard deviation, when multiplied by two, was less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996). Endpoint scores on scales often have a finite start and endpoint, and this rule can be applied to them. When continuous data are presented on a scale that includes a possibility of negative values (such as change on a scale), it is impossible to tell whether data are non‐normally distributed (skewed) or not. It is thus preferable to use scale endpoint data, which typically cannot have negative values. If endpoint data were not available, we chose to use change data, because the statistics used in MetaView are rather robust towards skewness. If a scale starts from a positive value (such as Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210), the calculation described above in (b) should be modified to take the scale starting point into account. In these cases, skewness is present if 2 SD > (S‐S min), where S is the mean score and S min is the minimum score.
4.3.2.2 Intention‐to‐treat versus completer analyses
We assumed that for incomplete continuous data, intention‐to‐treat analyses would be impossible. In these cases, we analysed data as they were presented in the original publications.
4.3.2.3 Summary statistic
For continuous outcomes, we estimated a weighted mean difference (WMD) between groups. Again, we used a fixed‐effect model for homogeneous outcomes and a random‐effects model for heterogeneous outcomes. Whenever possible, we took the opportunity to make direct comparisons between trials that used the same measurement instrument to quantify specific outcomes. Where continuous data were presented from different scales rating the same effect, we presented both sets of data and inspected the general direction of effect.
4.3.2.4 Rating scales
A wide range of instruments is available to measure mental health outcomes. These instruments vary in quality and many are not valid or even ad hoc. For outcome instruments, some minimum standards have to be set. We only included continuous data from rating scales if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000), the instrument was either a self report or completed by an independent rater or relative (not the therapist), and the instrument could be considered a global assessment of an area of functioning. However, as it was expected that therapists would frequently also be the rater, we commented on such data as 'prone to bias'.
4.4 Data display
We entered data into RevMan in such a way that the area to the left of the line of no effect indicated a favourable outcome for lithium alone or lithium augmentation.
4.5 Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals are unduly narrow, and statistical significance is overestimated causing type I errors (Bland 1997; Gulliford 1999). Secondly, RevMan does not currently support meta‐analytic pooling of clustered dichotomous data, even when these are correctly analysed by the authors of primary studies, since the 'design effect' (a statistical correction for clustering) cannot be incorporated.
Where clustering was not accounted for in primary studies, we presented data in a table, with an asterisk (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies, to seek intraclass correlation coefficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, we presented these data in a table. We will attempt no further secondary analysis (including meta‐analytic pooling) until there is consensus on the best methods of doing so and until RevMan, or any other software, allows this. A Cochrane Statistical Methods Workgroup is currently addressing this issue. In the interim,we very crudely classified individual studies as positive or negative, according to whether a statistically significant result (P < 0.05) was obtained for the outcome in question, using an analytic method that allowed for clustering.
5. Heterogeneity
After considering the likelihood of clinical heterogeneity based on comparisons of the included studies, we visually inspected graphs to investigate the possibility of statistical heterogeneity. We calculated the value of the I² statistic to provide an estimate of the percentage of variability due to heterogeneity rather than chance alone. We interpreted an I² statistic value of 50% or greater as indicating high levels of heterogeneity (Higgins 2003). When the results were statistically significantly heterogeneous, we sought reasons for the heterogeneity, and if these were identified, we excluded these studies. If reasons were not found, we combined the studies using a random‐effects model.
6. Publication bias
We entered data from all included trials into a funnel graph (trial effect versus trial size or 'precision') in an attempt to investigate the likelihood of overt publication bias. We undertook a formal test of funnel plot asymmetry (suggesting potential publication bias) where appropriate (Egger 1997). We set significance levels of P < 0.1 a priori to accept the presence of asymmetry.
7. Sensitivity analysis
We examined whether the exclusion of schizoaffective participants led to a significant change of the primary outcomes by carrying out a sensitivity analysis. We also used a sensitivity analysis to test whether the exclusion of trials with attrition rates of higher than 50% significantly changed the results of the primary outcome parameters.
8. Posthoc amendment to the protocol
For a number of studies, we were able to examine individual participant data. This enabled us to dichotomise many scale‐derived data. As explained above (see 4.3.1), we used a clinical rating of 'at least much improved' or a 50% reduction of a rating scale as a cut‐off for a clinically significant response whenever the original authors did not present a definition of their own. However, since there is an uncertainty about which cut‐off is best, we also analysed the data using a relatively low threshold (at least 20% reduction) and an intermediate threshold (at least 35% reduction) as in the review on carbamazepine (Leucht 2014). Furthermore, when original participant data were available, we used the Kolmogornov‐Smirnoff test to assess normal distribution (SPSS 2001). Therefore, continuous data could sometimes be used for meta‐analytic calculations, although these data did not meet the criteria stipulated in 4.3.2.1 above.
Appendix 2. Previous searches
2.1 Search in 2002 and 2006
We searched the Cochrane Schizophrenia Group Trials Study‐Based Register in November 2006 using the following phrase: [((lithium* or lithicarb* or eskalith* or lithobid* or lithane* or cibalith‐s* or quilonum* or hypnorex*) in title, abstract and index fields in REFERENCE) OR (lithium* in interventions field in STUDY)]. This register is compiled by systematic searches of major databases, handsearches, and conference proceedings (see Group's Module).
In March 2002, we also searched the Cochrane Schizophrenia Group's Trials Study‐Based Register with the same phrase.
2.2 2012 search
2.2.1 Abstract section
We searched the Cochrane Schizophrenia Group's Trials Study‐Based Register (July 6, 2012). This register is compiled by methodical searches of MEDLINE, EMBASE, BIOSIS, CINAHL (Cumulative Index to Nursing and Allied Health Literature), Dissertation Abstracts, LILACS (Latin American and Caribbean Health Science Information database), PSYNDEX, PsycINFO, RUSSMED, and Sociofile, supplemented with handsearching of relevant journals and numerous conference proceedings. For the first version of the review, we also contacted pharmaceutical companies and authors of relevant studies to identify further trials and to obtain original participant data.
2.2.2 Electronic searches
2.2.2.1 Cochrane Schizophrenia Group Trials Study‐Based Register
The Trials Search Co‐ordinator searched the Cochrane Schizophrenia Group's Trials Study‐Based Register (July 6, 2012):
1.1 Intervention search
The 'Intervention' field was searched using the following phrase:
(lithium* or lithicarb* or eskalith* or lithobid* or lithane* or cibalith‐s* or quilonum* or hypnorex*)
The Cochrane Schizophrenia Group's Trials Study‐Based Register is compiled by systematic searches of major resources (including MEDLINE, EMBASE, AMED (the Allied and Complementary Medicine Database), BIOSIS, CINAHL, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates; handsearches; grey literature; and conference proceedings. The searches do not have language limitation (for details of databases searched by the Schizophrenia Group, please see their Group's Module).
2.2.3 Searching other resources
2.2.3.1 Reference searching
We inspected references of all included studies for further relevant studies.
2.2.3.2 Personal contact
For the original search, we contacted the first author of each included study for information regarding unpublished trials and additional information (however, this was not done in this update).
Data and analyses
Comparison 1. Lithium as sole treatment versus placebo as sole treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 2 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.44, 1.55] |
| 2 Leaving the study early | 3 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.29, 4.44] |
| 3 Mental state: 1. General ‐ at least 20% MS or NH improvement | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 At least 20% MS improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.61, 1.40] |
| 3.2 At least 20% NH improvement | 1 | 15 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.13, 3.53] |
| 4 Mental state: 2. General ‐ at least 35% MS or NH improvement | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 At least 35% MS improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.47, 1.58] |
| 4.2 At least 35% NH improvement | 1 | 15 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.13, 3.53] |
| 5 Mental state: 3. General ‐ at least 50% MS or NH improvement | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 At least 50% MS improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.44, 1.69] |
| 5.2 At least 50% NH improvement | 1 | 15 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.13, 3.53] |
| 6 Mental state: 4. General ‐ mean MS global score at endpoint (high = poor) | 1 | 39 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐2.88, 2.88] |
| 7 Mental state: 5. General ‐ unable to use (skewed data) | Other data | No numeric data | ||
| 8 Mental state: 6. Specific ‐ depression ‐ various degrees of MADRS improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 At least 20% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.70, 2.19] |
| 8.2 At least 35% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.57, 1.94] |
| 8.3 At least 50% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.65, 3.16] |
| 9 Mental state: 7. Specific ‐ mania ‐ various degrees of Bech‐Rafaelsen Mania Scale improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 At least 20% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.62, 1.67] |
| 9.2 At least 35% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.49, 2.68] |
| 9.3 At least 50% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.38, 2.81] |
| 10 Mental state: 8. Specific ‐ negative symptoms ‐ various degrees of MS‐negative subscore improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 At least 20% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.49, 2.68] |
| 10.2 At least 35% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.41, 2.44] |
| 10.3 At least 50% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.41, 2.44] |
| 11 Mental state: 9. Specific ‐ positive symptoms ‐ various degrees of MS‐positive subscore improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 At least 20% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.42, 1.08] |
| 11.2 At least 35% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.44, 1.69] |
| 11.3 At least 50% improvement | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.44, 1.69] |
| 12 Adverse events: 1. Movement disorder ‐ unable to use (skewed data) | Other data | No numeric data |
1.1. Analysis.
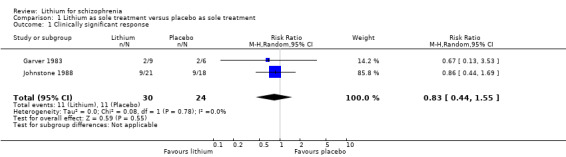
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 1 Clinically significant response.
1.2. Analysis.
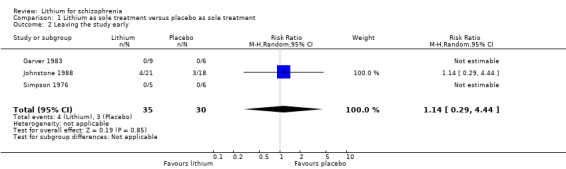
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 2 Leaving the study early.
1.3. Analysis.
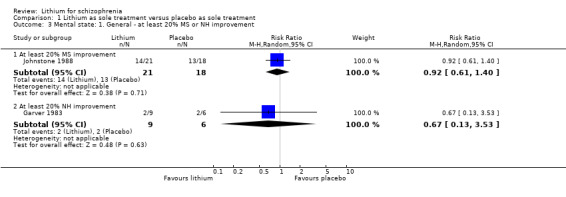
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 3 Mental state: 1. General ‐ at least 20% MS or NH improvement.
1.4. Analysis.

Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 4 Mental state: 2. General ‐ at least 35% MS or NH improvement.
1.5. Analysis.
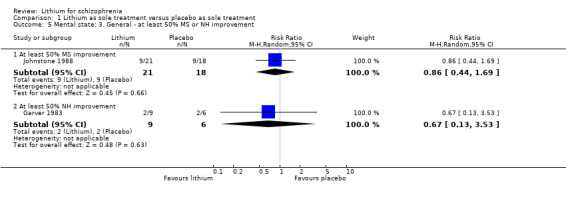
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 5 Mental state: 3. General ‐ at least 50% MS or NH improvement.
1.6. Analysis.
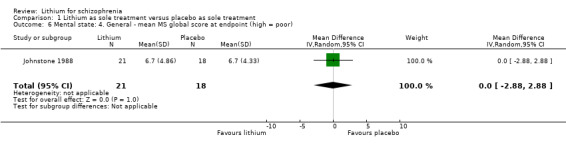
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 6 Mental state: 4. General ‐ mean MS global score at endpoint (high = poor).
1.7. Analysis.
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 7 Mental state: 5. General ‐ unable to use (skewed data).
| Mental state: 5. General ‐ unable to use (skewed data) | |
|---|---|
| Study | |
| Garver 1983 | NH global score at endpoint ‐ high = poor. Lithium: mean = 13.8, SD = 7.2. N = 9. Placebo: mean = 13.2, SD = 7.8. N = 6. |
1.8. Analysis.
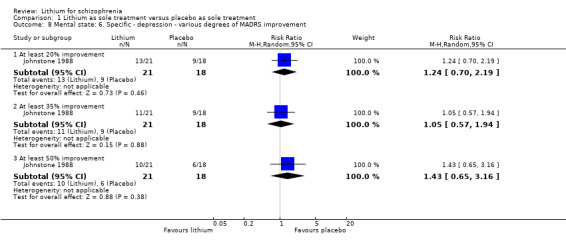
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 8 Mental state: 6. Specific ‐ depression ‐ various degrees of MADRS improvement.
1.9. Analysis.
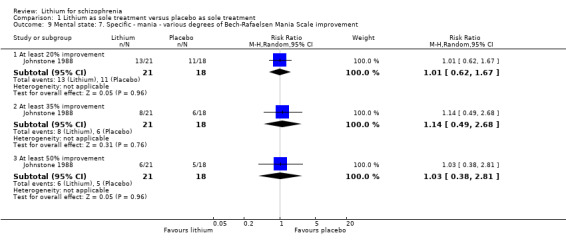
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 9 Mental state: 7. Specific ‐ mania ‐ various degrees of Bech‐Rafaelsen Mania Scale improvement.
1.10. Analysis.
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 10 Mental state: 8. Specific ‐ negative symptoms ‐ various degrees of MS‐negative subscore improvement.
1.11. Analysis.
Comparison 1 Lithium as sole treatment versus placebo as sole treatment, Outcome 11 Mental state: 9. Specific ‐ positive symptoms ‐ various degrees of MS‐positive subscore improvement.
Comparison 2. Lithium versus antipsychotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 3 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.28, 1.84] |
| 2 Leaving the study early | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Overall | 8 | 270 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.01, 3.11] |
| 2.2 Due to adverse events | 4 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.08, 19.21] |
| 2.3 Due to inefficacy of treatment | 4 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 2.89 [1.09, 7.67] |
| 3 Global state: 1. Improved CGI | 2 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.06, 5.98] |
| 4 Global state: 2. Relapse | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5 Mental state: 1. General ‐ various degrees of MS global score improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 At least 20% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.53, 1.01] |
| 5.2 At least 35% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.37, 1.00] |
| 5.3 At least 50% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.33, 1.01] |
| 6 Mental state: 2. General ‐ BPRS/MS global score at endpoint (high = poor) | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Mean BPRS at endpoint | 3 | 92 | Mean Difference (IV, Random, 95% CI) | 10.23 [6.32, 14.15] |
| 6.2 Mean MS at endpoint | 1 | 44 | Mean Difference (IV, Random, 95% CI) | 3.0 [0.38, 5.62] |
| 6.3 Mean SCI at endpoint | 1 | 11 | Mean Difference (IV, Random, 95% CI) | ‐2.0 [‐81.29, 77.29] |
| 7 Mental state: 3. Specific ‐ depression ‐ various degrees of MADRS improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 At least 20% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.53, 1.18] |
| 7.2 At least 35% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.44, 1.14] |
| 7.3 At least 50% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.43, 1.25] |
| 8 Mental state: 4. Specific ‐ mania ‐ various degrees of Bech‐Rafaelsen Mania Scale improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 At least 20% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.58, 1.37] |
| 8.2 At least 35% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.35, 1.29] |
| 8.3 At least 50% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.24, 1.09] |
| 9 Mental state: 5. Specific ‐ negative symptoms ‐ various degrees of MS‐negative subscore improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 At least 20% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.23, 0.69] |
| 9.2 At least 35% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.20, 0.68] |
| 9.3 At least 50% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.20, 0.68] |
| 10 Mental state: 6. Specific ‐ positive symptoms ‐ various degrees of MS‐positive subscore improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 At least 20% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.36, 0.83] |
| 10.2 At least 35% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.29, 0.83] |
| 10.3 At least 50% improvement | 1 | 44 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.29, 0.83] |
| 11 Adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Blurred vision | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.12, 3.21] |
| 11.2 Dry mouth | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.28, 1.66] |
| 11.3 Constipation | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.07, 1.38] |
| 12 Adverse events: 2. Central nervous system/neurologic | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 Ataxia | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.12, 3.21] |
| 12.2 Dizziness | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.12, 3.21] |
| 12.3 Hyperactive reflexes | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 8.66 [0.46, 162.49] |
| 12.4 Muscle weakness | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.18, 8.41] |
| 12.5 Slurred speech | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.15, 4.70] |
| 12.6 Somnolence | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.04, 0.73] |
| 12.7 Toxic confusion | 2 | 104 | Risk Ratio (M‐H, Random, 95% CI) | 8.83 [1.16, 67.38] |
| 13 Adverse events: 3. Dermatologic ‐ pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14 Adverse events: 4. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 Dehydration | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 3.71 [0.16, 88.51] |
| 14.2 Nausea | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.12, 3.21] |
| 14.3 Vomiting | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.15, 4.70] |
| 15 Adverse events: 5. Movement disorder | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Parkinsonism | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.01, 5.00] |
| 15.2 Tremor | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.69, 6.87] |
| 15.3 Use of antiparkinson medication | 1 | 21 | Risk Ratio (M‐H, Random, 95% CI) | 0.10 [0.01, 1.68] |
| 16 Adverse events: 6. Unable to use (skewed data) | Other data | No numeric data | ||
| 17 Laboratory abnormalities | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 Decreased white blood cell count | 1 | 21 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.11] |
| 17.2 Increased white blood cell count | 1 | 21 | Risk Ratio (M‐H, Random, 95% CI) | 17.42 [1.14, 265.34] |
| 17.3 Increased blood uric acid level | 1 | 21 | Risk Ratio (M‐H, Random, 95% CI) | 6.42 [0.37, 110.71] |
| 17.4 Proteinuria | 1 | 21 | Risk Ratio (M‐H, Random, 95% CI) | 4.58 [0.25, 85.33] |
| 18 Mental state: 2. General ‐ BPRS/MS global score at endpoint (high = poor) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 18.1 Mean BPRS at endpoint | 3 | 92 | Std. Mean Difference (IV, Random, 95% CI) | 1.02 [0.16, 1.89] |
| 18.2 Mean MS at endpoint | 1 | 44 | Std. Mean Difference (IV, Random, 95% CI) | 0.67 [0.06, 1.28] |
2.1. Analysis.
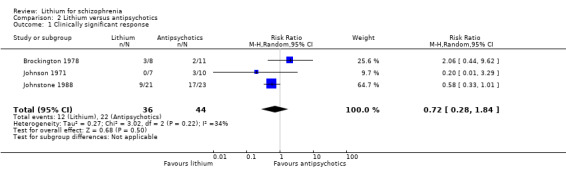
Comparison 2 Lithium versus antipsychotics, Outcome 1 Clinically significant response.
2.2. Analysis.
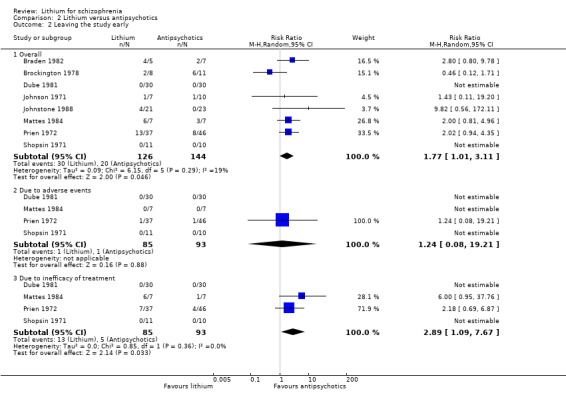
Comparison 2 Lithium versus antipsychotics, Outcome 2 Leaving the study early.
2.3. Analysis.
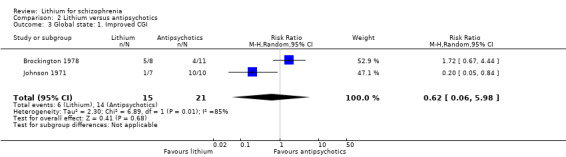
Comparison 2 Lithium versus antipsychotics, Outcome 3 Global state: 1. Improved CGI.
2.4. Analysis.
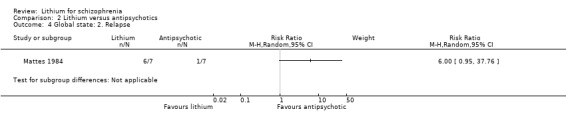
Comparison 2 Lithium versus antipsychotics, Outcome 4 Global state: 2. Relapse.
2.5. Analysis.
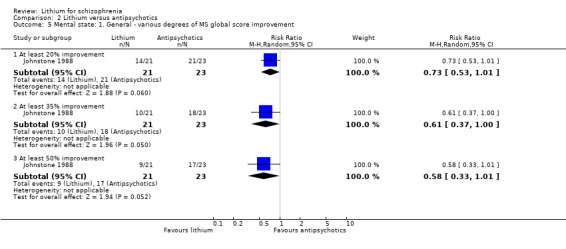
Comparison 2 Lithium versus antipsychotics, Outcome 5 Mental state: 1. General ‐ various degrees of MS global score improvement.
2.6. Analysis.
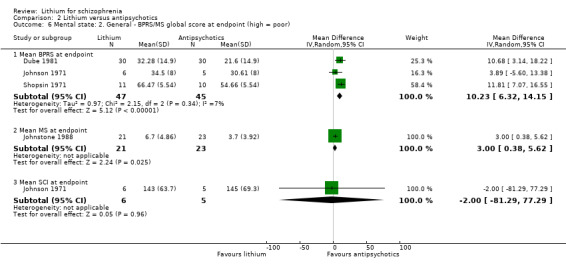
Comparison 2 Lithium versus antipsychotics, Outcome 6 Mental state: 2. General ‐ BPRS/MS global score at endpoint (high = poor).
2.7. Analysis.
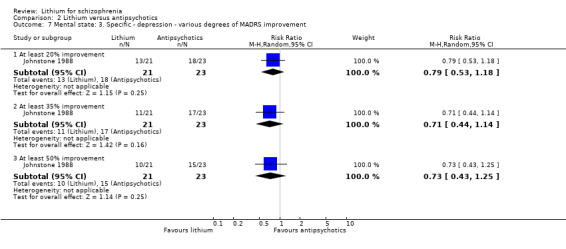
Comparison 2 Lithium versus antipsychotics, Outcome 7 Mental state: 3. Specific ‐ depression ‐ various degrees of MADRS improvement.
2.8. Analysis.
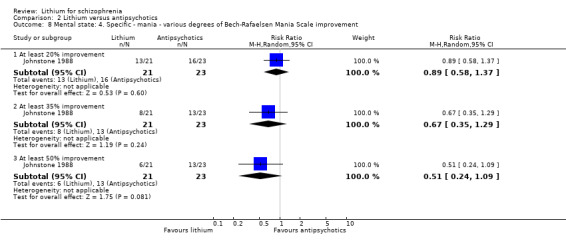
Comparison 2 Lithium versus antipsychotics, Outcome 8 Mental state: 4. Specific ‐ mania ‐ various degrees of Bech‐Rafaelsen Mania Scale improvement.
2.9. Analysis.
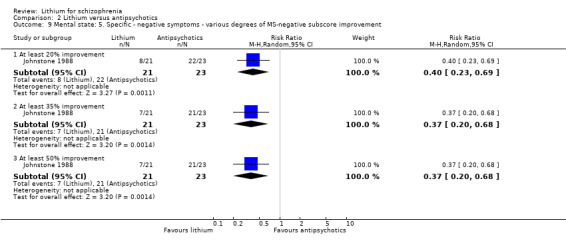
Comparison 2 Lithium versus antipsychotics, Outcome 9 Mental state: 5. Specific ‐ negative symptoms ‐ various degrees of MS‐negative subscore improvement.
2.10. Analysis.
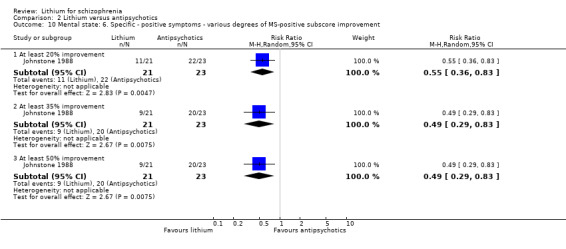
Comparison 2 Lithium versus antipsychotics, Outcome 10 Mental state: 6. Specific ‐ positive symptoms ‐ various degrees of MS‐positive subscore improvement.
2.11. Analysis.
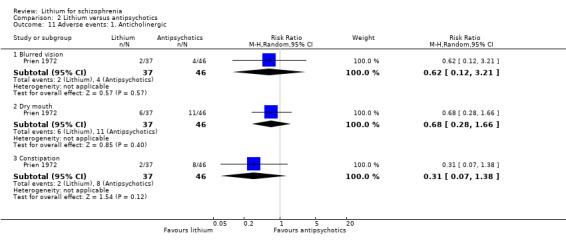
Comparison 2 Lithium versus antipsychotics, Outcome 11 Adverse events: 1. Anticholinergic.
2.12. Analysis.
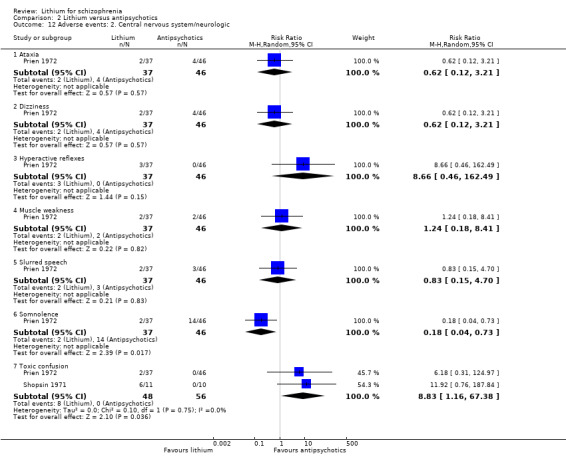
Comparison 2 Lithium versus antipsychotics, Outcome 12 Adverse events: 2. Central nervous system/neurologic.
2.13. Analysis.
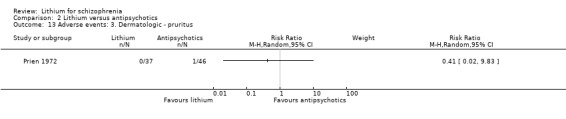
Comparison 2 Lithium versus antipsychotics, Outcome 13 Adverse events: 3. Dermatologic ‐ pruritus.
2.14. Analysis.

Comparison 2 Lithium versus antipsychotics, Outcome 14 Adverse events: 4. Gastrointestinal.
2.15. Analysis.
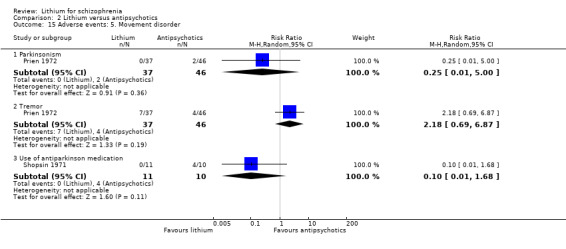
Comparison 2 Lithium versus antipsychotics, Outcome 15 Adverse events: 5. Movement disorder.
2.16. Analysis.
Comparison 2 Lithium versus antipsychotics, Outcome 16 Adverse events: 6. Unable to use (skewed data).
| Adverse events: 6. Unable to use (skewed data) | ||
|---|---|---|
| Study | Mean + SD | Mean + SD |
| Johnstone 1988 | AIMS global score at endpoint ‐ high = poor. Pimozide: mean = 3.6, SD = 3.2. N = 23. Lithium: mean = 3.1, SD = 2.7. N = 21. | TAKE global score at endpoint ‐ high = poor. Pimozide: mean = 5.5, SD = 4.0. N = 23. Lithium: mean = 3.2, SD = 2.4. N = 21. |
2.17. Analysis.
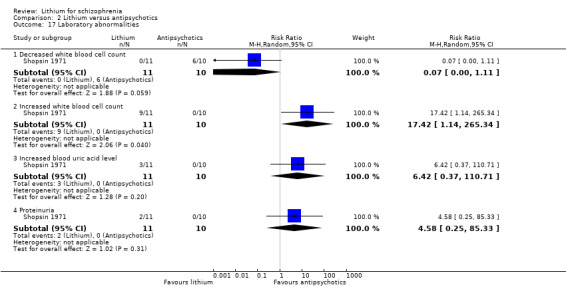
Comparison 2 Lithium versus antipsychotics, Outcome 17 Laboratory abnormalities.
2.18. Analysis.
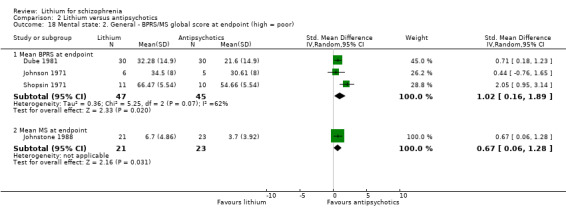
Comparison 2 Lithium versus antipsychotics, Outcome 18 Mental state: 2. General ‐ BPRS/MS global score at endpoint (high = poor).
Comparison 3. Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 10 | 396 | Risk Ratio (M‐H, Random, 95% CI) | 1.81 [1.10, 2.97] |
| 2 Leaving the study early | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Overall | 11 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 1.89 [0.93, 3.84] |
| 2.2 Due to adverse events | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 4.32 [0.22, 84.48] |
| 3 Global state: 1. Improved CGI | 4 | 115 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.57, 4.02] |
| 4 Global state: 2. Relapse | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Mental state: 1. General ‐ number of participants with at least 20% BPRS/MS improvement | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 At least 20% BPRS improvement | 5 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.83, 1.76] |
| 5.2 At least 20% MS improvement | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.71, 1.13] |
| 6 Mental state: 2. General ‐ number of participants with at least 35% BPRS/MS improvement | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 At least 35% BPRS improvement | 5 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.02, 3.10] |
| 6.2 At least 35% MS improvement | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.67, 1.30] |
| 7 Mental state: 3. General ‐ number of participants with at least 50% BPRS/MS/PANSS improvement | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 At least 50% BPRS improvement | 5 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 2.52 [1.17, 5.42] |
| 7.2 At least 50% MS improvement | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.69, 1.40] |
| 7.3 At least 50% PANSS improvement | 2 | 152 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.16, 1.90] |
| 8 Mental state: 4. General ‐ BPRS/MS/PANSS total score at endpoint (high = poor) | 7 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 BRPS at endpoint | 4 | 102 | Mean Difference (IV, Random, 95% CI) | ‐3.26 [‐7.10, 0.57] |
| 8.2 MS at endpoint | 1 | 45 | Mean Difference (IV, Random, 95% CI) | 0.70 [‐1.53, 2.93] |
| 8.3 PANSS at endpoint | 2 | 152 | Mean Difference (IV, Random, 95% CI) | ‐8.53 [‐11.44, ‐5.62] |
| 9 Mental state: 5.1 Specific ‐ depression ‐ at least 20% MADRS/BPRS‐depression score improvement | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Montgomery‐Asberg | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.72, 1.35] |
| 9.2 BPRS depression | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.48, 1.95] |
| 10 Mental state: 5.2 Specific ‐ depression ‐ at least 35% MADRS/BPRS‐depression score improvement | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Montgomery‐Asberg | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.66, 1.45] |
| 10.2 BPRS depression | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.53, 2.55] |
| 11 Mental state: 5.3 Specific ‐ depression ‐ at least 50% MADRS/BPRS‐depression score improvement | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Montgomery‐Asberg | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.69, 1.58] |
| 11.2 BPRS depression | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.53, 2.55] |
| 12 Mental state: 5.4 Specific ‐ depression‐ average HS or MAS at endpoint (high = poor) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12.1 Hamilton Depression Scale (HS) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 3.40 [‐7.66, 14.46] |
| 12.2 Montgomery‐Asberg Scale (MAS) | 1 | 45 | Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐1.64, 0.96] |
| 13 Mental state: 6.1 Specific ‐ mania ‐ at least 20% Bech‐Rafaelsen Mania Scale improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14 Mental state: 6.2 Specific ‐ mania ‐ at least 35% Bech‐Rafaelsen Mania Scale improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15 Mental state: 6.3 Specific ‐ mania ‐ at least 50% Bech‐Rafaelsen Mania Scale improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16 Mental state: 6.4 Specific ‐ mania ‐ mean PANSS‐EC at endpoint | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17 Mental state: 7.1 Specific ‐ negative symptoms ‐ at least 20% SANS/MS/PANSS‐negative score improvement | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 SANS | 3 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.66, 3.10] |
| 17.2 MS negative subscore | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.81, 1.11] |
| 17.3 PANSS negative | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.12, 2.14] |
| 18 Mental state: 7.2 Specific ‐ negative symptoms ‐ at least 35% SANS/MS/PANSS‐negative score improvement | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 SANS | 3 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.64, 2.04] |
| 18.2 MS negative subscore | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.83, 1.20] |
| 18.3 PANSS negative | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 13.87] |
| 19 Mental state: 7.3 Specific ‐ negative symptoms ‐ at least 50% SANS/MS/PANSS‐negative score improvement | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 SANS | 3 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.50, 2.17] |
| 19.2 MS negative subscore | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.83, 1.20] |
| 19.3 PANSS negative | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 65.90] |
| 20 Mental state: 7.4 Specific ‐ negative symptoms ‐ BPRS, MS, SANS, or PANSS negative score at endpoint (high = poor) | 7 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.1 BPRS negative subscore | 2 | 61 | Mean Difference (IV, Random, 95% CI) | 0.56 [‐1.31, 2.43] |
| 20.2 MS negative subscore | 1 | 45 | Mean Difference (IV, Random, 95% CI) | 0.29 [‐0.31, 0.89] |
| 20.3 PANSS negative subscore | 2 | 112 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐2.85, 2.72] |
| 20.4 SANS | 2 | 41 | Mean Difference (IV, Random, 95% CI) | ‐4.50 [‐8.81, ‐0.19] |
| 21 Mental state: 8.1 Specific ‐ positive symptoms ‐ at least 20% BPRS/MS/PANSS‐positive score improvement | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 21.1 BPRS positive subscore | 2 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.87, 1.82] |
| 21.2 MS positive subscore | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.93, 1.18] |
| 21.3 PANSS positive | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.42, 2.40] |
| 22 Mental state: 8.2 Specific ‐ positive symptoms ‐ at least 35% BPRS/MS/PANSS‐positive score improvement | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 22.1 BPRS positive subscore | 2 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.66, 1.98] |
| 22.2 MS positive subscore | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.73, 1.21] |
| 22.3 PANSS positive | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.34, 2.93] |
| 23 Mental state: 8.3 Specific ‐ positive symptoms ‐ at least 50% BPRS/MS/PANSS‐positive score improvement | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 23.1 BPRS positive subscore | 2 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.71, 2.28] |
| 23.2 MS positive subscore | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.62, 1.13] |
| 23.3 PANSS positive | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.40, 4.49] |
| 24 Mental state: 8.4 Specific ‐ positive symptoms ‐ mean BPRS, MS, or PANSS‐positive subscore (high = poor) | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 24.1 BPRS positive subscore | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 0.14 [‐3.45, 3.73] |
| 24.2 Manchester Scale positive subscore | 1 | 45 | Mean Difference (IV, Random, 95% CI) | 0.23 [‐1.34, 1.80] |
| 24.3 PANSS positive subscore | 2 | 112 | Mean Difference (IV, Random, 95% CI) | ‐3.15 [‐4.91, ‐1.39] |
| 25 Aggressive behaviour ‐ mean PANSS supplemental aggression risk profile at endpoint | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 25.1 PANSS Supplemental at endpoint | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐1.34 [‐2.76, 0.08] |
| 26 Medication use: 1. Mean haloperidol dose (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 27 Medication use: 2. Mean dose of antipsychotics (unable to use ‐ skewed data) | Other data | No numeric data | ||
| 28 Medication use: 3. Number of particpants taking benzodiazepines | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 29 Adverse events: 1. Central nervous system ‐ delirium | 2 | 61 | Risk Ratio (M‐H, Random, 95% CI) | 2.56 [0.28, 23.28] |
| 30 Adverse events: 2. Movement disorder ‐ dichotomous data | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 30.1 At least one extrapyramidal side effect | 2 | 113 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [0.22, 9.40] |
| 30.2 Use of antiparkinson medication | 2 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.58, 1.63] |
| 30.3 Akathisia | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.28, 1.99] |
| 30.4 Tremor | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.35, 2.21] |
| 31 Adverse events: 3. Movement disorder ‐ average SAS score at endpoint (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 32 Adverse events: 4. Movement disorder ‐ unable to use (skewed data) | Other data | No numeric data | ||
| 33 Adverse events: 5. Non‐specific dyscomfort | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 34 Adverse events: 6. UKU at endpoint ‐ unable to use (skewed data) | Other data | No numeric data | ||
| 35 Adverse events: Specific | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 35.1 Constipation | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.50, 3.54] |
| 35.2 Dizziness | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.21, 4.70] |
| 35.3 Dry mouth | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.42, 3.21] |
| 35.4 Electroencephalogram abnormalities | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.91, 1.39] |
| 35.5 Insomnia | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.50, 2.48] |
| 35.6 Lactation | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.32] |
| 35.7 Liver function abnormalities | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 35.8 Nausea | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.38, 2.62] |
| 35.9 QTC/ECG abnormalities | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.74, 1.69] |
| 35.10 Somnolence | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.37, 1.79] |
| 35.11 Tachycardia | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.26, 8.56] |
| 35.12 Weight gain | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.38, 1.57] |
3.1. Analysis.
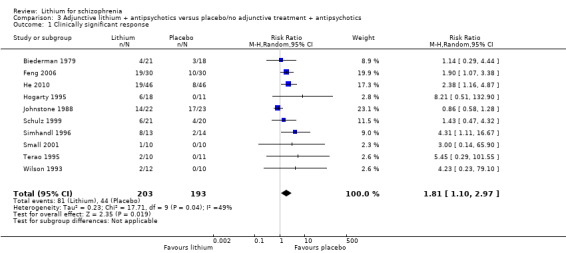
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 1 Clinically significant response.
3.2. Analysis.
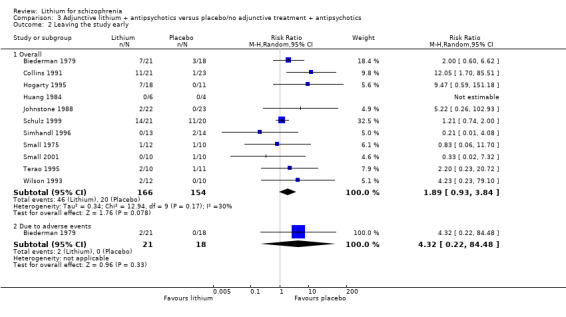
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 2 Leaving the study early.
3.3. Analysis.
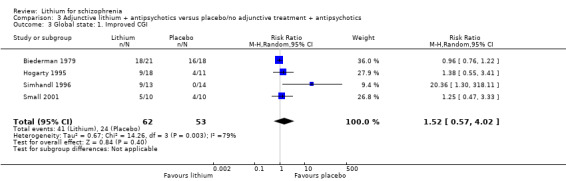
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 3 Global state: 1. Improved CGI.
3.4. Analysis.

Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 4 Global state: 2. Relapse.
3.5. Analysis.
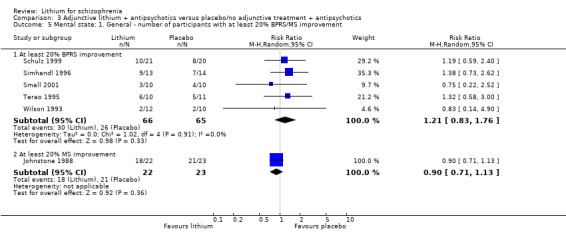
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 5 Mental state: 1. General ‐ number of participants with at least 20% BPRS/MS improvement.
3.6. Analysis.
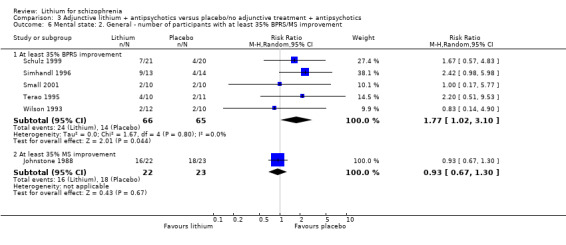
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 6 Mental state: 2. General ‐ number of participants with at least 35% BPRS/MS improvement.
3.7. Analysis.
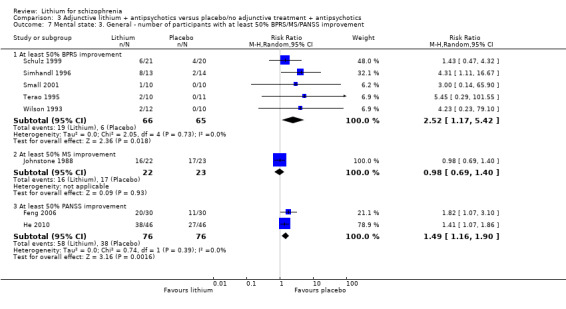
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 7 Mental state: 3. General ‐ number of participants with at least 50% BPRS/MS/PANSS improvement.
3.8. Analysis.
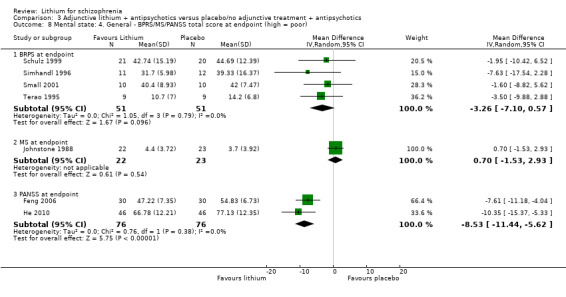
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 8 Mental state: 4. General ‐ BPRS/MS/PANSS total score at endpoint (high = poor).
3.9. Analysis.
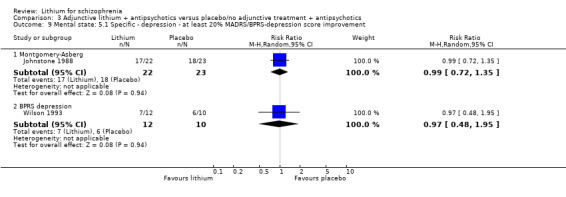
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 9 Mental state: 5.1 Specific ‐ depression ‐ at least 20% MADRS/BPRS‐depression score improvement.
3.10. Analysis.
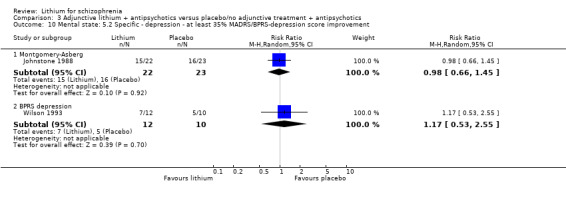
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 10 Mental state: 5.2 Specific ‐ depression ‐ at least 35% MADRS/BPRS‐depression score improvement.
3.11. Analysis.
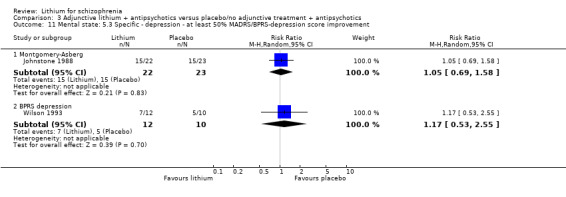
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 11 Mental state: 5.3 Specific ‐ depression ‐ at least 50% MADRS/BPRS‐depression score improvement.
3.12. Analysis.
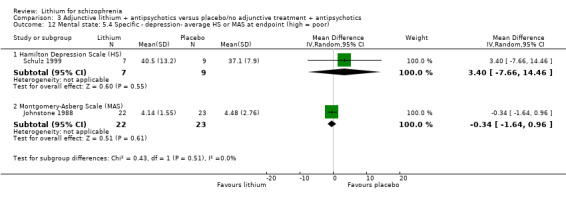
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 12 Mental state: 5.4 Specific ‐ depression‐ average HS or MAS at endpoint (high = poor).
3.13. Analysis.

Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 13 Mental state: 6.1 Specific ‐ mania ‐ at least 20% Bech‐Rafaelsen Mania Scale improvement.
3.14. Analysis.

Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 14 Mental state: 6.2 Specific ‐ mania ‐ at least 35% Bech‐Rafaelsen Mania Scale improvement.
3.15. Analysis.
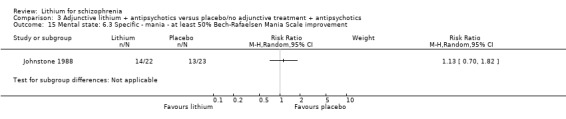
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 15 Mental state: 6.3 Specific ‐ mania ‐ at least 50% Bech‐Rafaelsen Mania Scale improvement.
3.16. Analysis.
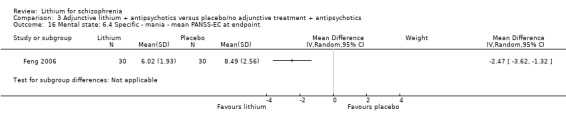
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 16 Mental state: 6.4 Specific ‐ mania ‐ mean PANSS‐EC at endpoint.
3.17. Analysis.
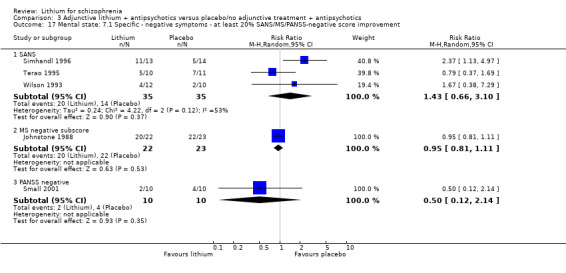
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 17 Mental state: 7.1 Specific ‐ negative symptoms ‐ at least 20% SANS/MS/PANSS‐negative score improvement.
3.18. Analysis.
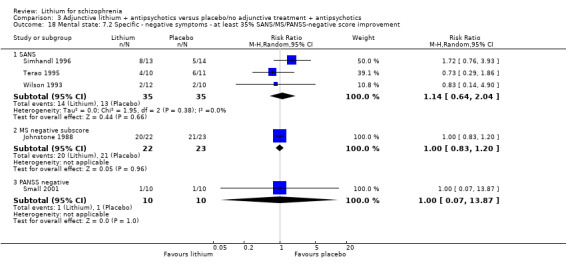
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 18 Mental state: 7.2 Specific ‐ negative symptoms ‐ at least 35% SANS/MS/PANSS‐negative score improvement.
3.19. Analysis.
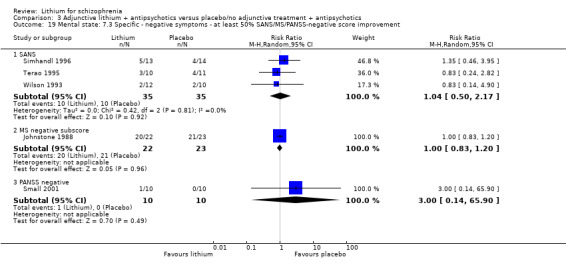
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 19 Mental state: 7.3 Specific ‐ negative symptoms ‐ at least 50% SANS/MS/PANSS‐negative score improvement.
3.20. Analysis.
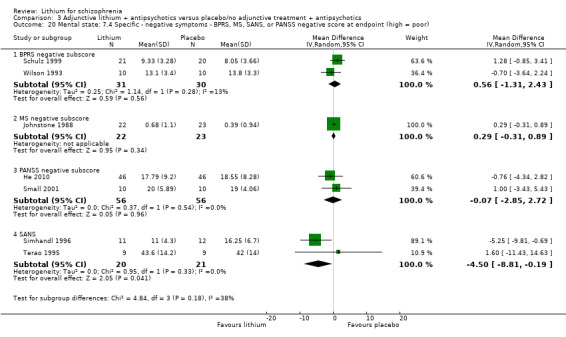
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 20 Mental state: 7.4 Specific ‐ negative symptoms ‐ BPRS, MS, SANS, or PANSS negative score at endpoint (high = poor).
3.21. Analysis.
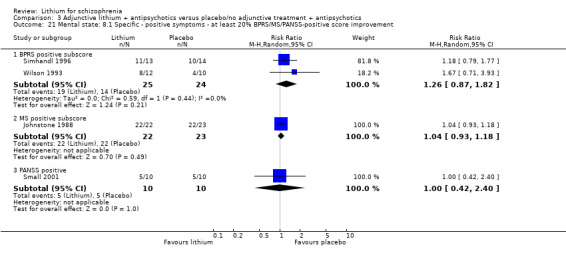
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 21 Mental state: 8.1 Specific ‐ positive symptoms ‐ at least 20% BPRS/MS/PANSS‐positive score improvement.
3.22. Analysis.
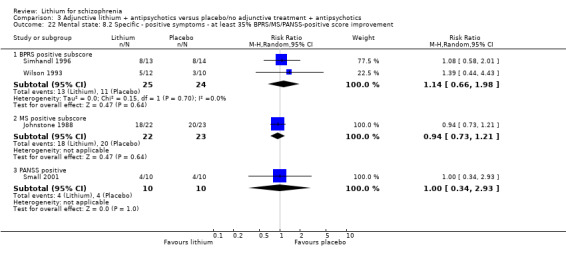
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 22 Mental state: 8.2 Specific ‐ positive symptoms ‐ at least 35% BPRS/MS/PANSS‐positive score improvement.
3.23. Analysis.
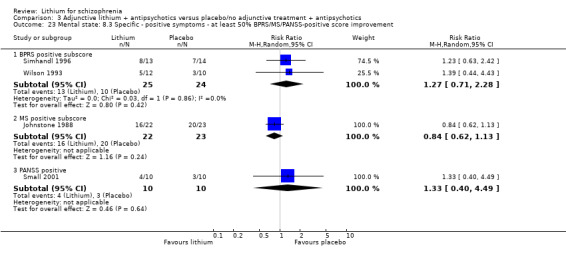
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 23 Mental state: 8.3 Specific ‐ positive symptoms ‐ at least 50% BPRS/MS/PANSS‐positive score improvement.
3.24. Analysis.
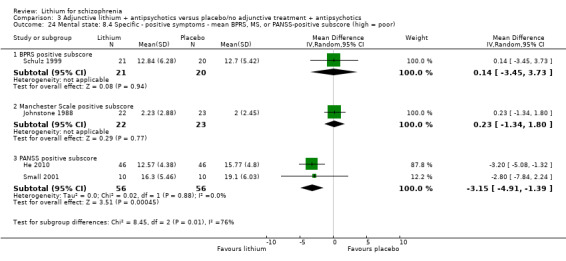
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 24 Mental state: 8.4 Specific ‐ positive symptoms ‐ mean BPRS, MS, or PANSS‐positive subscore (high = poor).
3.25. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 25 Aggressive behaviour ‐ mean PANSS supplemental aggression risk profile at endpoint.
3.26. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 26 Medication use: 1. Mean haloperidol dose (high = poor).
3.27. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 27 Medication use: 2. Mean dose of antipsychotics (unable to use ‐ skewed data).
| Medication use: 2. Mean dose of antipsychotics (unable to use ‐ skewed data) | |
|---|---|
| Study | |
| Biederman 1979 | 1. Haloperidol + lithium: at week 5 mean (range) dose = 28 (10 to 60) mg/day, SD ?. N = 15. 2. Haloperidol + placebo: at week 5 mean (range) dose = 35 (8 to 70) mg/day, SD ?. N = 18. No statistically significant difference. |
| Collins 1991 | 1. Antipsychotics + lithium: at week 4 mean dose in chlorpromazine equivalents = 1452 mg/day, SD ?. N = 21. 2. Antipsychotics + placebo: at week 4 mean dose in chlorpromazine equivalents = 1257 mg/day, SD ?. N = 23. |
| Feng 2006 | 1. Antipsychotics + lithium: clozapine mean dose = 425.66 ± 201.65 mg/d. N = 30. 2. Antipsychotics + placebo: clozapine mean dose = 438.70 ± 238.12 mg/d. N = 30. |
| He 2010 | 1. Antipsychotics + lithium: risperidone flexible dose, mean = 3.8 ± 1.4 mg/d. N = 46. 2. Antipsychotics + placebo: risperidone flexible dose, mean = 4.2 ± 1.6 mg/d. N = 46. |
3.28. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 28 Medication use: 3. Number of particpants taking benzodiazepines.
3.29. Analysis.
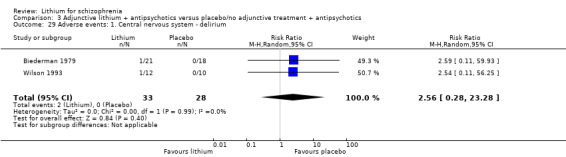
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 29 Adverse events: 1. Central nervous system ‐ delirium.
3.30. Analysis.

Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 30 Adverse events: 2. Movement disorder ‐ dichotomous data.
3.31. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 31 Adverse events: 3. Movement disorder ‐ average SAS score at endpoint (high = poor).
3.33. Analysis.
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 33 Adverse events: 5. Non‐specific dyscomfort.
3.35. Analysis.
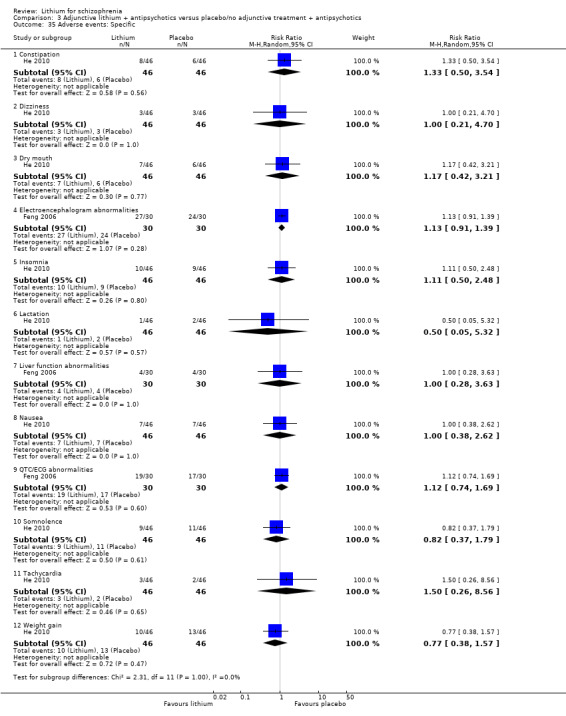
Comparison 3 Adjunctive lithium + antipsychotics versus placebo/no adjunctive treatment + antipsychotics, Outcome 35 Adverse events: Specific.
Comparison 4. Subgroup analyses: antipsychotics + lithium versus antipsychotics + placebo/no adjunctive treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response ‐ participants with or without schizoaffective or prominent affective symptoms | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Participants without schizoaffective disorder or prominent affective symptoms | 7 | 272 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [0.95, 2.81] |
| 1.2 Participants with schizoaffective disorder or prominent affective symptoms | 4 | 114 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.64, 2.74] |
| 2 Clinically significant response ‐ participants with or without treatment‐resistant schizophrenia | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Treatment‐resistant participants | 4 | 199 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [0.90, 4.01] |
| 2.2 Non‐treatment‐resistant participants | 6 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.68, 2.71] |
Comparison 5. Sensitivity analysis 1: participants with affective symptoms excluded.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 7 | 272 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [0.95, 2.81] |
Comparison 6. Sensitivity analysis 2: studies with attrition > 50% excluded.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 9 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [0.93, 3.00] |
Comparison 7. Sensitivity analysis 3: non‐double‐blind studies excluded.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 7 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 1.82 [0.84, 3.96] |
Comparison 8. Sensitivity analysis 4: fixed‐effect model.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinically significant response | 10 | 396 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [1.33, 2.37] |
8.1. Analysis.
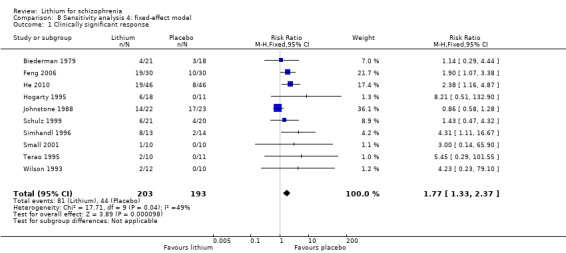
Comparison 8 Sensitivity analysis 4: fixed‐effect model, Outcome 1 Clinically significant response.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Biederman 1979.
| Methods | Allocation: randomised (no further details were provided). Blinding: double (no further details were provided). Duration: 5 weeks. Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia, schizoaffective, and affective schizoaffective (RDC). N = 36*. Age: mean ˜ 30 years.* Sex: 25 M, 11 F.* History: elevated mood or motor hyperactivity, acute admissions, number of previous admissions, and mean duration of the illness was not indicated. | |
| Interventions | 1. Adjunctive lithium (flexible dose of haloperidol, which was started at admission) + flexible lithium dose, which was started at 1200 mg/day and then adjusted according to therapeutic effect and side effects. N = 21. 2. Adjunctive placebo (flexible dose of haloperidol, which was started at admission) + flexible placebo dose, which was adjusted according to therapeutic effect and side effects. N = 18. |
|
| Outcomes | Leaving the study early. Global state: CGI. Adverse events: toxic symptoms. Unable to use Mental state: mean BPRS, mean mania scale (no SD was given). |
|
| Notes | *The data of 3 participants who left before the end of week 3 were not considered in the description by the original authors. In total, there were 39 participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 328): "Assignment (...) according to a prearranged random order." |
| Allocation concealment (selection bias) | Low risk | Quote (page 328): "Assignment of code numbers to each sequential patient admitted to the study was done by the control psychiatrist." |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 328): "Double blind design", "identical capsules", "neither the patient (...), nor the nursing staff were aware of [the manipulation]." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 328): "Double blind design", "identical capsules", "neither the patient (...), nor the nursing staff were aware of [the manipulation]." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 329): "The blood was submitted for serum lithium level determination to the control psychiatrist, who was physically removed from the study location and had no contact with the patients or nursing staff." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 328): "Clinical psychiatrist (clinical rater) [was not] aware of [the manipulation]." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote (page 329): "Three patients discontinued [and were excluded form analysis ‐ Lithium]", "[another] seven patients discontinued [and] were included in all data analyses that used improvement scores to the end of treatment [from placebo]" ‐ last observation carried forward was used; there was an attrition rate of more than 25%; the participants were not evenly distributed (3/18 to 7/21); reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | The paper reported no SDs. |
| Other bias | Low risk | We found no other bias. |
Braden 1982.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: 3 weeks. Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia (12)*, schizoaffective disorder, mania, and other psychotic disorders (RDC). N = 78. Age: this was not clearly indicated. Sex: this was not clearly indicated. History: drug‐free, at least 2 manic symptoms. | |
| Interventions | 1. Lithium: target plasma level = 1.6 mEq/L (mean level of all participants: 1.16 mEq/l). N = 5*. 2. Chlorpromazine up to 2000 mg/day (mean of all participants: 796 mg/day). N = 7*. |
|
| Outcomes | Leaving the study early. Unable to use Mental state: BPRS, GAS, structured interview, clinician's overall ratings (no SD was given, only P values). |
|
| Notes | *Data could be extracted only for those with schizophrenia. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 70): "Patients were assigned randomly to treatment." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 69): "Double‐blind." Quote (page 70): "Identical appearing capsules." Quote (page 70): "The attending psychiatrist was informed of the identity of the drug; nursing stuff and raters were not." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 69): "Double‐blind." Quote (page 70): "Identical appearing capsules." Quote (page 70): "The attending psychiatrist was informed of the identity of the drug; nursing stuff and raters were not." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 70): "Raters were not [informed about the identity of the drug]." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 70): "Raters were not [informed about the identity of the drug]." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote (page 72): "[There was a] high rate of early terminations. There was no overall significant difference in frequency of participants leaving early, of number of days in the study between the lithium and chlorpromazine group" ‐ the attrition rate was 50%; participants were not evenly distributed between groups (4/5 and 2/7); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | The paper provided no SD for the BPRS and other ratings. |
| Other bias | Unclear risk | Quote (page 71): "Nonblind attending psychiatrist made an emergency decision based on serious worsening of patient's condition." |
Brockington 1978.
| Methods | Allocation: randomised (sealed envelopes were used; allocations were made within the trial, from a table of random numbers). Blindness: double (no further details were provided). Duration: 4 weeks. Design: parallel. Setting: this was not indicated. | |
| Participants | Diagnosis: schizomania (PSE). N = 19. Age: mean ˜ 35 years. Sex: 30 M, 12 F. History: "chronic", non‐response to > 3 neuroleptics (2 different chemical classes) in the last 2 years, duration of the illness ˜ 10 years. | |
| Interventions | 1. Lithium: flexible dose (maximum 2500 mg/day) to achieve therapeutic level (the range was not indicated). N = 8. 2. Chlorpromazine: flexible dose (400 to 1000 mg/day). N = 11. |
|
| Outcomes | Leaving the study early. Global state: judgement of rater. Unable to use Mental state: BPRS, PSE (no SD was given). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "Table of random numbers." |
| Allocation concealment (selection bias) | Low risk | Quote (letter): "Sealed envelopes." |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 162): "Double blind." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 162): "Double blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 162): "Double blind." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 162): "Double blind." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The was an attrition rate of over 40%; the participants were not evenly distributed (2/8 and 6/11); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | There was no SD given for the BPRS. |
| Other bias | Low risk | We found no other bias. |
Collins 1991.
| Methods | Allocation: randomised (no further description was given). Blinding: single (rater). Duration: 4 weeks. Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia (DSM‐III‐R and PSE). N = 44. Age: mean ˜ 39 years. Sex: all male. History: all persistent psychotic symptoms for a minimum period of 6 months prior to the study despite adequate neuroleptic treatment, current hospital stay of 1 to 19 years, number of previous hospitalisations and duration of illness was not indicated. | |
| Interventions | 1. Adjunctive lithium (starting dose of 800 mg, which was then adjusted to keep plasma levels within 0.4 to 1.0 mml/L) + flexible dose of antipsychotics (mean chlorpromazine equivalent at week 4 = 1452 mg). N = 21. 2. No additional treatment + flexible dose of antipsychotics (mean chlorpromazine equivalent at week 4 = 1257 mg). N = 23. |
|
| Outcomes | Leaving the study early. Unable to use Mental state: MS, SANS (no mean or SD was given). Medication use: mean antipsychotic dose (no SD was given). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 150): "Randomized." Quote (letter): "Random number list." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Unclear risk | Quote (page 150): "Single‐blind." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 150): "Single‐blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 150): "Single‐blind." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 150): "Single‐blind." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote (page 152): "Eleven patients [out of 21] in the treatment group failed to complete the treatment protocol" [as compared to 1 out of 23 in the control group] ‐ the attrition rate was 27%; participants were not evenly distributed between groups (11/21 and 1/23); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | There was no SD given for the rating scales. |
| Other bias | Low risk | We found no other risk. |
Dube 1981.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: 7 weeks (1 week of placebo, 4 weeks of CPZ or lithium, 2 weeks of placebo). Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia (ICD‐9). N = 60. Age: this was not indicated. Sex: this was not indicated. History: no details. | |
| Interventions | 1. Lithium: flexible dose, adjusted to achieve therapeutic serum levels (the range was not indicated), mean dose = 827 mg/day. N = 30. 2. Chlorpromazine: flexible dose, mean dose = 992 mg/day. N = 30. |
|
| Outcomes | Leaving the study early. Unable to use Global state: CGI (only a P value was given, not a SD). Mental state: BPRS (only a P value was given, not a SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 194): "Randomly administered." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 193): "Double blind." Quote (page 194): "Identical capsules were used." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 193): "Double blind." Quote (page 194): "Identical capsules were used." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 193): "Double blind." Quote (page 194): "Identical capsules were used." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 193): "Double blind." Quote (page 194): "Identical capsules were used." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no attrition. |
| Selective reporting (reporting bias) | High risk | There was no SD given for the BPRS and CGI. |
| Other bias | Low risk | We found no other bias. |
Feng 2006.
| Methods | Allocation: randomised. Blindness: open study. Duration: 8 weeks. Setting: inpatients. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3) and treatment‐resistant form of schizophrenia. N = 60. Age: 21.95 ± 9.26 years. Sex: 34 M and 26 F. History: duration of illness was 5.95 ± 4.15 years; number of previous hospitalisations: 4.93 ± 1.97, not all participants were in their first episode. | |
| Interventions | 1. Adjunctive lithium (mean dose: 1.0 to 1.5 g/day; the starting dose was 0.5 g/day, which increased to 1.0 g/day the next week) + dose of antipsychotics (mean clozapine dose: 425.66 ± 201.65 mg/day; the starting dose was 50 mg/day, which increased to 50 to 100 mg/day the next day; maximum: 300 to 600 mg/day). N = 30. 2. No additional treatment + dose of antipsychotics (clozapine mean dose: 438.70 ± 238.12 mg/day). N = 30. |
|
| Outcomes | Mental state: PANSS total score, PANSS Excited Component, PANSS Supplemental Aggression Risk profile. Adverse events. Unable to use Leaving the study early: not reported. Adverse events: TESS score (no means and SDs were reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Divided into two groups randomly" ‐ no further details were provided. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | No information about blinding was provided. The study was open‐label. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding was provided. The study was open‐label. |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | No information about blinding was provided. The study was open‐label. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding was provided. The study was open‐label. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about attrition was provided. |
| Selective reporting (reporting bias) | High risk | TESS: no means or SDs were reported. |
| Other bias | Low risk | We found no other bias. |
Garver 1983.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: flexible according to response. Design: cross‐over. Setting: hospital. | |
| Participants | Diagnosis: acute schizophrenia, schizophreniform disorder, schizoaffective disorder, major depressive episode (1*) (DSM‐III and RDC). N = 16. Age: this was not indicated. Sex: this was not indicated. History: acute admissions, all had a red blood cell/lithium ratio < 0.31 or > 0.37. | |
| Interventions | 1. Lithium: the dose was calculated so that the level was between 0.8 to 1.4 mEq/L, N = 9. 2. Placebo, N = 7. |
|
| Outcomes | Leaving early. Mental state: NH. Unable to use Global state: CGI (no individual numbers or means were given). |
|
| Notes | *This participant with a major depressive episode was excluded from all analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 58): "Design of random initial assignment." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 57): "Double blind." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 57): "Double blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 57): "Double blind." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 57): "Double blind." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no attrition. |
| Selective reporting (reporting bias) | High risk | CGI was used (there were no individual numbers or means given). |
| Other bias | Low risk | We found no other bias. |
He 2010.
| Methods | Allocation: random. Blindness: open study. Duration: 12 weeks. Setting: outpatients and inpatients. | |
| Participants | Diagnosis: participants meeting CCMD‐3 criteria for schizophrenia; specifically, they also met the criteria for treatment‐resistant schizophrenia. N = 92. Age: 29.05 ± 4.6 years. Sex: 54 M and 38 F. History: the mean duration of illness was 4.4 ± 2.9 years. | |
| Interventions | 1. Adjunctive lithium (flexible dose, mean: 0.6 ± 0.15 g/d, maximum: 1.0 g/d) + dose of antipsychotics (risperidone, flexible dose, mean: 3.8 ± 1.4 mg/d, range: 4 to 6 mg/d). N = 46. 2. No additional treatment + dose of antipsychotics (risperidone, flexible dose, mean: 4.2 ± 1.6 mg/d, range: 6 to 8 mg/d, twice per day). N = 46. |
|
| Outcomes | Mental state: PANSS total, PANSS positive symptoms, PANSS negative symptoms, PANSS general pathology. Adverse events. Unable to use Adverse events: TESS score (no means and SDs were reported). Leaving the study early: this was not reported. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomly divided" ‐ no further information was provided. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | No information about blinding was provided. The study was open‐label. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | No information about blinding was provided. The study was open‐label. |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | No information about blinding was provided. The study was open‐label. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | No information about blinding was provided. The study was open‐label. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about attrition was provided. |
| Selective reporting (reporting bias) | High risk | The TESS score was measured, but no means and SDs were reported. |
| Other bias | Low risk | We found no other bias. |
Hogarty 1995.
| Methods | Allocation: randomised ‐ lithium:placebo 2:1 (no further details were provided). Blindness: double (no further details were provided). Duration: 2 weeks (the intervention was withdrawn at week 6). Design: parallel. Setting: outpatients. | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (RDC). N = 29. Age: mean ˜ 36 years. Sex: M 15, F 14. History: "persistent distress by anxiety for at least 3 months prior to the study". The participants had not sufficiently responded in 2 preceding trials of anticholinergic challenge and fluphenazine dose reduction. The mean duration of the illness was ˜ 10 years*; the mean number of previous hospitalisations was ˜ 4*. | |
| Interventions | 1. Adjunctive lithium (the dose was adjusted to maintain plasma levels between 0.4 to 0.8 mmol/L) + constant dose of fluphenazine decanoate (median biweekly dose: 10 mg). N = 18. 2. Placebo + constant dose of fluphenazine decanoate (median biweekly dose: 10 mg). N = 11. |
|
| Outcomes | Response: no overall improvement, relapse.
Leaving the study early Unable to use Mental state: BPRS subscores, BDI, anxiety rating scale, personal comfort scale (no SD was given). Adverse events: Barnes Akathisia Scale (no SD was given). |
|
| Notes | *Data refer to the whole group of distressed participants who had entered the 2 previous trials. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 31): "Randomized." Quote (letter): "Table of random numbers." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 29): "Double blind." Quote (page 35): "LI or placebo administration was (...) unknown to the treating clinicians." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 29): "Double blind." Quote (page 35): "LI or placebo administration was (...) unknown to the treating clinicians." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 29): "Double blind." Quote (page 35): "LI or placebo administration was (...) unknown to the treating clinicians." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 29): "Double blind." Quote (page 35): "LI or placebo administration was (...) unknown to the treating clinicians." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 24%; participants were not evenly distributed (7/18 and 0/11); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | No SDs were given for various scales (BDI, BPRS subscores, etc.) |
| Other bias | Unclear risk | Data refer to the whole group of distressed participants who had entered the 2 previous trials. |
Huang 1984.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: 10 weeks (the intervention was withdrawn at week 6). Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia (10*), bipolar depression, mania (DSM‐III‐R). N = 27. Age: mean ˜ 34 years*. Sex: 9 M, 1 F*. History of the schizophrenia participants: all had chronic paranoid type; the duration of the illness was not indicated. | |
| Interventions | 1. Adjunctive lithium (the dose was adjusted to maintain levels between 0.6 to 1.2 mEq/L) + haloperidol (the dose was not indicated). N = 6*. 2. Adjunctive placebo + haloperidol (the dose was not indicated). N = 4*. |
|
| Outcomes | Leaving the study early. Unable to use Mental state: GAS (no mean or SD was given). |
|
| Notes | *Only data of those 10 participants with schizophrenia were used for the review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 327): "Double blind." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 327): "Double blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 327): "Double blind." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 327): "Double blind." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no dropouts. |
| Selective reporting (reporting bias) | High risk | No mean and SD was given for the GAS. |
| Other bias | Low risk | We found no other bias. |
Johnson 1971.
| Methods | Allocation: randomised (a table with random numbers was used). Blindness: double (the participant and rater were blind, not the treating psychiatrist). Duration: 3 weeks. Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: acute schizophrenia and acute schizoaffective disorder. N = 17. Age: mean ˜ 39 years. Sex: this was not indicated. History: the duration of the illness was not indicated, but all participants were in an "excited" state at the time of the study. | |
| Interventions | 1. Lithium: flexible dose, increased until response or toxic effect; all participants had levels higher than 1 mEq/L. N = 7. 2. Chlorpromazine: dosage increased until response or toxic effect. N = 10. |
|
| Outcomes | Response: no improvement.
Leaving the study early. Mental state: BPRS, SCI. Unable to use Adverse events and laboratory parameters (no clear numbers were given). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "Randomised (table with random numbers)." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 267): "Controlled double blind study." Quote (page 268): "The raters were blind throughout", "identical capsules." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 267): "Controlled double blind study." Quote (page 268): "The raters were blind throughout", "identical capsules." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 267): "Controlled double blind study." Quote (page 268): "The raters were blind throughout", "identical capsules." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 267): "Controlled double blind study." Quote (page 268): "The raters were blind throughout", "identical capsules." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 11%; the participants were evenly distributed (1/7 and 1/10); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | No numerical results for CGI, TRAM, and NOSIE were reported. |
| Other bias | Low risk | We found no other bias. |
Johnstone 1988.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: 8 weeks (this was for the initial study, then there was an open extension of up to 6 years for the responders). Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: "functional psychoses" (schizophrenia, schizophreniform disorder, schizoaffective disorder, atypical psychoses paranoid disorder, mania, or depression)* (DSM‐III and PSE). N = 120. Age: the mean age was unclear. Sex: 64 M, 56 F. History: 38 were neuroleptic‐naive, and 22 had no neuroleptics for at least 1 year; the duration of the illness was unclear. | |
| Interventions | 1. Pimozide alone: 16 mg/day. N = 23*. 2. Lithium alone, levels adjusted to 0.5 to 1.2 mmol/l. N = 21*. 3. Pimozide + lithium (same doses as in the monotherapy group). N = 22.* 4. Placebo. N = 18*. |
|
| Outcomes | Leaving the study early. Mental state: Krawiecka Scale, Montgomery‐Asberg Depression Scale, Bech‐Rafaelsen Mania Scale. Adverse events: AIMS, TAKE. Unable to use Response: relapse rates and other long‐term data (the numbers were unclear). |
|
| Notes | *The data of only those with schizophrenia or similar psychoses are used in the analyses (schizophrenia, schizophreniform disorder, schizoaffective disorder, atypical psychosis, paranoid disorder). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "The randomisation was done using computer generated lists of random numbers." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 120): "Allocated (...) blindly", "to preserve blindness it was arranged for the clinical chemistry department to send false results in the case of patients on placebo lithium." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 120): "Allocated (...) blindly", "to preserve blindness it was arranged for the clinical chemistry department to send false results in the case of patients on placebo lithium." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 120): "Allocated (...) blindly", "to preserve blindness it was arranged for the clinical chemistry department to send false results in the case of patients on placebo lithium." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 120): "Allocated (...) blindly", "to preserve blindness it was arranged for the clinical chemistry department to send false results in the case of patients on placebo lithium." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 9%; participants were not evenly distributed (4/21 and 0/23); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | No SDs were given for the scales. |
| Other bias | Low risk | We found no other bias. |
Mattes 1984.
| Methods | Allocation: randomised (a table with random numbers was used). Blindness: double (no further details were provided). Duration: 1 year. Design: parallel. Setting: outpatients. | |
| Participants | Diagnosis: schizophrenic schizoaffective disorder (RDC). N = 14. Age: mean ˜ 26 years. Sex: this was not indicated. History: 5 participants were chronic, 8 were subchronic, and 1 was subacute according to RDC criteria; on average, the participants had not worked 50% of the time they could have worked in the last 5 years; on average, each had 3.7 previous episodes; the mean duration of the illness was not indicated. | |
| Interventions | 1. Lithium: flexible dose, plasma levels > 0.6. N = 7. 2. Fluphenazine: 5 to 20 mg/day orally or 12.5 to 50 mg biweekly. N = 7. |
|
| Outcomes | Response: relapse Leaving the study early. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "Randomised (table with random numbers)." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 446): "Double blind", "lithium blood levels were (...) monitored monthly by nontreating psychiatrist." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 446): "Double blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 446): "Double blind." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 446): "Double blind." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 64%; participants were not evenly distributed (6/7 and 3/7); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | Low risk | There was no selective reporting. |
| Other bias | Unclear risk | Quote (page 446): "Regardless of prior medication, eligible patients were openly changed to combined lithium (...) and fluphenazine." |
Prien 1972.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (identical capsules were used). Duration: 3 weeks. Design: parallel. Setting: hospital, multicentre. | |
| Participants | Diagnosis: schizoaffective disorder, excited type (DSM‐II). N = 83. Age: mean ˜ 39 years. Sex: 59 M, 24 F. History: the duration of the illness was not indicated; the median number of previous hospitalisation was 3. | |
| Interventions | 1. Lithium carbonate: the starting dose was 750 mg, which was then adjusted according to the participants's clinical condition and side effects (plasma levels < 2.0 mEq/L). N = 37. 2. Chlorpromazine: the starting dose was 600 mg/day, which was then adjusted according to the participant's clinical condition and side effects. N = 46. |
|
| Outcomes | Leaving the study early. Adverse events. Unable to use Mental state: BPRS, IMPS, PIP (only P values were given). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 182): "Randomly assigned." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 184): "Identically appearing capsules", "only the treating psychiatrist knew the identity of the patient's study medication. Other treatment personnel [were] the clinical raters [who] operated under double blind conditions." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 184): "Identically appearing capsules", "only the treating psychiatrist knew the identity of the patient's study medication. Other treatment personnel [were] the clinical raters [who] operated under double blind conditions." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 184): "Identically appearing capsules", "only the treating psychiatrist knew the identity of the patient's study medication. Other treatment personnel [were] the clinical raters [who] operated under double blind conditions." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 184): "Identically appearing capsules", "only the treating psychiatrist knew the identity of the patient's study medication. Other treatment personnel [were] the clinical raters [who] operated under double blind conditions." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 25.3%, and participant distribution was 13/37 and 8/46. Quote (page 186): "Both early terminators and patients who completed the full three weeks of treatment were included into the analysis" ‐ last observation carried forward analysis was used; no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | No SDs were given for BPRS, IMPS, and PIP. |
| Other bias | Low risk | We found no other bias. |
Schulz 1999.
| Methods | Allocation: randomised (stratified for gender and diagnoses). Blindness: double (the participant and independent rater was blind, not the therapist). Duration: 8 weeks. Design: parallel. Setting: outpatients. | |
| Participants | Diagnosis: schizophrenia, schizophreniform, and schizoaffective disorder (DSM‐III‐R). N = 41. Age: mean ˜ 29 years. Sex: 34 M, 7 F. History: severely ill (mean BPRS = 47) despite treatment with depot antipsychotics for 6 months before the trial. The mean duration of the illness and mean number of previous episodes was not indicated. | |
| Interventions | 1. Adjunctive lithium (plasma levels were maintained between 0.8 to 1.0 mEq/L) + constant dose of fluphenazine. N = 21. 2. Placebo + constant dose of fluphenazine. N = 20. |
|
| Outcomes | Leaving the study early. Mental state: BPRS, HAMD. Unable to use Mental state: CGI, SAS (no mean or SD was given). Adverse events: these were monitored but not reported. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 368): "Assignment to lithium of placebo was randomised, but stratified to account for gender differences and differences between schizophrenia and schizoaffective illness group." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 368): "Double blind", "a non‐blinded physician assessed side effects and monitored lithium levels", "baseline assessment and weekly assessments of symptoms were performed by blinded psychiatrist and study nurse." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 368): "Double blind", "a non‐blinded physician assessed side effects and monitored lithium levels", "baseline assessment and weekly assessments of symptoms were performed by blinded psychiatrist and study nurse." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 368): "Double blind", "a non‐blinded physician assessed side effects and monitored lithium levels", "baseline assessment and weekly assessments of symptoms were performed by blinded psychiatrist and study nurse." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 368): "Double blind", "a non‐blinded physician assessed side effects and monitored lithium levels", "baseline assessment and weekly assessments of symptoms were performed by blinded psychiatrist and study nurse." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 60%; participants were evenly distributed (14/21 and 11/20). Quote (page 366): "Intent‐to‐treat analysis." No reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | No SDs were given for CGI and SAS; adverse effects were monitored but not reported. |
| Other bias | Low risk | We found no other bias. |
Shopsin 1971.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (identical capsules were used ‐ the participant and rater were blinded, not the therapist). Duration: 5 weeks (2‐week placebo run‐in, 3 weeks of lithium or chlorpromazine, and 2 weeks of placebo). Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: acute schizophrenia (undifferentiated type and paranoid type), schizoaffective disorder (clinical diagnose by at least 2 psychiatrists). N = 21. Age: 21 to 62 years. Sex: this was not indicated. History: included within 3 days of admission, having their first episode (N = 2) or exacerbation of chronic illness (N = 19); the mean duration of illness was not indicated. | |
| Interventions | 1. Lithium: the starting dose was 750 to 1000 mg/day, maximum dose = 3000 mg/day (plasma levels < 1.5 mEq/L). N = 11. 2. Chlorpromazine: the starting dose was 300 to 400 mg/day, maximum dose = 1200 mg/day. N = 10. |
|
| Outcomes | Leaving the study early. Adverse events: use of antiparkinson medication, specific events. Laboratory: white blood cell count, blood uric acid, proteinuria. Unable to use Global state: CGI (only a P value was given). Mental state: BPRS, IMPS, SCI, SRSS (only P values were given). Behaviour: NOSIE (only a P value was given). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 435): "Subjects were assigned to either drug in random fashion." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 435): "Identical pink capsules", "a third psychiatrist, uninvolved in patient rating, assigned and regulated treatment, he was also charged with patient care." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 435): "Identical pink capsules", "a third psychiatrist, uninvolved in patient rating, assigned and regulated treatment, he was also charged with patient care." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 435): "Identical pink capsules", "a third psychiatrist, uninvolved in patient rating, assigned and regulated treatment, he was also charged with patient care." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 435): "Identical pink capsules", "a third psychiatrist, uninvolved in patient rating, assigned and regulated treatment, he was also charged with patient care." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no attrition. |
| Selective reporting (reporting bias) | High risk | No SDs were given for BPRS, IMPS, SCI, SRSS, and NOSIE. |
| Other bias | Low risk | We found no other bias. |
Simhandl 1996.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: 8 weeks (the intervention was withdrawn at week 6). Design: parallel. Setting: this was not indicated. | |
| Participants | Diagnosis: schizophrenia (DSM‐III‐R). N = 42. Age: mean ˜ 35 years. Sex: 30 M, 12 F. History: "chronic", non‐response to > 3 neuroleptics of 2 different chemical classes in the last 2 years, the mean duration of illness was ˜ 10 years. | |
| Interventions | 1. Adjunctive carbamazepine (the dose increased during weeks 1 to 2 until plasma levels were 15 to 42 micromol/L) + constant dose of antipsychotics. N = 15. 2. Lithium (the dose increased during weeks 1 to 2 until plasma levels were 0.6 to 1.2 myml/L) + constant dose of antipsychotics. N = 13. 3. Placebo + constant dose of antipsychotics. N = 14. |
|
| Outcomes | Leaving the study early. Mental state: BPRS, SANS. |
|
| Notes | *The data of this group were not used in the analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomised." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 317): "Double blind." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Quote (page 317): "Double blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 317): "Double blind." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Quote (page 317): "Double blind." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 7%, but only in 1 group (0/13 and 2/14); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | No SD was given for BPRS. |
| Other bias | Low risk | We found no other bias. |
Simpson 1976.
| Methods | Allocation: randomised (a coin toss was used). Blindness: double (raters and participants were blind, not the therapists). Duration: 18 weeks (6 weeks' baseline for withdrawal of antipsychotics, then 2 6‐week cross‐over phases). Design: cross‐over. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia. N = 11. Age: mean ˜ 72 years. Sex: 3 M, 8 F. History: long‐term hospitalised, all with tardive dyskinesia. The mean duration of hospitalisation was ˜ 30 years; the mean duration of illness was not indicated. |
|
| Interventions | 1. Lithium: flexible dose to maintain plasma levels between 0.6 to 1.0 MEq/L. N = 5. 2. Placebo. N = 6. |
|
| Outcomes | Leaving the study early. Unable to use Global state: CGI (only a P value was given). Adverse events: Tardive dyskinesia rating scale (only a P value was given). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "Randomized by toss of coin." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 76): "Double blind." Quote (page 78): "Placebo identical to the study medication", "the ward personnel (...) was blind." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 76): "Double blind." Quote (page 78): "Placebo identical to the study medication", "the ward personnel (...) was blind." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 76): "Double blind." Quote (page 78): "Psychiatrist (...) responsible for the evaluation (...) was blind as to the design and the patients' medication (active or placebo)." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 76): "Double blind." Quote (page 78): "Psychiatrist (...) responsible for the evaluation (...) was blind as to the design and the patients' medication (active or placebo)." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no attrition. |
| Selective reporting (reporting bias) | High risk | No numerical values for CGI were given, and adverse effects were reported. |
| Other bias | Unclear risk | Different washouts (gradual versus abrupt) (page 78). |
Small 1975.
| Methods | Allocation: randomised (no further details were provided). Blindness: double (no further details were provided). Duration: 16 weeks (4 phases of 4 weeks). Design: cross‐over. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia, schizoaffective disorder (Feighner's criteria). N = 22. Age: mean ˜ 36 years. Sex: 4 M, 18 F. History: "very chronically ill", all had failed to respond to various previous treatment approaches including at least 1 allocation of phenothiazine, haloperidol, and thiothixene; the mean duration of hospitalisation was ˜ 9 years; the mean duration of illness was ˜ 10 years. | |
| Interventions | 1. Adjunctive lithium (flexible dose to maintain plasma levels between 0.6 to 1.2 MEq/L) + constant dose of antipsychotics. N = 12. 2. Placebo + constant dose of antipsychotics. N = 10. |
|
| Outcomes | Leaving the study early. Unable to use Global state: CGI (there were no data for the first cross‐over phase). Mental state: BPRS (there were no data for the first cross‐over phase). Behaviour: NOSIE (there were no data for the first cross‐over phase). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1316): "Random assignment." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 1315): "Double blind." Quote (page 1316): "Double‐blind conditions were maintained throughout the study for patients and for the psychiatrists, nurses and psychometrists who gathered the data. White capsules of identical appearance..." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 1315): "Double blind." Quote (page 1316): "Double‐blind conditions were maintained throughout the study for patients and for the psychiatrists, nurses and psychometrists who gathered the data. White capsules of identical appearance..." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 1315): "Double blind", "blind psychiatric and nursing ratings." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 1315): "Double blind", "blind psychiatric and nursing ratings." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 9%; participants were evenly distributed (1/12 and 1/10); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | For CGI, BPRS, and NOSIE, there were no data for the first cross‐over phase; no SD was given. |
| Other bias | Low risk | We found no other bias. |
Small 2001.
| Methods | Allocation: randomised (a table with random numbers was used). Blindness: double (lithium and placebo tablets had the same taste). Duration: 16 weeks (4 phases of 4 weeks). Design: cross‐over. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia, schizoaffective disorder (DSM‐IV). N = 20. Age: mean ˜ 37 years. Sex: 14 M, 6 F. History: all had failed to respond satisfactorily to adequate treatment with at least 2 different antipsychotics; the mean duration of hospitalisation and mean duration of illness was not indicated. | |
| Interventions | 1. Adjunctive lithium (flexible dose to maintain plasma levels > 0.4 MEq/L) + constant dose of clozapine (mean dose: ˜ 400 mg/day). N = 10. 2. Placebo + constant dose of clozapine (mean dose: ˜ 400 mg/day). N = 10. |
|
| Outcomes | Leaving the study early. Mental state: BPRS. Unable to use Adverse events: there were no data from the first cross‐over phase. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial was randomised using a table with random numbers. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not indicated. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 2): "Raters and patients were blinded; clinicians were not." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | Quote (page 2): "Raters and patients were blinded; clinicians were not." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 2): "Raters and patients were blinded; clinicians were not." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 2): "Raters and patients were blinded; clinicians were not." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 5% (1 participant from 1 group (0/10 and 1/10)); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | High risk | There were no data from the first cross‐over phase for the adverse effects reported. |
| Other bias | Unclear risk | We found no other bias. |
Terao 1995.
| Methods | Allocation: randomised (a table with random numbers was used). Blindness: double (identical capsules were used). Duration: 19 weeks (the intervention was withdrawn between the 2 8‐week cross‐over phases). Design: cross‐over. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia (DSM‐III‐R). N = 21. Age: mean ˜ 47 years. Sex: all male. History: the mean duration of illness was ˜ 21 years; the mean number of hospitalisations was ˜ 2.6; the duration of current hospitalisation was ˜ 6.7 years. | |
| Interventions | 1. Adjunctive lithium (the dose was calculated to reach a plasma level of at least 0.4 mEq/L) + constant dose of antipsychotics. N = 10. 2. Placebo + constant dose of antipsychotics. N = 11. Mean daily antipsychotic dose in haloperidol equivalents 27.1 mg (both groups combined). |
|
| Outcomes | Leaving the study early. Mental state: BPRS, SANS. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "We used [a] random number list to allocate subjects randomly." |
| Allocation concealment (selection bias) | Low risk | Quote (page 221): "Randomisation by a controller." |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 220): "Double blind." Comment: Clinicians and raters were blind. Quote (page 221): "Placebo capsules with appearance identical to that of the lithium capsules." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 220): "Double blind." Comment: Clinicians and raters were blind. Quote (page 221): "Placebo capsules with appearance identical to that of the lithium capsules." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 220): "Double blind." Comment: Clinicians and raters were blind. Quote (page 221): "Placebo capsules with appearance identical to that of the lithium capsules." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 220): "Double blind." Comment: Clinicians and raters were blind. Quote (page 221): "Placebo capsules with appearance identical to that of the lithium capsules." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 14%: 20% in 1 group and 9% in the second (2/10 and 1/11); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | Low risk | There was no selective reporting. |
| Other bias | Low risk | We found no other bias. |
Wilson 1993.
| Methods | Allocation: randomised (blocks of 6 participants, sealed envelopes were used derived from a table of random numbers). Blinding: double (identical placebo capsules were prepared by a pharmacist). Duration: 6 weeks' of baseline, followed by an 8‐weeks experimental phase. Design: parallel. Setting: hospital. | |
| Participants | Diagnosis: schizophrenia without a concurrent major affective disorder (DSM‐III‐R). N = 29* Age: this was not indicated. Sex: this was not indicated. History: all with persistent psychosis for at least 8 weeks despite antipsychotic treatment with at least 800 mg chlorpromazine equivalent; the mean current hospital stay was ˜ 20 months; the mean previous hospitalisations was ˜ 10 times; the mean duration of illness was ˜ 14 years. | |
| Interventions | Baseline phase before randomisation (6 weeks): All participants were put on haloperidol at "best clinical dose". Experimental phase (8 weeks): 1. Lithium (flexible dose adjusted to reach plasma level of 1.0 mEq/L) + haloperidol (the dose was not indicated). N = 12. 2. Placebo + haloperidol (the dose was not indicated). N = 10. |
|
| Outcomes | Leaving the study early. Mental state: BPRS, SANS. Adverse events: number of participants with delirium. medication use (mean haloperidol dose, use of benzodiazepines and anticholinergics). Unable to use Adverse events: EPS (AIMS and Barnes Akathisia Scale ‐ data were skewed). |
|
| Notes | *7 participants left the study during the baseline phase before randomisation. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (letter): "Assigned randomly (...) based on a computer generated random number". |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes were used. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Quote (page 359): "Double blind." Comment: Clinicians and raters were blind. Quote (page 360): "Identical appearing placebo capsules." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Quote (page 359): "Double blind." Comment: Clinicians and raters were blind. Quote (page 360): "Identical appearing placebo capsules." |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Quote (page 359): "Double blind." Comment: Clinicians and raters were blind. Quote (page 360): "Identical appearing placebo capsules." |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | Quote (page 359): "Double blind." Comment: Clinicians and raters were blind. Quote (page 360): "Identical appearing placebo capsules." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition rate was 9%; participants were unevenly distributed (2/12 and 0/10); no reasons for discontinuation were given. |
| Selective reporting (reporting bias) | Low risk | There was no selective reporting. |
| Other bias | Low risk | We found no other bias. |
General abbreviations CPZ = chlorpromazine. EPS = extrapyramidal side effects. F = females. ICD‐9 = Ninth Revision of the International Classification of Diseases. L = litre. M = males. mg = milligram. mEq = milliequivalents. N = number. SD = standard deviation. TAKE = Scale for Targeting Abnormal Kinetic Effects TRAM = Treatment response assessment method Diagnostic tools CCMD‐3 = Chinese Classification of Mental Disorders. DSM‐II = Diagnostic and Statistical Manual of Mental disorders, second edition. DSM‐III = Diagnostic and Statistical Manual of Mental disorders, third edition. DSM‐III‐R = Diagnostic and Statistical Manual of Mental disorders, third edition, revised. DSM‐IV = Diagnostic and Statistical Manual of Mental disorders, fourth edition. RDC = Research Diagnostic Criteria. Global effect scales CGI = Clinical Global Impression. Mental state scales BDI = Beck Depression Inventory. BPRS = Brief Psychiatric Rating Scale. GAS = Global Assessment of Symptoms Scale. HAMD = Hamilton Depression Scale. IMPS = Inpatient Multidimensional Psychiatric Scale. PANSS = Positive and Negative Syndrome Scale. PIP = Psychotic Inpatient Profile. PSE = Present State Examination. MS = Manchester Scale. MMSE = Mini‐Mental State Examination. MSRS = Manic State Rating Scale. NHSI = (Modified) New Haven Schizophrenia Index. NOSIE = Nurses' Observation Scale for Inpatient Evaluation. SANS = Scale for the Assessment of Negative Symptoms. SCI = Structured Clinical Interview. SRSS = Self‐Rating Symptom Scale. TESS = Treatment Emergent Symptom Scale. Side effect scales AIMS = Abnormal Involuntary Movement Scale. Barnes Akathisia Scale. SAS = Simpson‐Angus Scale. SAFTEE = Systematic Assessment for Treatment Emergent Events.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alexander 1979 | Allocation: not randomised ("within patient cross‐over"). |
| Aronoff 1970 | Allocation: randomised. Participants: the majority were those with manic depressive illness, not schizophrenia. |
| Baastrup 1967 | Allocation: not randomised. This was an observational study with no control group. |
| Bellaire 1990 | Allocation: randomised. Participants: the majority were bipolar, not schizophrenic. |
| Berger 2006 | Allocation: placebo‐controlled ‐ it was unclear if the study was randomised. Participants: people at high risk of a first episode. |
| Bigelow 1981 | Allocation: not randomised. The study was double‐blind, with the cross‐over of a single participant. |
| Bowers 1983 | Allocation: not randomised ("within patient cross‐over"). |
| Braden 1980 | Allocation: randomised. Participants: newly admitted adult inpatients who manife manic behavior with or without schizophrenic symptoms participated in this study. Interventions: chlorpromazine versus lithium carbonate. Outcomes: no useable data were presented. |
| Brewerton 1983 | Allocation: randomised. Participants: the majority of participants were bipolar, not schizophrenic. |
| Cabrera 1986 | Allocation: randomised. Participants: the majority were not schizophrenic. |
| Campbell 1972 | Allocation: not randomised (matched groups). |
| Campbell 1982 | Allocation: not a RCT. This was an overview of literature. |
| Carman 1981 | Allocation: randomised, double‐blind, cross‐over study. Participants: those with schizophrenia or schizoaffective disorder. Interventions: lithium versus placebo added to antipsychotic drugs. Outcomes: no single outcome could be analysed. |
| Chen 2001 | Allocation: alternate allocation by hospital ID number (not randomised). |
| Dinsmore 1972 | Allocation: not randomised. This was an observational study. |
| Edelstein 1981 | Allocation: not randomised. This was a controlled clinical study. |
| Gao 2002 | Allocation: allocated according to odd or even number of date and ID of admission. |
| Garver 1988 | Allocation: not randomised ("within patient cross‐over"). |
| Gerlach 1975 | Allocation: randomised, double‐blind. Participants: diagnostically mixed group, all showing tardive dyskinesia, most had schizophrenia. Interventions: lithium or placebo added to ongoing treatment. Outcomes: focus on tardive dyskinesia, but no data could be extracted. |
| Gram 1971 | Allocation: randomised, double‐blind. Participants: unclear diagnosis ("psychotic" children and adolescents). |
| Greil 1997 | Allocation: randomised. Participants: those with schizoaffective disorder. Interventions: lithium versus carbamazepine as sole agents, there was no placebo/no intervention group. |
| Growe 1979 | Allocation: it was unclear whether the study was randomised (it was double‐blind). Participants: those with schizophrenia or schizoaffective disorder. Interventions: lithium versus placebo added to antipsychotic drugs. Outcomes: no single outcome could be used. |
| Haastrup 1973 | Allocation: this was a controlled clinical trial; it was unclear whether the study was randomised. Participants: those with schizophrenia. Interventions: unclear. Outcomes: not specified. |
| Harrison 1980 | Allocation: not randomised. This was a retrospective chart analysis. |
| Hofmann 1970 | Allocation: not randomised. This was a case series. |
| Hullin 1975 | Allocation: not randomised. This was a case series. |
| Jus 1978 | Allocation: randomised, double‐blind. Participants: schizophrenia, all with tardive dyskinesia. Interventions: lithium, deanol, or placebo as sole treatments. Outcomes: focus on tardive dyskinesia, but no data could be extracted. |
| Kahn 1990 | Allocation: randomised. Participants: people with schizophrenia. Interventions: haloperidol alone and, subsequently, haloperidol in combination with carbamazepine or lithium carbonate, measuring the change in haloperidol serum levels. |
| Lenzi 1985 | Allocation: randomised, double‐blind. Participants: mixed group, 8 out of 30 had schizophrenia‐like disorders. Interventions: placebo or carbamazepine added to antipsychotic drugs, no placebo, or no intervention control group. |
| Lerner 1988 | Allocation: not adequately randomised (alternate allocation). |
| Liebowitz 1976 | Allocation: not randomised. This was a case report. |
| Liu 2006 | Allocation: randomised. Participants: not people with schizophrenia (periodic psychosis). |
| Mackkay 1980 | Allocation: not randomised, pairs were matched then there was "within patient cross‐over". |
| Marken 1994 | Allocation: randomised. Participants: the majority were bipolar, not schizophrenic. |
| Martorano JT | Allocation: not randomised. This was a paper on case reports. |
| Miller 1979 | Allocation: not randomised. This was a review with no original data. |
| Nct 2005 | Allocation: randomised. Participants: mania, it was unclear if the majority were schizophrenic. |
| Nemes 1986 | Allocation: not randomised ("within patient cross‐over"). |
| Paykel 2000 | Allocation: randomised. Participants: women with post‐partum psychoses. Interventions: lithium versus haloperidol versus placebo. Notes: we contacted the authors who confirmed that the study was never undertaken. |
| Placidi 1986 | Allocation: randomised, double‐blind. Participants: mixed group with "major affective, schizoaffective or schizophreniform disorders". Interventions: lithium versus carbamazepine as sole agents, no placebo, or no intervention group. |
| Prange 1973 | Allocation: not randomised. This was a case series. |
| Prien 1972a | Allocation: randomised, double‐blind. Participants: those with mania; there were no participants with schizophrenia or schizoaffective disorder. |
| Rice 1956 | Allocation: not randomised. This was a case series. |
| Rosenthal 1980 | Allocation: not randomised. This was a case series. |
| Schnexnayder 1995 | Allocation: not randomised, there was no control group. This was a case series. |
| Schou 1954 | Allocation: randomised. Participants: those with mania. |
| Shalif 1981 | Allocation: not randomised. The authors used data from previous studies to assess a non‐parametric statistical technique for multidimensional evaluation of ratings. |
| Shaw 1974 | Allocation: not randomised, controlled clinical trial. Participants: those with chronic schizophrenia. Interventions: 2 different sustained‐release preparations of lithium carbonate, there was no placebo or intervention group. |
| Shopsin 1975 | Allocation: randomised, double‐blind. Participants: those with mania. |
| Smulevitch 1974 | Allocation: not randomised. This was a controlled clinical trial. |
| Sun 2008 | Allocation: not randomised, allocation was according to the order of hospitalisation. |
| Taylor 1974 | Allocation: not randomised. This was a case series. |
| Van Kammen 1978 | Allocation: the allocation of drug intervention (lithium or pimozide) was not randomised, but participants were randomised to receive an additional amphetamine or placebo infusion. |
| Van Kammen 1980 | Allocation: not randomised, there was no control group. |
| Van Kammen 1985 | Allocation: randomised, double‐blind. Participants: those with schizophrenia. Interventions: amphetamine + placebo versus amphetamine + lithium. |
| Van Putten 1975 | Allocation: not randomised ("within patient cross‐over"). |
| Vieweg 1989 | Allocation: non‐randomised comparison of participants with healthy volunteers. |
| Volavka 1986 | Allocation: randomised. Participants: chronic psychiatric inpatients with tardive dyskinesia. Interventions: lithium versus lecithin, there was no placebo or intervention group. |
| Wang 1995 | Allocation: randomised cross‐over study, no further details were provided. Participants: people with schizophrenia. Interventions: chlorpromazine + lithium versus chlorpromazine alone, but clozapine could be added to both groups. |
| White 1966 | Allocation: not randomised. This was a case series. |
| Wilner 1996 | Allocation: randomised. Participants: healthy volunteers. |
| Wilson 1994 | Allocation: not randomised, follow up of clozapine treatment on a cohort of participants in a previous lithium trial. |
| Zemlan 1984 | Allocation: not randomised. This was a case series. |
| Zhou 2011 | Allocation: randomised. Participants: people with schizophrenia. Interventions: paroxetine versus paroxetine + lithium. |
ID = identification. RCT = randomised controlled trial.
Characteristics of studies awaiting assessment [ordered by study ID]
Kamisada 1988.
| Methods | Allocation: random (there were no details of the randomisation). Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Setting: hospital. |
| Participants | Diagnosis: schizophrenic psychosis (ICD‐9). N = 30. Age: 21 to 57 years. Sex: this was not indicated. History: participants were included if they were rated as moderate or more in "tention", "mannerisms and posturing" or "excitement" of BPRS. The number of previous admissions and mean duration of illness was not indicated. |
| Interventions | 1. Lithium: fixed‐flexible method, the dose was started at 400 mgs/d, with an upper limit of 1200 mgs/d. N = unknown. 2. Carbamazepine: fixed‐flexible method, the dose was started at 400 mgs/d, with an upper limit of 1200 mgs/d. N = unknown. 3. Placebo: N = unknown. |
| Outcomes | Adverse events. Unable to use General Improvement Rating (only a P value was provided). Global Usefulness Rating (only a P value was provided). Overall Safety Rating (only a P value was provided). |
| Notes | Insufficient data were provided to identify the original group size (prior to dropouts). |
McGorry 2002.
| Methods | Allocation: randomised. |
| Participants | Diagnosis: first episode of psychosis. |
| Interventions | 1. Risperidone 2 mg versus risperidone 4 mg versus risperidone plus lithium. |
| Outcomes | No useable data were presented; this was a conference proceeding only. |
| Notes | Authors were contacted in an attempt to get further information. |
Mosolov 1998.
| Methods | Allocation: unclear. |
| Participants | Diagnosis: affective and schizoaffective disorder. |
| Interventions | 1. Lithium carbonate versus contemnol. |
| Outcomes | Unclear. |
| Notes | Translation of the paper is needed. |
Differences between protocol and review
We have updated the methods section to reflect changes in Cochrane methodology since publication of the original version of this review.
Contributions of authors
Stefan Leucht: protocol development, searching, data extraction, analysis, data interpretation, and writing the final report (all versions) John McGrath: protocol development, data checking, and data interpretation (2008). Werner Kissling: protocol development and data interpretation (2008). Markus Dold: update searches (2012, 2015) data extraction, analysis, and data interpretation. Bartosz Helfer: update of the review (2012, 2015), data extraction, analysis, data interpretation, and writing the final report.
Sources of support
Internal sources
Queensland Health, Australia.
Freistaat Bayern, Germany.
External sources
German Research Network on Schizophrenia, German Federal Ministry of Education and Research BMBF (grant 01 GI 993x), Germany.
Declarations of interest
Stefan Leucht: in the last three years Stefan Leucht has received honoraria for lectures from EliLilly, Lundbeck (Institute), Pfizer, Janssen, BMS, Johnson and Johnson, Otsuka, Roche, SanofiAventis, ICON, Abbvie, AOP Orphan, Servier; for consulting/advisory boards from Roche, Janssen, Lundbeck, EliLilly, Otsuka, TEVA; for the preparation of educational material and publications from Lundbeck Institute and Roche. EliLilly has provided medication for a clinical trial led by SL as principal investigator. John McGrath: John McGrath is a member of the following advisory boards: Eli Lilly Australia, Lundbeck Australia, and Pfizer Australia. In addition, JM has been a co‐investigator on studies of neuroleptic medications produced by the following companies: Astra, Janssen‐Cilag, Eli Lilly, Zeneca (ICI), Sandoz, and Pfizer. The same companies have provided travel and accommodation expenses for John McGrath to attend relevant investigator meetings and scientific symposia. No funds have been paid directly to Dr McGrath. Payments related to participation in drug trials and board attendances have been paid to a government‐audited trust account to support schizophrenia research. Werner Kissling: Werner Kissling has received honoraria or research support or both from Janssen‐Cilag, Sanofi‐Aventis, Johnson & Johnson, Pfizer, Bristol‐Myers Squibb, AstraZeneca, Lundbeck, Novartis, and Eli Lilly. Markus Dold: nothing to declare. Bartosz Helfer: nothing to declare.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Biederman 1979 {published data only}
- Biederman J, Lerner Y, Belmaker RH. Combination of lithium carbonate and haloperidol in schizo‐affective patients. Archives of General Psychiatry 1979 Mar;36:327‐33. [DOI] [PubMed] [Google Scholar]
Braden 1982 {published data only}
- Braden W, Fink EB, Qualls CB, Ho CK, Samuels WO. Lithium and chlorpromazine in psychotic inpatients. Psychiatry Research 1982;7:69‐81. [DOI] [PubMed] [Google Scholar]
Brockington 1978 {published data only}
- Brockington I, Kendell R, Kellett J, Curry S, Wainwright S. Trials of lithium, chlorpromazine and amitriptyline in schizoaffective patients. British Journal of Psychiatry 1978 Aug;133:162‐8. [DOI] [PubMed] [Google Scholar]
Collins 1991 {published data only}
- Collins EJ, Larkins EP, Shubsacs AP. Lithium carbonate in chronic schizophrenia ‐ a brief trial of lithium carbonated added to neuroleptics for treatment resistant schizophrenic patients. Acta Psychiatrica Scandinavica 1991;84:150‐4. [DOI] [PubMed] [Google Scholar]
Dube 1981 {published data only}
- Dube S, Sethi BB. Efficacy of lithium in schizophrenia. Indian Journal of Psychiatry 1981;23:193‐9. [PMC free article] [PubMed] [Google Scholar]
Feng 2006 {published data only}
- Feng Z, Yang K‐J, Gao H, Wu D‐H, Wu H‐A. The effect of lithium carbonate combining clozapine for the resistant schizophrenics with agitation and aggressive behaviour. Medical Journal of Chinese People's Health 2006;18(6):427‐8. [Google Scholar]
Garver 1983 {published data only}
- Garver DL, Hirschowitz J, Fleishmann R, Djuric P. Lithium response and psychoses: a double‐blind, placebo‐controlled study. Psychiatry Research 1984 May;12:57‐68. [DOI] [PubMed] [Google Scholar]
He 2010 {published data only}
- He Y‐F, Xie W‐C, Li X‐Y. Risperidone combined with lithium in treatment of refractory schizophrenia. Guangzhou Medical Journal 2010;2:6‐8. [Google Scholar]
Hogarty 1995 {published data only}
- Hogarty GE, McEvoy JP, Ulrich RF, DiBarry AL, Bartone P, Cooley S, Hammill K, Carter M, Munetz MR, Perel J. Pharmacotherapy of impaired affect in recovering schizophrenic patients. Archives of General Psychiatry 1995;52:29‐41. [DOI] [PubMed] [Google Scholar]
Huang 1984 {published data only}
- Huang LG BC. Platelet monoamine oxidase response to lithium treatment in psychiatric patients. Journal of Clinical Psychopharmacology 1984;4:326‐31. [PubMed] [Google Scholar]
Johnson 1971 {published data only}
- Johnson G. Differential response to lithium carbonate in manic depressive and schizo‐affective disorders. Diseases of the Nervous System 1970;31:613‐5. [PubMed] [Google Scholar]
- Johnson G, Gershon S, Burdock EI, Floyd A, Hekimian L. Comparative effects of lithium and chlorpromazine in the treatment of acute manic states. British Journal of Psychiatry 1971;119:267‐76. [DOI] [PubMed] [Google Scholar]
- Johnson G, Gershon S, Hekimian L. Controlled evaluation of lithium and chlorpromazine in the treatment of manic states: an interim report. Comprehensive Psychiatry 1968;9:563‐73. [DOI] [PubMed] [Google Scholar]
Johnstone 1988 {published data only}
- Johnstone EC, Crow TJ, Frith CD, Owens D. The Northwick Park "functional" psychosis study: diagnosis and treatment response. Lancet 1988;2:119‐25. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Crow TJ, Owens D, Frith C. The Northwick Park 'Functional' Psychosis Study. Phase 2 ‐ Maintenance treatment. Journal of Psychopharmacology 1991;5:388‐95. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Owens DGC. Does early treatment have an effect on outcome?. 10th ECNP (European College of Neuropsychopharmacology) Congress, Vienna, Austria. Sep 13 ‐ 17, 1997.
- Johnstone EC, Owens DGC, Crow TJ, Davis JM. Does a four week delay in the introduction of medication alter the course of functional psychosis?. Journal of Psychopharmacology 1999; Vol. 13, issue 3:238‐44. [MEDLINE: ] [DOI] [PubMed]
Mattes 1984 {published data only}
- Mattes JA, Nayak D. Lithium versus fluphenazine for prophylaxis in mainly schizophrenic schizoaffectives. Biological Psychiatry 1984;19:445‐9. [PubMed] [Google Scholar]
Prien 1972 {published data only}
- Pokorny AD, Prien RF. Lithium in treatment and prevention of affective disorder: A VA NIMH collaborative study. Diseases of the Nervous System 1974;35:327‐33. [PubMed] [Google Scholar]
- Prien R, Caffey EJ, Klett C. A comparison of lithium carbonate and chlorpromazine in the treatment of excited schizo‐affectives. Report of the Veterans Administration and National Institute of Mental Health collaborative study group. Archives of General Psychiatry 1972;27:182‐9. [DOI] [PubMed] [Google Scholar]
Schulz 1999 {published data only}
- Schultz SC, Glick D, Group TSSCS. Lithium augmentation fails to reduce symptoms in poorly responsive schizophrenic outpatients. Schizophrenia Research 1995; Vol. 15, issue 1‐2:151. [MEDLINE: ] [PubMed]
- Schulz SC, Thompson PA, Jacobs M, Ninan PT, Robinson D, Weiden PJ, Yadalam K, Glick ID, Odbert CL. Lithium augmentation fails to reduce symptoms in poorly responsive schizophrenic outpatients. Journal of Clinical Psychiatry 1999;60:366‐72. [PubMed] [Google Scholar]
Shopsin 1971 {published data only}
- Shopsin B, Kim S, Gershon S. A controlled study of lithium vs. chlorpromazine in acute schizophrenics. British Journal of Psychiatry 1971;119:435‐40. [DOI] [PubMed] [Google Scholar]
Simhandl 1996 {published data only}
- Simhandl C, Meszaros K, Denk E, Thau K, Topitz A. Adjunctive carbamazepine or lithium carbonate in therapyresistant chronic schizophrenia [2]. Canadian Journal of Psychiatry/ Revue Canadienne de Psychiatrie 1996;41(5):317. [DOI] [PubMed] [Google Scholar]
Simpson 1976 {published data only}
- Simpson GM, Branchey MH, Lee JH, Voitashevsky A, Zoubok B. Lithium in tardive dyskinesia. Pharmakopsychiatrie‐Neuropsychopharmakologie 1976;9:76‐80. [DOI] [PubMed] [Google Scholar]
Small 1975 {published data only}
- Kellams J, Small J, Milstein V, Perex H. Lithium combined with neuroleptics in the treatment of chronic schizophrenia. Psychopharmacology Bulletin 1976;12:27‐30. [PubMed] [Google Scholar]
- Schulz SC, Kahn EM, Baker RW, Conley RR. Lithium and carbamazepine augmentation in treatment‐refractory schizophrenia. The Neuroleptic‐Nonresponsive Patient: Characterization and Treatment. Washington, DC, USA: American Psychiatric Press Inc, 1990:111‐36. [MEDLINE: ]
- Small JG, Kellams JJ, Milstein V, Moore J. A placebo‐controlled study of lithium combined with neuroleptics in chronic schizophrenic patients. American Journal of Psychiatry 1975;132:1315‐7. [DOI] [PubMed] [Google Scholar]
Small 2001 {published and unpublished data}
- Small JG, Klapper MH, Malloy FW, Steadman TM. Clozapine combined with lithium in schizophrenia and schizoaffective disorder. Schizophrenia Research 2001;49 (suppl):246. [DOI] [PubMed] [Google Scholar]
- Small JG, Klapper MH, Malloy FW, Steadman TM. Tolerability and efficacy of clozapine combined with lithium in schizophrenia and schizoaffective disorder. Journal of clinical psychopharmacology. United States of America, 2003; Vol. 23, issue 3:223‐8. [MEDLINE: ] [DOI] [PubMed]
Terao 1995 {published data only}
- Terao T, Oga T, Nozaki S, Ohta A, Ohtsubo Y, Yamamoto S, Zamami M, Okada M. Lithium addition to neuroleptic treatment in chronic schizophrenia: a randomized, double‐blind, placebo‐controlled, cross‐over study. Acta Psychiatrica Scandinavica 1995;92:220‐4. [DOI] [PubMed] [Google Scholar]
- Terao T, Oga T, Nozaki S, Ohta A, Ohtsubo Y, Yamoto S, et al. L'associazione di Litio alla terapia neurolettica nel trattamento della schizofrenia cronica: uno studio in doppio cieco crociato randomizzato, controllato con placebo. Rivista di Psichiatria. Italy: Il Pensiero Scientifico Editore, 1995; Vol. 30, issue 6:42. [MEDLINE: ]
Wilson 1993 {published data only}
- Wilson WH. Addition of lithium to haloperidol in non ‐ affective, antipsychotic non ‐ responsive schizophrenia. Schizophrenia Research 1993; Vol. 9:256. [MEDLINE: ] [DOI] [PubMed]
- Wilson WH. Addition of lithium to haloperidol in non‐affective, antipsychotic non‐responsive schizophrenia ‐ A double blind, placebo controlled, parallel design clinical trial. Psychopharmacology 1993;111:359‐66. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Alexander 1979 {published data only}
- Alexander PE, Kammen DP, Bunney WE. Antipsychotic effects of lithium in schizophrenia. American Journal of Psychiatry 1979;136:283‐7. [DOI] [PubMed] [Google Scholar]
Aronoff 1970 {published data only}
Baastrup 1967 {published data only}
- Baastrup PC, Schou G, Schou M. Lithium as a prophylactic agent. Archives of General Psychiatry 1967;16:162‐72. [DOI] [PubMed] [Google Scholar]
Bellaire 1990 {published data only}
- Bellaire W, Demisch K, Stoll KD. Carbamazepine versus lithium. Application in the prophylaxis of recidivating affective and schizoaffective psychoses [Carbamazepin Vs. Lithium. Einsatz in der Prophylaxe Rezidivierender Affektiver und Schizoaffektiver Psychosen]. Munchener Medizinische Wochenschriftenschrift 1990; Vol. 132:82‐6. [MEDLINE: ]
Berger 2006 {published data only}
- Berger G. Lithium in patients at ultra high risk of developing a first psychotic episode. Stanley Foundation Research Programs 2006.
Bigelow 1981 {published data only}
- Bigelow LB, Weinberger DR, Wyatt R. Synergism of combined lithium‐neuroleptic therapy ‐ A double‐blind, placebo‐controlled case study. American Journal of Psychiatry 1981;138:81‐3. [DOI] [PubMed] [Google Scholar]
Bowers 1983 {published data only}
- Bowers MB, Heninger GR, Sternberg DE. Correlates of lithium response in psychotic‐affective syndromes. Comprehensive psychiatry 1983;24:469‐75. [DOI] [PubMed] [Google Scholar]
Braden 1980 {published data only}
- Braden W. Lithium versus chlorpromazine in acute psychosis. Psychopharmacology Bulletin 1980; Vol. 16:70‐1. [MEDLINE: ]
Brewerton 1983 {published data only}
Cabrera 1986 {published data only}
- Cabrera JF, Muhlbauer HD, Schley J, Stoll KD, Muller‐Oerlinghausen B. Long‐term randomized clinical trial on oxcarbazepine vs lithium in bipolar and schizoaffective disorders ‐ Preliminary results. Pharmacopsychiatry 1986; Vol. 19, issue 4:282‐3. [MEDLINE: ]
Campbell 1972 {published data only}
- Campbell M, Fish B, Korein J, Shapiro T, Collins P, Koh C. Lithium and chlorpromazine: a controlled cross‐over study of hyperactive severely disturbed young children. Journal of Autism and Childhood Schizophrenia 1972;2:234‐63. [DOI] [PubMed] [Google Scholar]
Campbell 1982 {published data only}
Carman 1981 {published data only}
- Carman J, Bigelow LB, Wyatt RJ. Lithium combined with neuroleptics in chronic schizophrenic and schizoaffective patients. Journal of Clinical Psychiatry 1981;42:124‐8. [PubMed] [Google Scholar]
Chen 2001 {published data only}
- Chen L, Zhang X, Li J. A controlled study between clozapine and lithium carbonate in treatment‐refractory schizophrenia. Sichuan Mental Health 2001;14:213‐4. [Google Scholar]
Dinsmore 1972 {published data only}
- Dinsmore PR, Ryback R. Lithium in schizo‐affective disorders. Diseases of the Nervous System 1972;33:771‐6. [PubMed] [Google Scholar]
Edelstein 1981 {published data only}
- Edelstein P, Schultz JR, Hirschowitz J, Kanter DR, Garver D. Physostigmine and lithium response in the schizophrenias. American Journal of Psychiatry 1981;138:1078‐81. [DOI] [PubMed] [Google Scholar]
Gao 2002 {published data only}
- Gao S, Ma J, Xu F, Yang S. Clozapine combined with lithium carbonate treatment for patients with chronic schizophrenia: controlled clinical trial. Nervous Diseases and Mental Hygiene 2002;2:338‐9. [Google Scholar]
Garver 1988 {published data only}
- Garver DL, Kelly K, Fried KA, Magnusson M, Hirschowitz J. Drug response patterns as a basis of nosology for the mood‐incongruent psychoses (the schizophrenias). Psychological Medicine 1988;18:873‐85. [DOI] [PubMed] [Google Scholar]
Gerlach 1975 {published data only}
- Gerlach J, Thorsen K, Munkvad I. Effect of lithium on neuroleptic‐induced tardive dyskinesia compared with placebo in a double‐blind cross‐over trial. Pharmakopsychiatrie Neuropsychopharmakologie 1975;8:51‐6. [DOI] [PubMed] [Google Scholar]
Gram 1971 {published data only}
- Gram LF, Rafaelsen OJ. Lithium treatment of psychotic children and adolescents. A controlled clinical trial. Acta Psychiatrica Scandinavica 1972;48:253‐60. [DOI] [PubMed] [Google Scholar]
Greil 1997 {published data only}
- Greil W, Ludwig Mayerhofer W, Erazo N, Engel RR. Lithium vs cambamazepine in the maintenance treatment of schizoaffective disorder: A randomised study. European Archives of Psychiatry and Clinical Neuroscience 1997;247:42‐50. [DOI] [PubMed] [Google Scholar]
Growe 1979 {published data only}
- Growe GA, Crayton JW, Klass DB, Evans H, Strizich M. Lithium in chronic schizophrenia. American Journal of Psychiatry 1979;136:454‐5. [PubMed] [Google Scholar]
Haastrup 1973 {published data only}
- Haastrup S, Rasmussen S, Kirk L. Lithium therapy of schizophrenics. A controlled study. Acta Psychiatrica Scandinavica Supplement 1973;243:68. [Google Scholar]
Harrison 1980 {published data only}
- Harrison GP, Lipinski JF, Cohen BM, Aselrod DT. "Schizoaffective disorder": an invalid diagnosis? A comparison of schizoaffective disorder, schizophrenia, and affective disorder. American Journal of Psychiatry 1980;137:921‐7. [DOI] [PubMed] [Google Scholar]
Hofmann 1970 {published data only}
- Hofmann G, Kremser M, Katschnig H, Scheiber V. Prophylaktische Lithiumtherapie bei manisch‐depressivem Krankheitsgeschehen und bei Legierungspsychosen. Internationale Pharmacopsychiatrie 1970;4:187‐93. [Google Scholar]
Hullin 1975 {published data only}
- Hullin RP, McDonald R, Allsopp MNE. Further report on prophylactic lithium in recurrent affective disorders. British Journal of Psychiatry 1975;126:281‐4. [DOI] [PubMed] [Google Scholar]
Jus 1978 {published data only}
- Jus A, Villeneuve A, Gautier J, Jus K, Villeneuve C, Pires P, Villeneuve R. Deanol, lithium and placebo in the treatment of tardive dyskinesia. A double‐blind crossover study. Neuropsychobiology 1978;4:140‐9. [DOI] [PubMed] [Google Scholar]
Kahn 1990 {published data only}
Lenzi 1985 {published data only}
- Lenzi A, Lazzerini F, Grossi E. Use of carbamazepine in acute psychosis ‐ A controlled study. Journal of International Medical Research 1986;14:78‐84. [DOI] [PubMed] [Google Scholar]
Lerner 1988 {published data only}
- Lerner Y, Mintzer Y, Schestatzky M. Lithium combined with haloperidol in schizophrenic patients. British Journal of Psychiatry 1988;153:359‐62. [DOI] [PubMed] [Google Scholar]
Liebowitz 1976 {published data only}
- Liebowitz JH, Rudy V, Gershon ES, Gillis A. A pharmacogenetic case report: Lithium‐responsive postpsychotic antisocial behavior. Comprehensive Psychiatry 1976;17:655‐60. [DOI] [PubMed] [Google Scholar]
Liu 2006 {published data only}
- Liu N. The lithium compound norethisterone periodic relapse prevention control study [碳酸锂与复方炔诺酮预防周期性精神病复发的对照研究]. Medical Journal of Chinese People's Health [中国民康医学] 2006; Vol. 18, issue 5:199‐200.
Mackkay 1980 {published data only}
- MacKay AV, Sheppard GP, Saha BK, Motley B, Johnson AL, Marsden CD. Failure of lithium treatment in established tardive dyskinesia. Psychological Medicine 1980;10:583‐7. [DOI] [PubMed] [Google Scholar]
Marken 1994 {published data only}
Martorano JT {published data only}
- Martorano JT. Target symptoms in lithium carbonate therapy. Comprehensive Psychiatry 1972;13:533‐7. [DOI] [PubMed] [Google Scholar]
Miller 1979 {published data only}
- Miller V. Lithium carbonate in the treatment of schizophrenia and schizo‐affective disorder: Review and hypothesis. Journal of Clinical Psychiatry 1979;14:705‐10. [PubMed] [Google Scholar]
Nct 2005 {published data only}
- Nct. Divalproex extended release and placebo, lithium, or quetiapine for mania. http://www.clinicaltrials.gov 2005.
Nemes 1986 {published data only}
- Nemes ZC, Volavka J, Cooper TB, O'Donnell J, Jaeger J. Lithium and haloperidol. Biological Psychiatry 1986;21:568‐9. [DOI] [PubMed] [Google Scholar]
Paykel 2000 {published data only}
- Paykel ES. Prophylaxis of puerperal psychosis. National Research Register 2000.
Placidi 1986 {published data only}
- Placidi GF, Lenzi A, Lazzerini F, Cassano G. The comparative efficacy and safety of carbamazepine versus lithium: A randomized, double blind 3 year trial in 83 patients. Journal of Clinical Psychiatry 1986;47:490‐4. [PubMed] [Google Scholar]
- Placidi GF, Lenzi A, Rampello E, Andreani MF, Cassano GB, Grossi E. Long term‐double blind prospective study on carbamazepine versus lithium in bipolar and schizoaffective disorders. Preliminary results. Anticonvulsants in Affective Disorders. Amsterdam, Netherlands: Elsevier, 1984:188‐97. [MEDLINE: ]
Prange 1973 {published data only}
- Prange A. Preliminary experience with tryptophan and lithium in the treatment of tardive dyskinesia. Psychopharmacology Bulletin 1973;9:36‐7. [Google Scholar]
Prien 1972a {published data only}
- Prien RF, Caffey EM, Klett J. Factors associated with treatment in lithium carbonate prophylaxis. Archives of General Psychiatry 1974;31:189‐92. [DOI] [PubMed] [Google Scholar]
- Prien RF, Point P, Caffey EM, Klett CJ. Comparison of lithium carbonate and chlorpromazine in the treatment of mania. Archives of General Psychiatry 1972;26:146‐53. [DOI] [PubMed] [Google Scholar]
Rice 1956 {published data only}
- Rice D. The use of lithium salts in the treatment of manic states. International Pharmacology 1956;3:604‐11. [DOI] [PubMed] [Google Scholar]
Rosenthal 1980 {published data only}
- Rosenthal NE, Rosenthal LN, Stallone F, Dunner DL, Fieve RR. Toward the validation of RDC schizoaffective disorder. Archives of General Psychiatry 1980;37:804‐10. [DOI] [PubMed] [Google Scholar]
Schnexnayder 1995 {published data only}
- Schnexnayder LW, Hirschowitz J, Sautter FJ, Garver DL. Predictors of response to lithium in patients with psychoses. American Journal of Psychiatry 1995;152:1511‐3. [DOI] [PubMed] [Google Scholar]
Schou 1954 {published data only}
- Schou M, Juel‐Nielsen N, Strömgen E, Voldby H. The treatment of manic psychoses by the administration of lithium salts. Journal of Neurology Neurosurgery and Psychiatry 1954;17:250‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shalif 1981 {published data only}
Shaw 1974 {published data only}
- Shaw DM, Hewland R, Johnson AL, Hilary‐Jones P, Howlett MR. Comparison of serum levels of two sustained‐release preparations of lithium carbonate. Current Medical Research Opinion 1974;2:90‐4. [DOI] [PubMed] [Google Scholar]
Shopsin 1975 {published data only}
- Shopsin B, Gershon S, Thompson H, Collins P. Psychoactive drugs in mania. Archives of General Psychiatry 1975;32:34‐42. [DOI] [PubMed] [Google Scholar]
Smulevitch 1974 {published data only}
- Smulevitch AB, Zavidovskaya GI, Igonin AL, Mikhailova NM. The effectiveness of lithium in affective and schizoaffective psychoses. British Journal of Psychiatry 1974;125:65‐72. [DOI] [PubMed] [Google Scholar]
Sun 2008 {published data only}
- Sun H, Jiao Y‐T, Zhang L‐X. A comparative study of clozapine and clozapine unite lithium carbonate in the treatment of male patients with treatment‐resistant schizophrenia. Journal of Sichuang Mental Health 2008;21(2):83‐5. [Google Scholar]
Taylor 1974 {published data only}
- Taylor MA, Abrams R. The phenomenology of mania. Archives of General Psychiatry 1973;29:520‐2. [DOI] [PubMed] [Google Scholar]
- Taylor MA, Gaztanaga P, Abrams R. Manic‐depressive illness and acute schizophrenia: a clinical, family history, and treatment‐response study. American Journal of Psychiatry 1974;131:678‐82. [DOI] [PubMed] [Google Scholar]
Van Kammen 1978 {published data only}
- Kammen DP, Alexander P. d Amphetamine raises serum prolactin in man: Evaluations after chronic placebo, lithium and pimozide treatment. Life Sciences 1978;23:1487‐92. [DOI] [PubMed] [Google Scholar]
Van Kammen 1980 {published data only}
- Kammen D, Alexander P, Bunney WJ. Lithium treatment in post‐psychotic depression. British Journal of Psychiatry 1980;136:479‐85. [DOI] [PubMed] [Google Scholar]
Van Kammen 1985 {published data only}
- Kammen DP, Docherty JP, Marder SR. Lithium attenuates the activation‐euphoria but not the psychosis induced by d‐amphetamine in schizophrenia. Psychopharmacology 1985;87:111‐5. [DOI] [PubMed] [Google Scholar]
Van Putten 1975 {published data only}
- Putten T, Sanders D. Lithium in treatment failures. Journal of Nervous and Mental disease 1975;161:255‐64. [DOI] [PubMed] [Google Scholar]
Vieweg 1989 {published data only}
- Vieweg V, Godleski L, Hundley P, Harrington D, Yank G. Antipsychotic drugs, lithium, carbamazepine, and abnormal diurnal weight gain in psychosis. Neuropsychopharmacology 1989;2:39‐43. [DOI] [PubMed] [Google Scholar]
Volavka 1986 {published data only}
- Volavka J, O'Donald J, Muragali R. Lithium and lecithin in tardive dyskinesia: An update. Psychiatry Research 1986;19:101‐4. [DOI] [PubMed] [Google Scholar]
Wang 1995 {published data only}
- Wang Q, Yuan F, Ai L, Yin D, Xiao Y. Chlorpromazine combined with lithium carbonate treatment for patients with refractory schizophrenia: a randomized double‐blind cross‐over clinical trial. Journal of Clinical Psychological Medicine 1995;5:185. [Google Scholar]
White 1966 {published data only}
- White RB, Schlagenhauf G, Tupin JP. The treatment of manic depressive states wtih lithium carbonate. Current Psychiatric Therapies 1966;11:230‐42. [Google Scholar]
Wilner 1996 {published data only}
- Wilner KD, Anziano RJ, Tensfeldt TG, Pelletier SN, Apseloff G, Gerber N. The effects of ziprasidone on steady‐state lithium levels and renal clearance of lithium. American Psychiatric Association, 149th conference. May 4‐9, 1996. [DOI] [PMC free article] [PubMed]
Wilson 1994 {published data only}
- Wilson WH. Open clozapine treatment following a controlled clinical trial of lithium augmentation of haloperidol for refractory schizophrenia. Lithium 1994; Vol. 5, issue 2:113‐4.
Zemlan 1984 {published data only}
- Zemlan FP, Hirschowitz J, Sautter FJ, Garver DL. Impact of lithium therapy on core psychotic symptoms of schizophrenia. British Journal of Psychiatry 1984;144:64‐9. [DOI] [PubMed] [Google Scholar]
Zhou 2011 {published data only}
- Zhou Guangyu, Rong Min. Paroxetine combined with lithium in patients with refractory schizophrenia.. Medical Journal of Chinese People's Health 2011;23(15):1899‐1900. [Google Scholar]
References to studies awaiting assessment
Kamisada 1988 {published data only}
- Kamisada M, Tateyama M, Nakano Y, Kawachi Y, FUjii Y, Tanoue A, Takamiya M, Nakajima S, Sakuma K, Oguchi E, Yagi G. Comparison of the clinical effects of lithium carbonate and carbamazepine on excited schizophrenics. Psychopharmacology 1988;96 (suppl):348. [Google Scholar]
McGorry 2002 {published data only}
- McGorry PD, Cocks J, Harrigan S, Elkins K, Lambert T, Owen S. Very low‐dose risperidone treatment in first‐episode psychosis: how effective is it?. Proceedings of the 3rd International Conference on Early Psychosis; 2002 Sep 25‐28; Copenhagen, Denmark 2002:48.
Mosolov 1998 {published data only}
- Mosolov SN, Kostiukova EG, Kuzavkova MV. The efficacy of lithium carbonate and contemnol in preventing affective and schizoaffective psychoses. Zhurnal Nevropatologii i Psikhiatrii Imeni S. S. Korsakova 1998; Vol. 98, issue 3:15‐8. [MEDLINE: ] [PubMed]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1989
- Andreasen NC. Scale for the assessment of negative symptoms. British Journal of Psychiatry 1989;155:53‐8. [PubMed] [Google Scholar]
Astrachan 1972
- Astrachan BM, Harrow M, Adler D, Brauer L, Schwartz A, Tucker G. A scale for the diagnosis of schizophrenia. British Journal of Psychiatry 1972;121:529. [DOI] [PubMed] [Google Scholar]
Atre‐Vaidya 1989
- Atre‐Vaidya N, Taylor MA. Effectiveness of lithium in schizophrenia: Do we really have an answer?. Journal of Clinical Psychiatry 1989;50(5):170‐173. [PubMed] [Google Scholar]
Barnes 1989
- Barnes TRE. A rating scale for drug‐induced akathisia. British Journal of Medicine 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Bech 1978
- Bech P, Rafaelsen OJ, Kramp P, Bolwig TG. The mania rating scale: scale construction and inter‐observer agreement. Neuropharmacology 1978;17:430‐1. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Burdock 1968
- Burdock EI, Hardesty, AS. A psychological test for psychopathology. Journal of Abnormal Psychology 1968;73:62‐9. [DOI] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT Jr, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Cheine 2001
- Cheine M, Ahonen J, Wahlbeck K. Supplementing standard drug treatment of those with schizophrenia with beta‐blocking medication. Cochrane Database of Systematic Reviews 2001, Issue 3. [DOI: 10.1002/14651858.CD000234] [DOI] [Google Scholar]
Christison 1991
- Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophrenia Bulletin 1991;17:217‐45. [DOI] [PubMed] [Google Scholar]
Citrome 2009
- Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence?. Expert Rev Neurother 2009;9(1):55‐71. [DOI] [PubMed] [Google Scholar]
Davey Smith 1998
- Davey Smith G, Egger M. Meta‐analysis: Unresolved issues and future developments. BMJ 1998;16:221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Abstracts of 8th International Cochrane Colloquium; 2000 Oct 25‐28th; Cape Town, South Africa. 2000.
Delva 1982
- Delva, NJ, Letemendia. FJ. Lithium treatment in schizophrenia and schizo‐affective disorders. The British Journal of Psychiatry 1982;141:387‐400. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Dold 2012
- Dold M, Li C, Tardy M, Khorsand V, Gillies D, Leucht S. Benzodiazepines for schizophrenia. Cochrane Database of Systematic Reviews 2012;11:CD006391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey mith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2009
- Essali A, Al‐Haj Haasan N, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database of Systematic Reviews 2009;1:CD000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
GRADE Profiler [Computer program]
- GRADE Working Group. GRADE Profiler. Version 3.2. GRADE Working Group, 2004.
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy U. ECDEU assessment manual for psychopharmacology. Revised. Rockville, Maryland, USA: National Institute of Mental Health, 1976. [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale of depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analysis. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. Available from www.cochrane‐handbook.org. [updated March 2011].
Jope 1999
- Jope RS. Anti‐bipolar therapy: mechanism of action of lithium. Molecular Psychiatry 1999;4(2):117‐28. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and negative syndrome scale (PANSS) manual. Positive and negative syndrome scale (PANSS) manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13(2):261‐75. [DOI] [PubMed] [Google Scholar]
Krawiecka 1977
- Krawiecka M, Goldberg D, Vaughan MA. Standardized assessment scale for rating chronic psychotic patients. Acta Psychiatrica Scandinavica 1977;55:299‐308. [DOI] [PubMed] [Google Scholar]
Lenox 2003
- Lenox RH, Wang L. Molecular basis of lithium action: integration of lithium‐responsive signaling and gene expression networks. Molecular Psychiatry 2003;8(2):135‐44. [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2013
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, Samara M, Barbui C, Engel RR, Geddes JR, Kissling W, Stapf MP, Lässig B, Salanti G, Davis JM. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple‐treatments meta‐analysis. The Lancet 2013;382(9896):951‐962. [DOI] [PubMed] [Google Scholar]
Leucht 2014
- Leucht S, Helfer B, Dold M, Kissling W, McGrath J. Carbamazepine for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 5. [DOI: 10.1002/14651858.CD001258.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marwaha S, Johnson S. Schizophrenia and employment ? A review.. Social Psychiatry and Psychiatric Epidemiology 2004;39:337‐49. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
NIMH 1970
- National Institute of Mental Health. Clinical global impression. In: Guy W, Bonato RR editor(s). Manual for the ECDEU Assessment Battery. Washington DC: NIMH, 1970. [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:790‐812. [Google Scholar]
Review Manager (RevMan) [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Schooler 1993
- Schooler NR, Keith SJ. Clinical research for the treatment of schizophrenia. Psychopharmacology Bulletin 1993;29:431‐46. [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schwarz 2008
- Schwarz C, Volz A, Li C, Leucht S. Valproate for schizophrenia (Cochrane review). Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD004028.pub3] [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Simpson 1970
- Simpson M, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212:11‐19. [DOI] [PubMed] [Google Scholar]
SPSS 2001 [Computer program]
- SPSS Inc. SPSS for Windows version 11.0.1. LEEDS technologies Inc., 2001.
Tharyan 2005
- Tharyan P, Adams CE. Electroconvulsive therapy for schizophrenia (Cochrane Review). Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD000076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality.. Archives of General Psychiatry 1978;35:153‐55. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Wahlbeck 2001
- Wahlbeck K, Tuunainen A, Ahokas A, Leucht S. Drop‐out rates in randomised antipsychotic drug trials. Psychopharmacology 2001;155:230‐3. [DOI] [PubMed] [Google Scholar]