

Abstract
Context
Hyperparathyroidism-jaw tumour (HPT-JT) syndrome is a rare autosomal dominant cause of familial hyperparathyroidism associated with ossifying fibromas (OF) of the maxillofacial bones and increased risk of parathyroid carcinoma, caused by inactivating germline mutation of the cell division cycle 73 (CDC73) gene.
Objective
To report the first Romanian family with HPT-JT and genetic screening of CDC73 gene.
Subjects and Methods
Mutational analysis of the CDC73 gene and genetic screening of the family of a proband with HPT-JT. Histological diagnosis of parathyroid tumors (WHO criteria) and immunohistochemistry (parafibromin) were performed.
Results
Three of the six screened family members had evidence of PHPT and surgically proven parathyroid tumours. Two of the three affected members had parathyroid carcinomas and one had two parathyroid adenomas. Genetic screening of CDC73 gene revealed that 4 of 6 patients showed a heterozygous germline deletion of one nucleotide: c.128-IVS1+1 delG. All the three affected patients, resulted to be carriers of the CDC73 mutation, but each one bearing a different CDC73 polymorphism.
Conclusions
We identified a new CDC73 germline mutation in a Romanian family of HPT-JT. Analysis of clinical phenotypes in the four mutated individuals confirmed the incomplete penetrance and the variable clinical expression of the disease.
Keywords: Familial Primary Hyperparathyroidism, Parathyroid Carcinoma, HPT-JT, Germline CDC73 mutation, Genetic screening
INTRODUCTION
Hyperparathyroidism-jaw tumor syndrome (HPT-JT) is a rare autosomal dominant cause of familial hyperparathyroidism associated with ossifying fibromas (OF) of the maxillofacial bones and increased risk of parathyroid carcinoma. The putative tumor suppressor gene CDC73 has been implicated in the syndrome, with a multitude of inactivating mutations identified, mostly comprised of point mutations and small insertions and deletions (1, 2). CDC73 is located in the autosomal chromosome locus 1q25- q32 and encodes parafibromin, which is a 17-exon and 531-amino acid protein, mainly expressed in the parathyroid glands, adrenal glands, kidney, heart, and skeletal muscle, and believed to function as a transcriptional regulator through interactions with the RNA-polymerase II – associated factor 1 (PAF-1) complex (3).
Because of its incomplete penetrance, patients with germline CDC73 mutation can present with a spectrum of phenotypes including seemingly sporadic parathyroid cancer, familial isolated hyperparathyroidism (FIHP) with or without parathyroid cancer, or full expression of HPT-JT (4). HPT is mostly due to a single parathyroid adenoma, but multigland disease may occur and parathyroid carcinoma is found in 15 to 37.5% in different case series (5-9).
Renal abnormalities occur in 15% of patients and include Wilms’ tumors, hamartomas, renal cell carcinoma, and polycystic disease (7). Uterine tumors affect up to 75% of female HPT-JT patients and may be benign (e.g., adenofibromas, leiomyomas) or malignant (e.g., adenosarcomas) (10).
The case presented here contributes to a relatively small body of literature devoted to HPT-JT.
SUBJECTS AND METHODS
Patients included in this study are 6 members of a HPT-JT family, evaluated at the Department of Endocrinology & Metabolism, National Institute of Endocrinology, Bucharest, Romania. The complete pedigree chart is shown in Figure 1. DNA samples were obtained and sent for mutation analysis to the Laboratory of Cellular and Molecular Biology, Department of Surgery and Translational Medicine, University of Florence, Florence, Italy.
Figure 1.
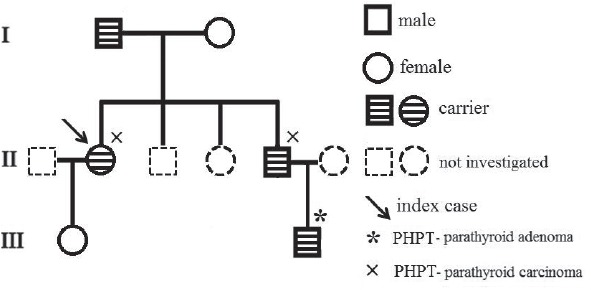
Pedigree chart of the family.
Informed consent for collection of clinical, biochemical and genetic data was obtained from all patients.
CASE PRESENTATION
The proband, female, has been diagnosed at the age of 35 with primary hyperparathyroidism: Ca=13.9 mg/dL; PTH=327 pg/mL; P= 1.8 mg/dL. Her past history was remarkable as she had been operated 4 yrs before for an OF of the mandible and an uterine fibroid; a kidney stone was eliminated 3 years before. She had nephrocalcinosis on renal US, normal renal function and normal BMD. A hypervascular tumor mass (2.2/1.8 cm) with cystic changes was detected by ultrasound (US) near the left lower pole of the thyroid (Fig. 2). A CT scan confirmed the left inferior parathyroid tumor, as well as the previous mandibular surgery. Surgery was performed and a parathyroid carcinoma was diagnosed, based on vascular and capsular invasion (Fig. 3). She was lost to follow-up but 5 yrs later the biochemistry was normal: Ca=9.85 mg/dL; PTH=44.4 pg/mL. Two years ago she presented to our clinic with her brother, 48 yrs, who had already been diagnosed with a parathyroid carcinoma, and was operated several times. He had persistent PHPT, Ca=11.3 mg/dL; PTH=193.7 pg/mL and on treatment with zoledronic acid, with normal renal function. He presented four years ago at another hospital with a large neck mass on the left side of the trachea with compression symptoms and laterocervical adenopathies. Tumor resection and subtotal thyroidectomy were performed based on the FNAB suggestion of a “clear cells” thyroid tumor; a repeated histological examination confirmed parathyroid carcinoma based on vascular and soft tissue invasion, and the appearance was of a clear cell parathyroid tumor (Fig. 4). As immunohistochemistry for TTF1 (thyroid transcription factor 1) was negative and serum calcium was 13 mg/dL, repeated surgeries for residual tumor and neck and mediastinal adenopathies were performed. The diagnosis of parathyroid carcinoma was established based on strong positive immunohistochemistry for PTH in mediastinal adenopathies (Fig. 5) and soft tissue invasion; Ki67 was positive at 10%. Several courses of chemotherapy and radiotherapy were performed as well as multiple doses of zoledronic acid. Serum calcium varied between 10.65 and 12.4 mg/dL, PTH from 167 pg/mL to 567 pg/mL for four years. In the fifth year calcium rose to 13.5 mg/dL and PTH to 1200 pg/mL and became refractory to iv bisphosphonates; denosumab was added as described (11, 12). No OF or renal lesions were identified. He had secondary osteoporosis by DXA.
Figure 2.
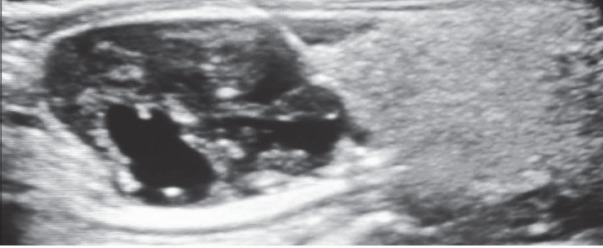
Tumor mass (2.2/1.8 cm) with cystic changes near the left lower pole of the thyroid (US).
Figure 3.
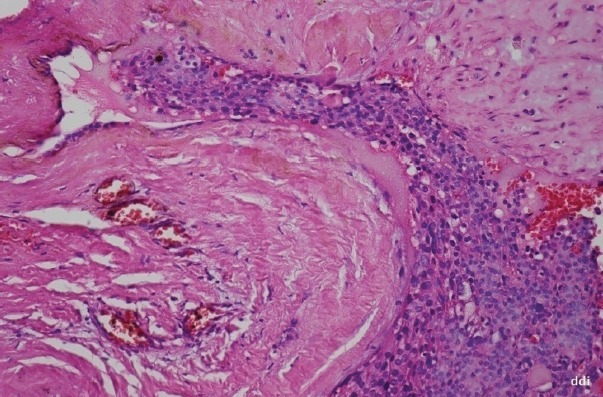
Parathyroid carcinoma- vascular invasion, thick capsule with evident vascular channel and tumoral thrombus (200 HE).
Figure 4.
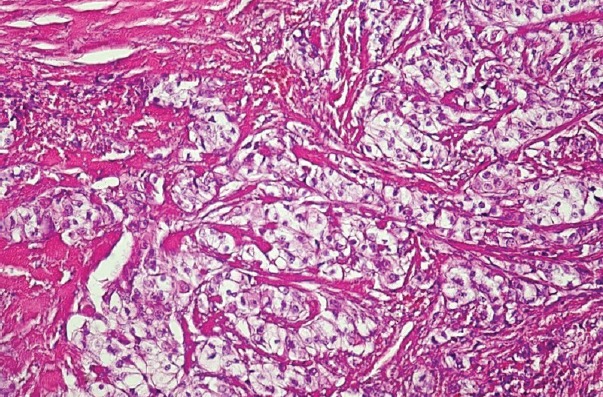
Parathyroid carcinoma – tumor detail (x 200 HE) - trabecular and nested pattern with clear cells.
Figure 5.
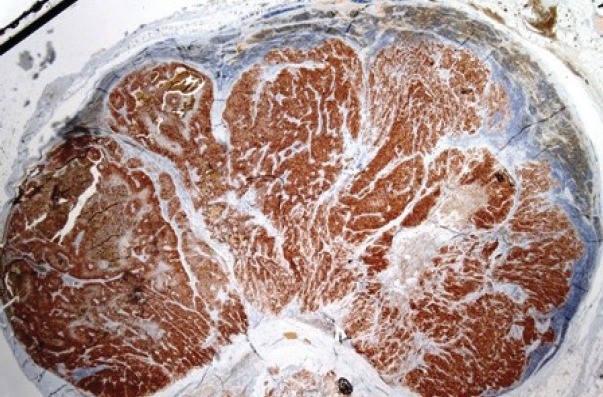
Positive staining for PTH in a metastatic lymph node (immunohistochemistry)(x12.5).
At the present time, a computerized tomography (CT) scan revealed a tumor in the left thyroid lobe (25/22 mm) with invasion in the thyroid and exthyroidal tissues; a necrotic adenopathy (11 mm) in the superior mediastinum and 4 secondary pulmonary tumors (the largest 10 mm). An 11-C methionine positron emission tomography (PET) scan showed residual metabolic activity on the left side of the trachea and inactive pulmonary nodules. The lesions showed a slow progression one year apart. The patient is currently enrolled in our follow-up regime dedicated to the control of hypercalcemia.
Other six members of the family were available for screening. The parents and other two siblings were asymptomatic, had normal serum calcium and no OF. The proband’s daughter (20 yrs old) was negative for OF and hyperparathyroidism. Instead, the son (24 years old) of the male patient with parathyroid carcinoma was recently diagnosed with primary hyperparathyroidism Ca=12.2 mg/dL; PTH=176 pg/mL. He was asymptomatic with normal renal, thyroid, adrenal and gonadal functions. Two parathyroid tumors were seen on neck US, in the right lower and left lower positions, which were removed by surgery. Surgical pathology demonstrated a well-circumscribed right lower parathyroid gland weighing 0.35 g consisting of an enlarged hypercellular nodule of chief cells devoid of cytologic atypia, invasion, and significant mitotic activity; the other tumor was described as an oxyphil adenoma of the lower left parathyroid of 0.25 g. These findings are consistent with double parathyroid adenoma and rule out atypical adenoma or carcinoma. Total serum calcium and PTH levels were normal at 8.8 mg/dL and 20.9 pg/mL, respectively, during follow-up at 3 months. Renal US showed microlithiasis, but otherwise negative; there was no OF on panoramic radiograph. He had low bone mass by DXA.
METHODS
Histological diagnosis
The histological diagnosis was confirmed according to the WHO guidelines. All surgical specimens were fixed in buffered formalin 10%, paraffin included, sectioned at 4 microns and HE stained.
Immunohistochemistry
The immunohistochemistry was performed on 3 µm sections from 10% formalin-fixed paraffin-embedded tissues from the 3 operated patients with CDC73 mutation (two adenomas and two carcinomas). The sections were incubated with primary antibody Parafibromin (Santa Cruz Biotechnology, 1:50, 2H1, SC-33638) at room temperature for 1 hour. The UltraVision Quanto Detection System HRP DAB (Thermo Fischer Scientific) was then applied for 20 min. Diaminobenzidine was used for detection, followed by a light hematoxylin counterstain. Tumors were classified as negative only if there were internal positive controls detected throughout the slide and adjacent areas.
The genetic screening of CDC73 gene
Genetic analysis to evaluate germinal mutations of the whole CDC73 coding sequence (17 exons, including exon-intron boundaries) was performed by PCR amplification and direct Sanger sequencing on genomic DNA extracted from peripheral blood leukocytes. Briefly, PCR reactions were carried out in a 50μL reaction volume containing 5 μL10X PCR Buffer (Thermo Scientific, Waltham, Massachusetts, USA), 0.2 nM dNTPs, 10 pmol of each primers,1.25 U DreamTaq DNA Polymerase (Thermo Scientific, Waltham, Massachusetts, USA) and 50 ng of DNA. PCR cycling conditions consisted of an initial 5 min denaturation step at 95°C, followed by 30 cycles of 95°C for 30 s annealing for 30 s, and extension at 72°C for 30 s, with final extension at 72°C for 5 min. Annealing temperatures were optimized for all primer sets. Purified PCR products were sequenced using the BigDye Terminator Cycle Sequencing Kit v. 1.1 (Applied Biosystems, Foster City, CA, USA). Purified sequencing reactions were separated on ABI PRISM 3100xl genetic analyzer capillary sequencer (Applied Biosystems, Foster City, CA, USA).
RESULTS
The mutational analysis of the CDC73 gene identified a germline heterozygous deletion of one nucleotide (c.128-IVS1+1 delG) (Fig. 6) in four of the six screened individuals (Table 1). Also CDC73 polymorphisms, with no known pathological role in HPT-JT, were found in five members of our family, as reported, in detail, in Table 1. All the three affected patients, with biochemical evidence of PHPT and surgically proven parathyroid tumors, resulted to be carriers of the CDC73 mutation, but each one bearing a different CDC73 polymorphism. Parafibromin staining was weakly positive in both adenomas and negative in both parathyroid carcinoma cases. The index case’s father resulted to have the mutation but no CDC73 polymorphisms; interestingly, he was still asymptomatic at the time of this study at the age of 81: serum calcium 9.4 mg/dL, serum PTH 52 pg/mL, no jaw tumor (X-ray), negative ultrasonographic screening for parathyroid and renal tumors.
Figure 6.
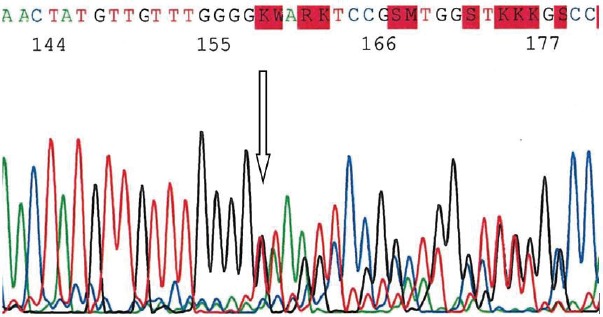
Electrophoregram of CDC73 gene exon 1 PCR amplicon shows germinal heterozygous c.128-IVs1+1 delG frameshift mutation.
Table 1.
Clinical characteristics and CDC73 mutational analysis results in the family
| Patients/ age at diagnosis | Parathyroid tumor | Associated diseases | Mutations | Polymorphisms |
|---|---|---|---|---|
| Index case/35 | Carcinoma | Jaw ossifying fibroma; uterine fibroid | Heterozygous deletion ex1 c.128-IVS1 +1 delG | Heterozygous IVS12 -86 C/T (rs41302543) |
| Father/81 | - | - | Heterozygous deletion ex1 c.128-IVS1 +1 delG | - |
| Mother/73 | - | - | - | Heterozygous IVS2 +28 del TCTA (rs80356645); IVS12 -86 C/T (rs41302543) |
| Brother/46 | Carcinoma | - | Heterozygous deletion ex1 c.128-IVS1 +1 delG | Heterozygous IVS2 +28 del TCTA (rs80356645) |
| Daughter/20 | - | - | - | Heterozygous IVS12 -86 C/T (rs41302543) |
| Nephew/24 | Double adenoma | - | Heterozygous deletion: ex1 c.128-IVS1 +1 delG. | Heterozygous IVS2 +28 C/T (rs4466634) |
Index case’s mother and daughter did not carry the mutation.
DISCUSSION
This is the first Romanian family diagnosed with HPT-JT and with a confirmed mutation of the CDC73 gene. To date approximately 200 patients in approximately 50 families carrying a germline mutation of this gene have been reported (13). Mutations in the CDC73 gene and alterations in the parafibromin protein have been established in 50%–75% of HPT-JT cases (1). The combination of findings of the proband (primary hyperparathyroidism, OF, uterine fibroid) was suggestive of HPT-JT and therefore we decided to perform CDC73 mutational analysis in this family. Four out of six patients showed a heterozygous germline deletion of one nucleotide c.128-IVS1+1 delG, previously described in an atypical adenoma, which had an overall cytogenetic profile resembling the carcinomas (14). This mutation is predicted to prematurely truncate parafibromin due to frameshift alterations (1, 2). A review of the literature identified 68 germline CDC73 mutations reported from patients with HPT-JT (n=38), FIHP (n=14), sporadic parathyroid carcinomas (n=10), and adenomas (n=4) and sporadic ossifying fibroma of the jaw (n=2) (1). Mutations in exons 1 (like in our patients), 2, and 7 were overrepresented accounting for 34, 17, and 21%, respectively (1). The search of a cytogenetic profile which may carry a higher risk of carcinoma is needed. The three affected patients manifest a different clinical phenotype, even in the presence of the same mutation; and one mutated subject had not yet developed any HPT-JT clinical manifestation at the age of 81. Five out of our six patients showed specific polymorphisms: the patient with the recurrent carcinoma had the same polymorphism as his mother, who did not carry the mutation; another polymorphism was shared by the proband, his mother and her daughter, both non-carriers of the mutation; the proband’ nephew (two adenomas) carries a unique polymorphism, different from those with carcinomas. The proband’s father, who transmitted the mutation, is asymptomatic and has no specific polymorphism. Our data confirms the autosomal dominant transmission, the variable penetrance and the absence of a direct genotype-phenotype correlation of the syndrome. Non penetrance of the CDC73 mutations in HPT-JT kindreds has been reported in >30% of mutation carriers (10); 25% in our family. Ossifying fibromas have been reported to occur in 25–50% of HPT-JT patients (5, 7), and are histologically distinct from the osteoclastic “brown” tumors of primary hyperparathyroidism and do not regress following parathyroid surgery (1). Only the proband had an OF in this family and no one had renal tumors.
PHPT in the context of HPT-JT has been previously characterized as a more aggressive disease relative to sporadic PHPT, with frequent multiglandular involvement, increased risk of persistent/recurrent disease, and a higher frequency of parathyroid carcinoma and metastasis (5); parathyroid carcinoma is found in 15 to 37.5% in different case series (5-9). Our cases confirm the high risk of parathyroid carcinoma in HPT-JT, as two out of three family members with HPT had carcinomas. The patient with persistent disease had a late diagnosis because his tumor has been misdiagnosed as a thyroid tumor. Misdiagnosis and confusion between parathyroid carcinoma and Hürthle cell thyroid lesion have been reported (15). As in our patient, immunohistochemical staining of parathyroid carcinoma is typically positive for PTH, and negative for calcitonin, thyroglobulin and parafibromin (15). Indeed, in our cases, parafibromin immunohistochemistry was negative in carcinomas and weakly positive in both adenomas, confirming other observations (5, 6). Quite recently, distinctive morphologic features in parafibromin deficient (CDC73 mutated) parathyroid tumors have been described (16). One patient had two adenomas and there is a wide variation reported in the frequency of synchronous multiglandular involvement (5, 6, 17). This is different from patients with MEN1, where all or most of the glands are involved simultaneously (4, 18).The high risk of carcinoma and a high rate of multiglandular involvement increase the risk of surgical failure (persistence or recurrence) in limited neck interventions. This is why some experts recommend en bloc resection of parathyroid tumors with bilateral neck exploration (5). Parathyroid carcinoma is not sensitive to radiation therapy or chemotherapy also suggested by our case; moreover radiation therapy increased the difficulty to reoperate through skin changes and tracheal deformation. Indeed an attempted surgery was ineffective in removing residual tumor in our male patient with parathyroid carcinoma.
The benefits of the genetic screening and identification of a CDC73 gene germline mutation in this family are multiple: the young patient with two adenoma is currently enrolled in our follow-up regime, as his father with parathyroid carcinoma; the proband’s daughter who is not a carrier of the mutation will not be enrolled; parathyroid carcinomas are rare, but the identification of germline CDC73 mutations in patients indicates that the risks for this disorder to the relatives are similar to those for an autosomal dominant disease, as outlined for HPT-JT; the siblings of the proband and their children who were not available for this screening have been invited to do so.
Our study has some limitations: some family members are living abroad and were not included in the genetic screening. In the near future we intend to define the function of the identified mutation.
In conclusion, we identified a new CDC73 germline mutation in a Romanian family of HPT-JT. Analysis of clinical phenotypes in the four mutated individuals confirmed the incomplete penetrance and the variable clinical expression of the disease, suggesting a possible role for other, still unknown, modifying factors in the determination of individual disease development and presentation.
Conflict of interest
No potential conflicts of interest were disclosed.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Acknowledgment
This work was supported by FirmoLab and F.I.R.M.O. Foundation.
References
- 1.Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat. 2010;31(3):295–307. doi: 10.1002/humu.21188. [DOI] [PubMed] [Google Scholar]
- 2.Cardoso L, Stevenson M, Thakker RV. Molecular genetics of syndromic and non-syndromic forms of parathyroid carcinoma. Hum Mutat. 2017;38(12):1621–1648. doi: 10.1002/humu.23337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32(4):676–680. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Simonds WF. Endocrine neoplasms in familial syndromes of hyperparathyroidism. Endocrine-Related Cancer. 2016;23(6):R229–R247. doi: 10.1530/ERC-16-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta A, Patel D, Rosenberg A, Boufraqech M, Ellis RJ, Nilubol N, Quezado MM, Marx SJ, Simonds WF, Kebebew E. Hyperparathyroidism-jaw tumor syndrome: results of operative management. Surgery. 2014;156(6):1315–1324. doi: 10.1016/j.surg.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iacobone M, Masi G, Barzon L, Porzionato A, Macchi V, Ciarleglio FA, Palù G, De Caro R, Viel G, Favia G. Hyperparathyroidism-jaw tumor syndrome: a report of three large kindred. Langenbecks Arch Surg. 2009;394(5):817–825. doi: 10.1007/s00423-009-0511-y. [DOI] [PubMed] [Google Scholar]
- 7.Bradley KJ, Thakker RV. The Hyperparathyroidism-jaw tumour (HPT-JT) syndrome. Clin Cases Miner Bone Res. 2006;3(1):167–174. [Google Scholar]
- 8.DeLellis RA, Mangray S. Heritable forms of primary hyperparathyroidism: a current perspective. Histopathology. 2018;72(1):117–132. doi: 10.1111/his.13306. [DOI] [PubMed] [Google Scholar]
- 9.van der Tuin K, Tops CMJ, Adank MA, Cobben JM, Hamdy NAT, Jongmans MC, Menko FH, van Nesselrooij BPM, Netea-Maier RT, Oosterwijk JC, Valk GD, Wolffenbuttel BHR, Hes FJ, Morreau H. CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2017;102(12):4534–4540. doi: 10.1210/jc.2017-01249. [DOI] [PubMed] [Google Scholar]
- 10.Bradley KJ, Hobbs MR, Buley ID, Carpten JD, Cavaco BM, Fares JE, Laidler P, Manek S, Robbins CM, Salti IS, Thompson NW, Jackson CE, Thakker RV. Uterine tumors are a phenotypic manifestation of the hyperparathyroidism-jaw tumor syndrome. Intern Med. 2005;257(1):18–26. doi: 10.1111/j.1365-2796.2004.01421.x. [DOI] [PubMed] [Google Scholar]
- 11.Grigorie D, Sucaliuc A. A single-dose, open-label, prospective clinical study of denosumab in patients with primary hyperparathyroidism. Acta Endocrinologica (Buc) 2014;10(3):1–8. [Google Scholar]
- 12.Hu MI, Glezerman IG, Leboulleux S, Insogna K, Gucalp R, Misiorowski W, Yu B, Zorsky P, Tosi D, Bessudo A, Jaccard A, Tonini G, Ying W, Braun A, Jain RK. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab. 2014;99(9):3144–3152. doi: 10.1210/jc.2014-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pichardo-Lowden AR, Manni A, Saunders BD, Baker MJ. Familial hyperparathyroidism due to a germline mutation of the CDC73 gene: implications for management and age-appropriate testing of relatives at risk. Endocr Pract. 2011;17(4):602–609. doi: 10.4158/EP10337.RA. [DOI] [PubMed] [Google Scholar]
- 14.Sulaiman L, Haglund F, Hashemi J, Obara T, Nordenström J, Larsson C, Juhlin CC. Genome-Wide and Locus Specific Alterations in CDC73/HRPT2-Mutated Parathyroid Tumors. PLOS ONE. 2012;7(9) doi: 10.1371/journal.pone.0046325. e46325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sriphrapradang C, Sornmayura P, Chanplakorn N, Trachoo O, Sae-Chew P, Aroonroch R. Fine-needle aspiration cytology of parathyroid carcinoma mimic hurthle cell thyroid neoplasm. Case Rep Endocrinol. 2014;2014:680876. doi: 10.1155/2014/680876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill AJ, Lim G, Cheung VKY, Andrici J, Perry-Keene JL, Paik J, Sioson L, Clarkson A, Sheen A, Luxford C, Elston MS, Meyer-Rochow GY, Nano MT, Kruijff S, Engelsman AF, Sywak M, Sidhu SB, Delbridge LW, Robinson BG, Marsh DJ, Toon CW, Chou A, Clifton-Bligh RJ. Parafibromin-deficient (HPT-JT Type, CDC73 Mutated) Parathyroid Tumors Demonstrate Distinctive Morphologic Features. Am J Surg Pathol. 2019;43(1):35–46. doi: 10.1097/PAS.0000000000001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masi G, Barzon L, Iacobone M, Viel G, Porzionato A, Macchi V, De Caro R, Favia G, Palù G. Clinical, genetic, and histopathologic investigation of CDC73-related familial hyperparathyroidism. Endocr Relat Cancer. 2008;15(4):1115–1126. doi: 10.1677/ERC-08-0066. [DOI] [PubMed] [Google Scholar]
- 18.Mele M, Rolighed L, Jespersen ML, Rejnmark L, Christiansen P. Recurrence of Hyperparathyroid Hypercalcemia in a Patient With the HRPT-2 Mutation and a Previous Parathyroid Carcinoma in Hyperparathyroidism-Jaw Tumor Syndrome. Int J Endocrinol Metab. 2016;14(2):e35424. doi: 10.5812/ijem.35424. [DOI] [PMC free article] [PubMed] [Google Scholar]