

Abstract
Penicillin-binding protein 5 (PBP 5) of Escherichia coli hydrolyzes the terminal D-Ala-D-Ala peptide bond of the stem peptides of the cell wall peptidoglycan. The mechanism of PBP 5 catalysis of amide bond hydrolysis is initial acylation of an active site serine by the peptide substrate, followed by hydrolytic deacylation of this acyl-enzyme intermediate to complete the turnover. The microscopic events of both the acylation and deacylation half-reactions have not been studied. This absence is addressed here by the use of explicit-solvent molecular dynamics simulations and ONIOM quantum mechanics/molecular mechanics (QM/MM) calculations. The potential-energy surface for the acylation reaction, based on MP2/ 6–31+G(d) calculations, reveals that Lys47 acts as the general base for proton abstraction from Ser44 in the serine acylation step. A discrete potential-energy minimum for the tetrahedral species is not found. The absence of such a minimum implies a conformational change in the transition state, concomitant with serine addition to the amide carbonyl, so as to enable the nitrogen atom of the scissile bond to accept the proton that is necessary for progression to the acyl-enzyme intermediate. Molecular dynamics simulations indicate that transiently protonated Lys47 is the proton donor in tetrahedral intermediate collapse to the acyl-enzyme species. Two pathways for this proton transfer are observed. One is the direct migration of a proton from Lys47. The second pathway is proton transfer via an intermediary water molecule. Although the energy barriers for the two pathways are similar, more conformers sample the latter pathway. The same water molecule that mediates the Lys47 proton transfer to the nitrogen of the departing D-Ala is well positioned, with respect to the Lys47 amine, to act as the hydrolytic water in the deacylation step. Deacylation occurs with the formation of a tetrahedral intermediate over a 24 kcal • mol−1 barrier. This barrier is approximately 2 kcal • mol−1 greater than the barrier (22 kcal • mol−1) for the formation of the tetrahedral species in acylation. The potential-energy surface for the collapse of the deacylation tetrahedral species gives a 24 kcal • mol−1 higher energy species for the product, signifying that the complex would readily reorganize and pave the way for the expulsion of the product of the reaction from the active site and the regeneration of the catalyst. These computational data dovetail with the knowledge on the reaction from experimental approaches.
Penicillin-binding proteins (PBPs) catalyze the transglycosylase, transpeptidase, and carboxypeptidase activities that are necessary for bacterial cell wall growth and septation during cell division. The coordinated activity of three PBP classes is used for peptidoglycan biosynthesis.1,2 The first class is the high-molecular-mass PBPs possessing both transglycosylase and transpeptidase activities. These PBPs are essential for cell viability. The second class is the high-molecular-mass PBPs involved in cell division.3 The third class is the low-molecular-mass PBP enzymes. While less critical for cell viability, the function(s) of this third class of PBPs is not fully understood. It is presumed that these enzymes moderate the degree of the cross-linking of cell wall, as evidenced by their contribution to the control of the shape of the bacterium.4 Escherichi coli has seven PBPs in this third class, and PBP 5 is the most abundant of these seven.5 PBP 5 is anchored into the outer leaflet of the cytoplasmic membrane by its C-terminus.6,7 The structure of PBP 5, as determined by X-ray crystallography of a soluble PBP 5 construct lacking the terminal anchor, shows two domains.8–10 The first domain is a β-sheet “stalk” that is believed to act to elevate the second, and catalytic, globular domain into contact with the cell wall peptidoglycan.11,12 The elegant studies of Spratt,13 de Pedro and Holtje,14 and Young and colleagues15 confirm a role for PBP 5 in the control of the E. coli rod shape (including the diameter, contour, and morphology of the rod), as well as a role for PBP 5 in cell wall septation during bacterial division.16
As is true for all PBPs at present, a deeper insight into the specific reactions that are catalyzed by PBP 5 is not available. In particular, the structure of the complex of PBP 5 with cell wall substrate(s) is unknown. The presumption that these substrates possess the complete acyl-L-Ala-γ-D-Glu-mDap-D-Ala-D-Ala (that is, uncross-linked; mDap for meso-diaminopimelate) peptide stem of the Gram-negative cell wall muropeptide is strongly implied by the ability of the β-lactam antibiotics to acylate the active site serine of PBP 5, and by the ability of PBP 5 to catalyze hydrolysis of the acyl-D-Ala-D-Ala susbtrate, with release of the terminal D-Ala residue.17 Most curiously, penams and cephems elaborated with such peptide stems are not PBP 5 substrates,18,19 and the active site requirements for PBP 5 acylation by D-Ala-D-Ala peptides is different (in some undetermined respect) from the requirements for catalytic acylation by β-lactams.7,20 Endogenous carboxypeptidase activity for PBP 5 is proven by muropeptide analyses of bacteria lacking this enzyme.16,21 The PBP 5 carboxypeptidase reaction has been thoroughly studied in vitro. With the soluble PBP 5 construct, and simple acyl-D-Ala-D-Ala derivatives as substrates, the hydrolytic cleavage of the terminal D-Ala residue occurs with an optimal pH of approximately 9.2 and a kcat/KM value of approximately 80 M−1 s−1.22,23 The kcat/KM value improves with more representative synthetic cell wall samples that include the full peptide and portions of the saccharide backbone.24
The detailed catalytic mechanism for amide bond hydrolysis by this PBP–nor of the PBPs in general–is not known. While the identity of PBP 5 as a serine peptidase is proven by its β-lactam reactivity, and from the structure of enzyme-inhibitor complexes, the PBP 5 active site is distinctly different when compared to the Asp-His-Ser catalytic triad of the classic serine proteases. Serine activation in PBP 5 uses the uncommon Lys-Ser dyad. The structural simplicity of this Lys47 and Ser44 dyad in PBP 5, and the unusual pH optimum (pH optimum for kcat of 9–10)22,23 of PBP 5 (now proven to coincide with the involvement of a Lys47 free amine in catalysis by experimental determination),23 prompted this computational evaluation of the mechanism of the acylation and deacylation half-reactions of PBP 5 catalysis.
Computational Methods
The PBP 5 carboxypeptidase reaction involves acylation and deacylation as discrete bond-making and bond-breaking steps, including both covalent and hydrogen bonds. We use the ONIOM quantum mechanics and molecular mechanics (QM/MM) method to analyze the microscopic events and nanosecond molecular dynamics (MD) simulations to observe active site conformational motion. The Amber program25 carries out molecular dynamics simulations that result in conformations occurring within (i) the PBP 5 Michaelis complex of N-acetyl-D-Ala-D-Ala as substrate, within the tetrahedral species obtained upon Ser44 addition to the N-acetyl-D-Ala-D-Ala amide, (ii) the acyl-enzyme, and (iii) within the tetrahedral species resulting from water addition to the acyl-enzyme. For each event, the MD simulation is combined with two-layer ONIOM QM/MM calculations. MD identifies a productive conformation for initiation of the reaction, and the QM/MM method evaluates the bond-forming and bond-breaking events.
The selection of the substrate for this computational study was made on the basis of experimental study.22,23 The substrate kcat/KM values for the carboxypeptidase activity of soluble PBP 5 is nearly independent of the N-terminal substituent of the D-Ala-D-Ala dipeptide.22 Hence, we selected the small N-acetyl substituent to complement the D-Ala-D-Ala dipeptide. After superimposition of the crystal structure of soluble PBP 5 (1.85 Å structure, PDB ID: 1NZO), with a second PBP 5 crystal structure (1Z6F) containing a deacylation mimetic,10 coordinates of the anticipated model for the N-acetyl-D-Ala-D-Ala substrate were extracted using SYBYL 7.1.26
The 1NZO crystal structure of PBP 5 encompasses 352 residues that are indexed from 4 to 355. The primary catalytic residues of the active site SXXK motif are Ser44 and Lys47. Standard procedures to computationally preprocess the protein were used, including the removal of uncritical atoms (such as water) and stipulation of the ɛ-tautomer for the histidine residues. Hydrogen atoms were added to the heavy atoms using the protonation program within the Amber (version 8) molecular dynamics package.
Substrate Docking into the PBP 5 Active Site.
The noncovalent enzyme–substrate complex was constructed with DOCK 5.27 Gasteiger–Hückel charges were applied to the substrate in preparation for docking. The terminal carboxylate was selected as the anchor set, and the scissile carbonyl (ensconced within the oxyanion hole) was selected as the rigid set. Following substrate preparation, Ser44 and Lys47 within the PBP5 binding site were energetically minimized using a 1000-step energy minimization and Powell algorithm, using the TRIPOS force field in SYBYL 7.1. Following energy minimization, the solvent-accessible molecular surface was computed to locate the spheres within the binding pocket. Subsequently, the substrate was docked onto the binding pocket. The receptor–substrate complexes were scored with an energy function that evaluates both van der Waals and Coulombic interactions. A 1000-step rigid-body simplex minimization was applied to the final receptor-substrate prior to more extensive explicit-solvent energy minimization and molecular dynamics simulation with the Amber package.
MD Simulations.
The complex of N-acetyl-D-Ala-D-Ala and PBP 5 was assigned a neutral charge using the Amber AddIons program. The Amber solvation program encompassed the active site with a rectangular parallelepiped solvent box. The dimensions of the box (approximately 94 Å × 85 Å × 159 Å) ensured that no atom within the protein–substrate active site complex is less than 10 Å from any face of the box. The Amber xleap program generated the topology file. A RESP two-step procedure was adopted for the force field parameters of the substrate.28 The resulting topology document integrates the charges, force fields, and connectivity information that are necessary for MD simulation. The topological data was incorporated into a previous MD protocol for the equilibration of the explicitly solvent structure.23,29 Under the restraints of the harmonic potentials of the atoms, the structure was energetically minimized in Cartesian space over 5000 steps, using the steepest decent method. A 4-ps MD simulation at a constant pressure of 1 atm was run by slowly increasing the temperature from 0 to 300 K. Subsequently, six steepest descent energy minimizations (each consisting of 1000 steps) were carried out by gradual relaxation of the restraints on the complex. The fully relaxed system was subjected to three MD simulations. Each simulation involved 12 ps heating from 0 to 100 K and 4 ps equilibration at a constant temperature of 100 K. Similar procedures were applied to two consecutive heating stages, from 100 to 200 K, and then 200 to 300 K. Lastly, 200 ps of equilibration MD simulation was carried out at 300 K.
QM/MM.
The system was partitioned into two layers, low and high. The atoms in the low layer are treated with molecular mechanics while the atoms in the high layer are treated with molecular orbital calculations. The low layer consists of 9,381 atoms, including the 1,000 water molecules closest to the bound ligand. Interactions among the atoms in the low region were treated with the molecular mechanics force fields in the Amber program. In determining atomic harmonic potentials and other force field parameters for the substrate, the parm99 and Gaff packages also were applied. The number of heavy atoms used in the QM layer for tetrahedral formation from the noncovalent enzyme–substrate complex was 34; for the collapse of this tetrahedral species, 38; for formation of the second tetrahedral species, 31; and for its collapse, 31. Potential-energy points in the QM/MM calculations were generated over a two-dimensional grid of two direct coordinates in each reaction step. The energy contour was generated using the program MATLAB.30
Results and Discussion
The spatial formulation of the complex between enzyme and substrate that is productive for catalysis is never a trivial undertaking. While this task is somewhat simplified for the serine proteases–the serine must position at an appropriate angle and distance from the amide carbonyl of the substrate, which is itself nestled within an oxyanion hole–the magnitude of the difficulty of assigning the correct Michaelis complex structure cannot be underestimated. In addressing this task for PBP 5, we recognized that a critical decision was the geometry of the scissile amide bond. There is now strong likelihood that several serine proteases recognize the cis-conformation of the scissile amide, and not the more populated trans-conformation seen in solution, for catalysis.31 As the acylation of the active site serine of PBP 5 by β-lactams occurs with a kcat/KM that is significantly greater than the corresponding value for acyl-D-Ala-D-Ala hydrolytic turnover, and as β-lactams possess a cis-amide (enforced by the β-lactam ring), the starting points for the computational evaluation of the PBP 5 carboxypeptidase reaction were evaluated for the competence of both the cis-and trans-conformations of N-acetyl-D-Ala-D-Ala in complex with PBP 5.
Preliminary evaluation indicated that both conformations were accommodated within the active site. Both showed similar positioning of the serine with respect to the scissile amide carbonyl, and similar positioning of the carbonyl in the oxyanion hole. We determined the potential-energy surfaces for the formation of the tetrahedral species for both the cis- and the trans-amide conformations of the scissile bond of the substrate. The energy barrier for tetrahedral formation for the cis-conformation was substantially higher in energy (by 10 kcal mole−1 see Figure 2S of the Supporting Information) than for the trans-conformation. This result is consistent with experimental observation that the catalytic residues used for serine acylation by D-Ala-D-Ala substrates are different than for β-lactams.20 Due to the large difference in energy for tetrahedral formation between the two conformations, further study of the acylation reaction used only the lower energy trans-amide substrate–PBP complex.
Conformation of the Trans-Amide Michaelis Complex.
Initial equilibration refined the N-acetyl-D-Ala-D-Ala trans-amide substrate contacts within the Lys47 and Ser44 dyad. Subsequently, the N-acetyl-D-Ala-D-Ala • PBP 5 complex was subjected to 50000 steps of energy minimization, followed by 1 ns of MD simulation. Two structures were extracted at 0.25 and 0.5 ns. Each structure served as a starting point for 3 ns of additional MD simulations. A total of 15000 snapshots were generated for conformational analyses. We note that in the crystal structure of the native enzyme, the Lys47 side chain amine and the hydroxyl of Ser44 form a hydrogen bond.9 This interaction was largely stable in the course of dynamics simulations at a typical distance of 2.7–2.8 Å. The geometry of this conformation was energetically optimized by the ONIOM two-layer method.32 Figure 1 shows the substrate of the Michaelis complex in the long and narrow groove between the β9 sheet (residues 205 to 217), and the loop between the α4 (residues 99 to 109) and α5 (residues 103 to 124).8 The oxyanion hole comprises hydrogen-bonding interactions between the amide carbonyl oxygen and the backbone amides of Ser44 (3.0 Å) and His216 (2.9 Å). Additional hydrogen bonds occur among the C-terminal substrate carboxylate and Arg198 Nζ (2.8 Å) and Ser110 Oγ (2.7 Å), and between the N-acetyl carbonyl oxygen and Thr217 Oγ (2.7 Å). The NH of the scissile amide is partially exposed to solvent through a hydrogen bond to water (3.0 Å). These distances are depicted in Figure 1A.
Figure 1.

(A) Stereo representation of the substrate acetyl-D-Ala-D-Ala bound as the trans-conformation to the active site of PBP 5 in the Michaelis complex. Hydrogen bonds are shown as dashed lines (distances in Å between heteroatoms). The substrate is nestled between β9 sheet and the loop connecting the α4 and α5 helices, with the protein depicted as the ribbon representation in green. Important active site residues and the substrate are represented in capped-stick. Carbon atoms are colored in gray, nitrogen in blue, and oxygen in red (hydrogens are not shown). Distances (Å) are rounded to the nearest tenth. (B) QM/MM potential-energy surface and the contour over reaction coordinates represented as a shadow. The reaction path is shown by red arrows from the Michaelis complex (I) to the tetrahedral species (II). (C) The structure of the tetrahedral species II is shown. The general description given for panel A applies here as well. Hydrogens are shown only for the Lys47 side chain ammonium group in cyan.
Tetrahedral Species from the Trans-Amide Conformation.
As formation of the tetrahedral species involves bond formation and cleavage, we used the aforementioned two-layer ONIOM ab initio molecular orbital calculations to study this reaction. This calculation treats the region surrounding the reaction center with quantum mechanics and the rest with molecular mechanics. The QM layer consists of atoms within 6 Å of the Ser44 Oγ.33 A total of 73 atoms (27 from the substrate) were included in the QM layer. The heavy atoms of the enzyme in the QM layer include the following: Lys47 (Cδ, Cɛ, and Nζ), Ser44 (Cα, Cβ, Oγ, N), Ala43 (carbonyl group), Gly215 (Cα, carbonyl group), His216 (backbone NH, Cα), Ser110 (Cα, Cβ, Oγ), and Lys213 (Cδ, Cɛ, and Nζ).
An HF/3–21G ONIOM QM/MM geometry optimization of the Michaelis complex was the starting point for subsequent QM/MM calculations for the formation of the tetrahedral species. Two reaction coordinates were defined (Figure 1B). The first reaction coordinate is the Ser44 side chain hydroxyl bond (d1). As d1 increases, a proton transfers to the Lys47 amine. The second reaction coordinate, the distance between the carbonyl carbon of the substrate and Ser44 Oγ (d2), describes the formation of the serine oxygen bond to the amide carbonyl. A two-dimensional potential-energy scan was conducted for both reaction coordinates at 0.1 Å intervals ranging from 1.0 to 1.8 Å for d1 and from 1.4 to 3.1 Å for d2. The potential-energy surface for tetrahedral formation is shown in Figure 1B. A total of 162 MP2/6–31+G(d) single point energy calculations define the surface. The locations of the Michaelis complex and the tetrahedral species are indicated on the surface and in the energy contour shown below the surface. The arrows indicate the progression of the reaction. The reference point for the reaction path is the energy minimum for the Michaelis complex (species I) at d1 = 1.0 Å and d2 = 2.5 Å. It is of note that the forward motion along the reaction path toward the tetrahedral species develops a nonstationary inflection point corresponding to species II. No discernible energy minimum is observed for II. Transition species II is located 22 kcal • mol−1 above the Michaelis complex.
During progress from the Michaelis complex to the tetrahedral species, the length of one of the hydrogen bonds in the oxyanion hole shortens by 0.2 Å (from 3.0 to 2.8 Å between the substrate carbonyl oxygen and the Ser44 backbone NH), consistent with the negative charge that builds on the formerly carbonyl oxygen atom.34 Although formation of the tetrahedral species results in a small rotational motion of the amine toward the Lys47 ammonium, the lone pair of nonbonding electrons of the amine is not yet predisposed for acceptance of a proton from the now-protonated Lys47. Hence, for Lys47 to act as the general acid for tetrahedral collapse, additional conformational change of the tetrahedral species is necessary to bring the nitrogen of the scissile bond into appropriate contact with the protonated Lys47.
Formation of the Acyl-Enzyme Intermediate from the Tetrahedral Species.
The necessary event for tetrahedral collapse is protonation of the substrate nitrogen of the tetrahedral species. The tetrahedral species derived from the trans-amide (Figure 1C) was examined over a 3 ns trajectory. Rotation of the amine, about the amide scissile bond, occurs concomitant with a smaller translational motion of the Lys47 ammonium during this MD simulation. The consequence of these two motions is alignment of the lone pair of electrons on the amine of the tetrahedral species with the Lys47 ammonium (parenthetically, see Figure 4A for a comparable alignment). The Lys47 motion during the course of the trajectory is driven by this alignment, and as well by improved electrostatic contact with the substrate carboxylate. We state parenthetically that in the tetrahedral species derived from the cis-amide, the protonated Lys47 is not proximal to the amine of the tetrahedral species. Rather, the methyl moiety of the C-terminus D-Ala hinders access to the scissile bond nitrogen by the Lys47 ammonium. It is therefore difficult to envision how proton transfer from Lys47 to the amine of the cis-amide-derived tetrahedral species could take place. This consideration, evaluated in concert with the higher energy barrier for formation of the tetrahedral species from the cis-amide (Figure 1S of the Supporting Information), implicates the transderived species as the productive Michaelis complex for tetrahedral formation and for collapse to the acyl-enzyme. Figure 2S (of the Supporting Information) contrasts the structures of the cis-and trans-amide-derived Michaelis complexes and tetrahedral species.
Figure 4.

(A) Stereoview representation of the tetrahedral intermediate in the reaction to acyl-enzyme through the water-mediated Lys47 protonation. Hydrogen bonds are shown as dashed lines (distances in Å between heteroatoms). The tetrahedral intermediate is nestled between β9 sheet and the loop connecting α4 and α5 helices, with the protein depicted as the ribbon representation in green. Important active site residues and the tetrahedral intermediate are represented in capped-stick. Carbon atoms are colored in gray, nitrogen in blue, oxygen in red, and hydrogen in cyan. Distances (Å) are rounded to the nearest tenth. (B) QM/MM potential-energy surface and the contour over reaction coordinates represented as a shadow. The reaction path is shown by red arrows from the tetrahedral intermediate (I) to the complex of D-Ala and acyl–enzyme (II). (C) The structure of the acyl-enzyme (III) has taken shape with progression of D-Ala away from that shown in structure II. The general description given for panel A applies here as well.
The increase in nitrogen basicity during tetrahedral formation enables the protonation step necessary for tetrahedral collapse to the acyl-enzyme species. The active site residues capable of proton donation to the tetrahedral species are Lys213 (which is present in the ammonium state in the Michaelis complex and in the native protein) and Lys47 (which is present as the free-base amine in both the Michaelis complex and the native enzyme, as per experimental data).23 Lys47 in its free-base form is the general base that promotes Ser44 for formation of the tetrahedral species. Hence, Lys47 is protonated at the stage of the tetrahedral species. The possibilities for proton transfer to the tetrahedral species include direct transfer from Lys47, direct transfer from Lys213, Ser110-mediated proton transfer (from either lysine), or active site water-mediated transfer (again, from either lysine). These possibilities were differentiated by analysis of the MD trajectory of the trans-amide derived tetrahedral species.
A 3 ns MD simulation using the geometry-optimized QM/MM structure of the tetrahedral species revealed movement of the Lys47 side chain to distance itself from the nitrogen of the tetrahedral species (Figure 2A). Similarly, Lys213 undergoes a small movement away from the tetrahedral species. Ser110 maintains its position, in contact with a water molecule that is hydrogen-bonded to the terminal carboxylate of the tetrahedral species. On the basis of this trajectory, participation of Lys213 and Ser110 in the protonation of the tetrahedral species is excluded. Accordingly, direct attention was given toward the specific role of Lys47 in this proton transfer. After adjustment of the Lys47 conformation during the first 1 ns, Lys47 is seen to interconvert (during the remaining 2 ns of the MD) between two primary conformations. These two conformations reflect intermittent formation (1.8 Å) and loss (3.4 Å) of a hydrogen bond between the Lys47 ammonium and the nitrogen of the tetrahedral species seen in Figure 2A (hydrogen bond lengths given in this figure are between the heteroatom and the hydrogen). Repetition of the MD simulation using a second trajectory (see Computational Methods) failed to show Lys47 motion into direct interaction with the nitrogen of the tetrahedral (Figure 2B). Instead, a water molecule moves into a straddling position between the two nitrogens (also see Figure 4A). As will be elaborated below, this water molecule subsequently serves the role of the hydrolytic water in the second half-reaction of this enzyme. The entry of the water molecule into the active site to interact with the basic Lys47 side chain is driven by the evolutionary process that created the hydrolytic activity. In addition, the trajectory shows a second water molecule in hydrogen-bonding interaction with the straddling water molecule. This pair of water molecules creates a water channel interconnecting Lys47 and bulk solvent. This water channel ends with the hydrolytic water (the “straddling” water molecule), which in turn is poised for facilitation of both the collapse of the first tetrahedral species, as well as participation in the ensuing hydrolysis of the acyl-enzyme. Close inspection of the trajectory data reveals that the triggering event leading (in the first 1 ns) to the formation of a water channel, between the hydrolytic water and solvent, is separation of the two nitrogen atoms. The hydrolytic water molecule makes two hydrogen bonds in its final position. The first involves donation of a hydrogen bond by water to the scissile bond nitrogen. The second involves the water accepting a hydrogen bond from the ammonium group of Lys47 (Figure 2B). The trajectory reveals that the first hydrogen bond (d5 in Figure 2B) forms at 800 ps of the simulation and is stable (at 1.7 to 2.0 Å distances between heteroatom and hydrogen) throughout the remaining 2 ns of the dynamics. The second hydrogen bond (d6) is less stable. It breaks periodically over the entire course of the trajectory, with two of the Lys47 η-hydrogens bonding to the water oxygen alternately with a range of bond distances of 2.2–2.6 Å (distances between heteroatom and hydrogen). Nonetheless, the preponderance of the MD snapshots shows the existence of this hydrogen bond between Lys47 and the water molecule. These observations imply that two paths are possible for the protonation of the tetrahedral species. The first is the direct migration of the proton from Lys47, while the second path uses the intermediary water molecule as a proton relay. Both paths were further examined by ONIOM QM/MM calculations.
Figure 2.

Distances collected from MD simulation supporting (A) the direct proton transfer (d3) from Lys47 and (B) for the water-mediated proton transfer route with dual transfers (d5 and d6), both over 3000 ps. Distances (Å) are the separation between heteroatom and hydrogen.
Evaluation of the direct proton transfer used the energy-minimized conformer collected from the MD simulations. In this conformation, direct hydrogen-bond contacts exist for each of the three ammonium hydrogens of Lys47. One of the hydrogens points to the substrate amide nitrogen, the second points to the Ser110 backbone carbonyl oxygen, and the third points to N-acetyl-carbonyl oxygen. These interactions ensconce the ammonium group in appropriate geometry for the transfer of the proton, with the tetrahedral nitrogen poised as the acceptor of Lys47 proton. Starting with this energy-minimized conformer, a potential-energy scan was conducted using the reaction coordinates defined by d3 and d4 (Figure 3). A total of 165 MP2/6–31+G(d) calculations were carried out using systematic variation of the distance between the carbon and nitrogen atoms of the scissile bond (d4, examined in 0.1 Å increments over the 1.2–2.6 Å interval) with respect to the distance between the Lys47 η-proton and the scissile bond nitrogen (d3, also in 0.1 Å increments over the 0.9–1.9 Å interval). Three minima (I, II and III) were found. Minimum I (d3 = 1.8 Å, d4 = 1.5 Å) corresponds to a well-defined tetrahedral species. The tetrahedral species converts to an intermediate (minimum II at d3 = 1.1 Å, d4 = 1.7 Å) over a reaction barrier of 10 kcal • mol−1. The outstanding feature of this transformation is that proton transfer (d3 = 1.1 Å) occurs with lengthening, rather than outright cleavage, of the C–N bond (d4). Hence (and in contrast to tetrahedral formation), proton transfer here proceeds asynchronously with respect to nucleofuge departure. Mimimum II, a zwitterionic tetrahedral species, transforms to species III with a barrier of 5 kcal • mol−1 As is evidenced by the contour of the surface in Figure 3B, the existence of the minimum III (d3 = 1.1 Å, d4 = 2.6 Å; Figure 3C) indicates that the complex of the acyl-enzyme species, in tight association of the released terminal D-Ala, is a discrete intermediate in the reaction profile. Species III is 13.4 kcal • mol−1 greater in energy than the tetrahedral species I and is 4.2 kcal • mol−1 higher in energy than species II (Figure 3).
Figure 3.

(A) Stereo representation of the tetrahedral intermediate in the reaction to acyl-enzyme through direct Lys47 protonation. Hydrogen bonds are shown as dashed lines (distances in Å between heteroatoms). The tetrahedral species is nestled between β9 sheet and the loop connecting α4 and α5 helices, with the protein depicted as the ribbon representation in green. Important active site residues and the tetrahedral intermediate are represented in capped-stick. Carbon atoms are colored in gray, nitrogen in blue, oxygen in red, and hydrogen in cyan. Distances (Å) are rounded to the nearest tenth. (B) QM/MM potential-energy surface and the contour over reaction coordinates represented as a shadow. The reaction paths are shown by red and green arrows from the tetrahedral intermediate (I) to an intermediate complex (II), and to the acyl-enzyme (III). The energy surface constructed using the complex coordinates (d3–dNH) vs d4 is qualitatively identical. The distance dNH is the distance between the lysine NH atoms. (C) The structure of the acyl-enzyme III is shown. The general description given for panel A applies here as well.
As stated earlier, MD simulations show the insertion of a water molecule between protonated Lys47 and the tetrahedral species. The possibility that this water molecule could serve as the conduit for the transfer of proton from Lys47 to the tetrahedral species was also evaluated. Starting with the structure of the tetrahedral species collected from MD, a QM/MM potential-energy surface of the water-mediated transition to the zwitterionic tetrahedral species was constructed. The reaction coordinates for this surface consisted of (i) the distance between the water hydrogen and the tetrahedral nitrogen (d5), which was set equal to d6 and (ii) the distance between Lys47 Hη and the water oxygen (Figure 4). A constraint on d5, setting it equal to d6, was used as QM/MM calculations (with coordinate d4 set at 1.6 Å) showed that the optimal energy path for the dual proton transfer, developed over d5 and d6, was nearly symmetric (Figure 3S of the Supporting Information). Therefore, the reaction coordinate is well defined by d5 alone. The second reaction coordinate (d4) is the distance between the tetrahedral carbon and the nitrogen atom of the scissile bond. A total of 165 calculations at the MP2/6–31+G(d) level of theory constructed the potential-energy surface. The resulting surface (Figure 4B) shows two energy minima. The first minimum at d4 = 1.5 Å and d5 = d6 = 1.8 Å is the anionic tetrahedral intermediate (I). The second minimum (d4 = 1.7 Å, d5 = d6 = 1.0 Å) is best described as the zwitterionic tetrahedral species, obtained via synchronous proton transfer to the tetrahedral nitrogen concurrent with acceptance by the water molecule of a proton from Lys47. The barrier for formation of the zwitterionic tetrahedral species from the anionic tetrahedral intermediate is 13 kcal • mol−1. Both the energy barrier for zwitterion formation and the energy level for the zwitterion are essentially identical for the two paths.
The potential-energy surfaces shown in Figure 3 (direct proton transfer from Lys47) and Figure 4 (water-mediated proton transfer from Lys47) show comparable barriers (10.4 compared to 13 kcal • mol−1) for the same transformation. Hence, both pathways have similar likelihoods. The difference in energy between reactant and product in each case was also similar (13.0 compared to 13.4 kcal • mol−1). The close similarity of the two sets of values indicates the two reactions to be thermodynamically comparable. However, it can be argued that the water-mediated pathway is dominant, on the grounds that during the MD simulations the hydrogen-bonding interactions mediated by water were more stable than the hydrogen-bonding interactions between Lys47 and the tetrahedral nitrogen. An additional argument in support of the water-mediated process as the more viable path is the possibility that the same water molecule that relays a proton from Lys47 to the tetrahedral species remains to participate as the nucleophilic water for acyl-enzyme hydrolysis.
As the bond defined by d4 in Figures 3 and 4 lengthens, it leads to the expulsion of the leaving group, the terminal D-Ala, from the active site. With the departure of D-Ala, the acyl-enzyme species is formed. It is this species that undergoes the final hydrolytic reaction with the water molecule coordinated to Lys47. The approach of the hydrolytic water is likely along the same trajectory that D-Ala uses as it moves away from the acyl-enzyme species. These issues were evaluated by further explicit solvent MD simulations and QM/MM calculations.
The second half-reaction is hydrolysis of the ester bond of the acyl-enzyme species. This reaction requires activation of a water molecule by an active site base. The acyl-enzyme resulting from the direct Lys47 protonation of the tetrahedral species is different from the acyl-enzyme resulting from the water-mediated Lys47 protonation event. Following tetrahedral collapse by the former pathway, no water molecule is found proximal to the carbonyl of the acyl-enzyme. MD simulations reveal the Lys47 side chain to move back, allowing entry of a water molecule to occupy a position suited for the second half-reaction. The water from the water-mediated tetrahedral collapse is already well positioned for acyl-enzyme hydrolysis. The separation of the oxygen of the hydrolytic water from the ester carbonyl carbon is 3.4 Å (Figure 5A). The only residue that can promote the hydrolytic water for nucleophilic attack at the acyl-enzyme carbonyl is Lys47, which is now present in the free-base form. Lys47 Nζ and the water molecule are strongly hydrogen-bonded to each other (2.7 Å side chain nitrogen to water oxygen). Hence Lys47, having served as the general base to initiate the acylation reaction, now is poised to act as the general base to initiate the deacylation reaction.
Figure 5.

(A) Stereo representation of the acyl-enzyme for the water hydrolysis. Hydrogen bonds are shown as dashed lines (distances in Å between heteroatoms). The acyl-enzyme is nestled between the β9 sheet and the loop connecting α4 and α5 helices, with the protein depicted as the ribbon representation in green. Important active site residues and the acyl-enzyme portion are represented in capped-stick. Carbon atoms are colored in gray, nitrogen in blue, oxygen in red, and hydrogen in cyan. Distances (Å) are rounded to the nearest tenth. (B) QM/MM potential-energy surface and the contour over reaction coordinates represented as a shadow. The reaction path is shown by red arrows from the acyl-enzyme (I) to the second tetrahedral species (II). (C) The structure of the tetrahedral species II is shown. The general description given for panel A applies here as well.
The starting point for these calculations was the acyl-enzyme species that emerged from the water-mediated proton transfer approach (Figure 4C), after removal of the departing D-Ala. MD equilibration was applied as described in the Computational Methods. To maintain the acyl-enzyme geometry for the water addition reaction, the acyl-enzyme was restrained in the energy minimization stage and in the heating and the 200 ps MD equilibrium stages (the acyl-enzyme atoms were restrained using a harmonic potential force constant of 100 kcal • mol−1 • Å−2). The final snapshot from the trajectory shows a 3.4 Å separation of the oxygen of the water from the carbonyl carbon of the acyl-enzyme, and a 2.5 Å separation of the water oxygen from the Lys47 Nζ nitrogen. Moreover, the water oxygen is located at 41° from the normal of the plane formed by the ester carbonyl carbon, oxygen, and Ser44 Oγ (that is, at an attack angle of 131° for nucleophilic addition to the carbonyl). A geometry optimization of this snapshot was performed with QM/MM using HF/3–21G for the quantum layer. The resultant structure (Figure 5A) shows a shortened distance of 3.0 Å between the oxygen of the water from the carbonyl carbon, a slightly longer distance of 2.6 Å between the water oxygen and the Lys47 Nζ, and a 118° attack angle.
Starting with this structure, we set out to gain a deeper understanding of this water addition through the construction of a potential-energy surface based on QM/MM calculations. As such, we define the reaction coordinates as the distance between the carbonyl carbon and the water oxygen atom (d7) and the complex coordinate defined by (d8 – d9), where the distance between the Lys47 Nζ atom and the Hη atom is d8 and the distance between the water oxygen and water hydrogen is d9. The resulting potential-energy surface, based on 136 MP2/6–31+G(d) points, is shown in Figure 5B. The surface has two energy minima. Minimum I (located at d8 = 1.6, d9 = 1.0, and d7 = 3.0 Å) is the acyl-enzyme species poised for the addition reaction. Minimum II (located at d8 = 1.0, d9 = 2.7, and d7 = 1.6 Å) is the tetrahedral intermediate. The addition reaction involves concerted proton migration from water to Lys47 and the formation of the C–O bond between water and the ester carbonyl carbon, en route to the formation of the second tetrahedral species. The barrier for this event is 24 kcal • mol−1. A stepwise process is unlikely, as no potential-energy minima are found for the proton migration at a fixed CO distance or vice versa. It is worth noting, however, that the proton migration is significantly less costly than the bond-making step to the ester carbonyl. The product of this event is nearly equal in energy to the reactant.
Tetrahedral Collapse to the Enzyme–Product Complex.
The pathway from the second tetrahedral species to the product complex is straightforward. Starting with the geometry of the tetrahedral species from the QM/MM calculations, a 50000 step forcefield-based energy minimization was carried out, followed by 1000 steps of energy minimization without restraints. The resulting structure was used for QM/MM calculations to construct an energy surface. The reaction coordinates for the collapse of the tetrahedral species are depicted in Figure 6. The pertinent distances d10 (distance between the protonated Lys47 Hη and the bound Ser44 Oγ and d11 (distance between the same Oγ atom and the tetrahedral carbon) were incremented at 0.1 Å intervals (d10 from 0.9 to 2.2 Å, and d11 from 1.3 to 2.6 Å). A total of 156 MP2/6–31+G(d) points defined the potential-energy surface of Figure 6A. The surface shows one potential-energy minimum (I) and a nonstationary inflection point (II). Minimum I is the tetrahedral species (d10 = 1.9 and d11 = 1.5 Å). The inflection point is located at d10 = 1.1 and d11 = 2.2 Å, at a higher energy point on the surface, and corresponds to the enzyme-product complex. In light of the presence of only one energy minimum and one inflection point, this step is likely to proceed in a concerted manner, whereby proton migration from Lys47 to Ser44 Oγ occurs concomitantly with the lengthening of the Ser44 Oγ in the scissile bond. The product species lies at 24 kcal • mol−1 greater than the starting point on the potential-energy surface.
Figure 6.

(A) QM/MM potential-energy surface and the contour over reaction coordinates represented as a shadow. The reaction path is shown by red arrows from the second tetrahedral species (I) to the enzyme–product complex. (B) Stereo representation of the enzyme–product complex (II). Hydrogen bonds are shown as dashed lines (distances in Å between heteroatoms). The complex is nestled between β9 sheet and the loop connecting α4 and α5 helices, with the protein depicted as the ribbon representation in green. Important active site residues and the substrate are represented in capped-stick. Carbon atoms are colored in gray, nitrogen in blue, oxygen in red, and hydrogen in cyan. Distances (Å) are rounded to the nearest tenth.
The lack of stability of the product species is likely necessary to promote the expulsion of the product from the active site in order to regenerate the native enzyme for the next cycle of catalysis. The conventional view is that once the carboxylic acid of the product is formed, it loses a proton to the milieu in the buffered environment and then repulsion of the nonbonding electron pairs among heteroatoms of the Ser44 side chain and the carboxylate oxygens would repel the product from the active site. Tetrahedral collapse in PBP 5 would appear to be initiated by a critical change in the enzyme-product complex, which takes place at the Ser44 backbone amide nitrogen and the product carboxylic acid oxygen. This is one of two hydrogen bonds involved in the oxyanion hole. This hydrogen bond lengthens by 0.3 Å (from 2.8 to 3.1 Å), whereas the other hydrogen bond (2.9 Å) does not change. This weakening of the hydrogen bond to the oxyanion hole is accompanied by a looser fit of the product within the active site, which sets the stage for the subsequent exit of the product from the active site. Hence, the lengthening of the oxyanion hydrogen bond may be a contributing factor to the destabilizing effect that the complex experiences, ultimately leading to the dissociation of the enzyme-product complex.
Concluding Remarks
The carboxypeptidase reaction of PBP 5 is a seemingly mundane reaction, in large part because proteases/peptidases have been known for a long time. In reality, hydrolysis of the amide bond is an extraordinary enzyme-catalyzed transformation, due to the stability of the amide to hydrolysis in neutral water. The typical amide bond undergoes hydrolysis under neutral (uncatalyzed) conditions with a remarkably long t1/2 of some several centuries.35,36 The breadth of catalytic motifs used by nature to hydrolyze peptides commonly includes nucleophilic catalysis by serine, threonine, cysteine, aspartyl, and zinc- and iron-activated water. As detailed in this study, the PBPs are pre-eminent members of an emerging serine subclass37 that uses the Lys-Ser dyad for catalysis of amide bond hydrolysis, and that is mechanistically distinct from the classical serine proteases that use an Asp-His-Ser catalytic triad for catalysis.37 Additional examples of Lys-Ser dyad peptidase/amidase enzymes are the ATP-dependent lon proteases,39 fatty acid amide hydrolase,40 and the signal peptidases.41 As the Lys-Ser organization of the active sites of these other enzymes is dissimilar from that of the PBPs, the mechanism seen for PBP 5 in this study likely does not pertain to these other enzymes of this class.
This concern over active site dissimilarity does not, however, extend to the PBP class. Examination of PBP transpeptidase active site structure shows the preservation of three signature motifs. While there is variation in the organization of these domains within the PBP structure, there is remarkable consistency in their spatial relationship within the active site. The first motif (...SXXK...) contains the nucleophilic serine (Ser44 in PBP 5) and catalytic lysine (Lys47); the second motif (...SXN...) contains a companion serine (Ser110) and an asparagine (Asn112) that provides a critical hydrogen bond to the substrate; and the third (...KTG...) provides a companion lysine (Lys213) and a histidine (H216) that contributes (with the chain amide of Ser44) one of the two hydrogen bonds of the oxyanion hole. The complexity of the possible interplay among these motifs has led to considerable discussion concerning the role of each in PBP substrate recognition and catalysis. In the case of PBP 5, the active site must also contain structural components that define its catalytic role as one of hydrolysis, and not transpeptidation. The specific structure(s) that confer this specificity are uncertain. All PBPs share in common, however, serine acylation as the first half-reaction. The likelihood that the Lys47-Ser44 dyad of PBP 5 was the catalytic dyad for serine acylation was suggested by the PBP active site structure,4,10,42,43 and further supported by the experimental determination that the Lys47 pKa aligns directly with the pH dependency of PBP 5 catalysis.23 This QM/MM study supports this conclusion. Lys47 acts as a general base, directly promoting Ser44 addition to the D-Ala-D-Ala amide. The resulting anionic tetrahedral species is predisposed to collapse to the acyl-enzyme as a result of rotational motions that enable the transfer of the proton removed by Lys47 from Ser44, to the amine of the tetrahedral intermediate. This proton transfer gives a zwitterionic tetrahedral intermediate that undergoes the low energy expulsion of the D-Ala leaving group as it progresses to the acyl-enzyme intermediate. Moreover, a direct role of a water molecule within the active site in this latter proton transfer, so as to juxtapose the hydrolytic water between Lys47 and the acyl-enzyme following tetrahedral collapse, is highly probable. QM/MM indicates that these events do not directly involve other residues of the active site.
Two very recent crystallographic studies, using closely related PBPs, provide structures of PBP D-Ala-D-Ala-derived D-Ala acyl-enzymes.44,45 Comparison of the computational PBP 5 acyl-enzyme, with the experimental D-Ala acyl-enzymes observed for B. subtillis PBP 4a (PDB accession code: 2J9P) and for S. pneumoniae PBP 1b* (2JCI) is rewarding (Figure 7). Gratifyingly, remarkably close overlay is seen for both acyl-enzymes with the computational structure, and for density assigned as the departing D-Ala seen in the PBP 4a structure. With one outstanding exception, there is also close overlay of the side chains of the active site residues. The exception is the position of Lys213. The mechanistic role of this companion lysine in PBPs (and in the serine β-lactamases) is a long-standing challenge. While QM/MM has clarified the role of the KTG motif lysine during β-lactamase catalysis (it has the critical task of modulating the electrostatic environment of the catalytic lysine, but is otherwise a spectator),46 the belief that the KTG lysine directly contributes to the activation of PBP acyl-enzymes for the transpeptidation/hydrolysis reactions, has persisted.2 Lys213 has a normal pKa and is therefore cationic throughout PBP 5 turnover chemistry.23 Malhotra and Nicholas, preparing the complete ensemble of position 213 mutants by saturation mutagenesis,20 observed that only the K213R mutant retained the ability to undergo acylation by penicillin. Furthermore, the rate constants for hydrolyses of the PBP 5 and K213R penicillin-derived acyl-enzymes were identical. Hence, a requirement exists for a cationic residue at 213 to enable enzyme acylation by penicillin, and Lys213 participation as the general base for the deacylation of the β-lactam-derived acyl-enzyme is excluded (as the Arg213 mutant is fully active for deacylation, and as arginine will remain protonated throughout the catalytic cycle and thus unable to act as the general base for hydrolytic activation of the water). Lastly, the K213R mutant is inactive as a carboxypeptidase, indicating an important role for Lys213 in conferring the ability to recognize N-acyl-D-Ala-D-Ala as a substrate and/or in conferring competence to D-Ala-D-Ala as a substrate capable of Ser44 acylation.
Figure 7.
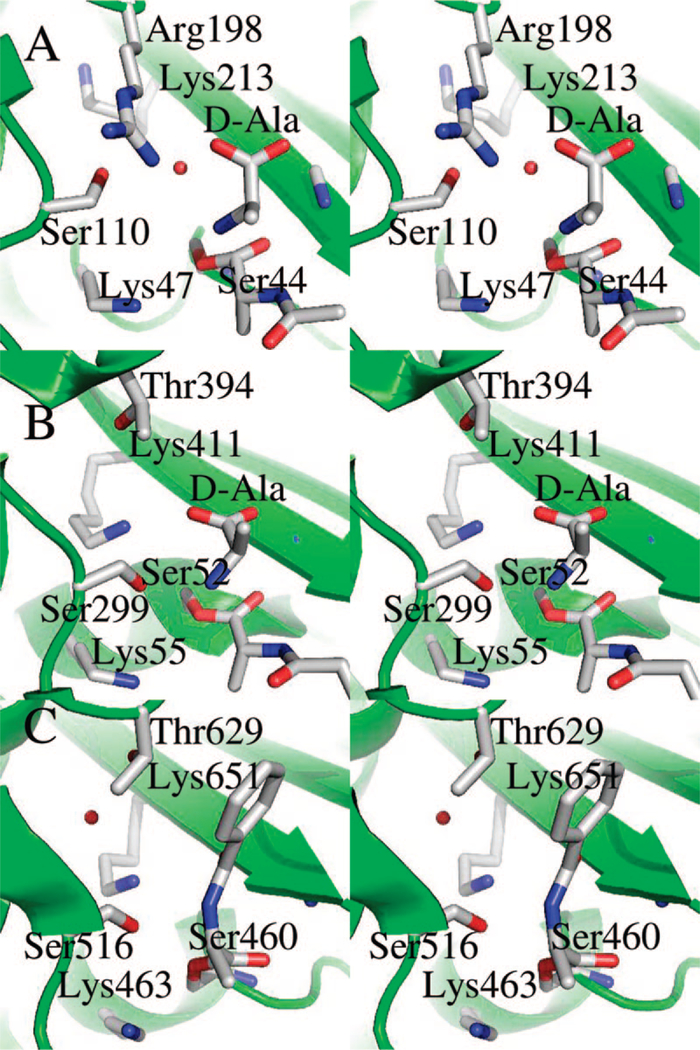
Stereo representation of Lys47, D-Ala-acylated Ser44, the departing D-Ala (A and B), Lys213, Ser110, and Arg198 in the optimized PBP 5 acyl-enzyme species (A, see also Figure 4) and the equivalent acyl-enzyme crystal structures of PBP 4a (B) and PBP 1b (C). The α helices, β sheets, and loops in the active site are colored in green. Residue side chains and acylated substrates are represented in capped-stick. Carbon atoms are colored in gray, oxygen in red, and nitrogen in blue. The water molecules involved in hydrogen bonds (to Ser110 and the carboxylate oxygen in A, and to Lys651 and Thr629 in C) are shown by small red spheres.
Our QM/MM study fails to identify a particular role for Lys213. In both of our PBP 5 Michaelis complex and acyl-enzyme computational structures, the Lys213 side chain is not in direct contact with the substrate. Rather, the role of ion pair engagement of the carboxylate of the terminal D-Ala of the substrate is undertaken by the guanidinium side chain of Arg198. The preference for arginine, rather than lysine, for the formation of the substrate ion pair is also seen for the cognate arginine of the R61 PBP,47 as discussed by Nicholas et al.9 Additional evidence points to this arginine as an important entity within the active site. Loss of Arg198 mobility has been suggested as a possible explanation for the loss of catalytic activity in the G105D PBP 5 mutant.9 Furthermore, recognizing that this arginine is not universally present in PBPs (in the transpeptidase PBPs, serine or threonine is found at this position), Sauvage et al. have presented the intriguing and insightful suggestion that the steric presence of Arg198 contributes to the inability of PBP 5 to catalyze transpeptidation.44 It is additionally possible that the use of the normally more hydrophilic arginine (as compared to lysine) here facilitates water entry into the active site, so as to enable the PBP 5 carboxypeptidase activity.
A return to experimental observation is essential to fully understand the active site circumstance accompanying the mechanistic transition from acylation to deacylation. As noted by Sauvage et al. in their successful isolation of a D-alanyl acyl-enzyme with B. subtilis PBP 4a, D-alanyl acyl-enzyme hydrolysis is likely the slower step (compared to acylation) in PBP DD-carboxypeptidase turnover.44 A detailed kinetic examination of E. coli PBP 5 hydrolysis of Ac2-L-Lys-D-Ala-D-Ala confirms this conclusion for this enzyme-substrate pair (unpublished data). If the hypothesis by Sauvage et al. that Arg198 of PBP 5 is an important steric barrier for the prevention of transpeptidation is correct, and given that the acyl-enzyme accumulates during steady-state turnover, then the expectation must be held that this arginine will maintain its place within the active site so as to protect the acyl-enzyme from transpeptidation. This is what we observe in the MD evaluation of our acyl-enzyme complex. After removal of the D-Ala product, a 2 ns MD indicates that both Arg198 and Lys213 hold their positions. Hence, Lys213 is left in a position where it cannot contribute directly to the deacylation mechanism, consistent with the observation that the K198R PBP 5 mutant successfully hydrolyzes the penicillin-derived acyl-enzyme. Nor is it at all evident from this MD simulation that Lys213 is in any way required for deacylation. Both computational pathways to the acyl-enzyme juxtapose a water molecule, in an optimal trajectory for carbonyl addition, between the free amine of Lys47 and the acyl-enzyme. General base activation of this water by Lys47 to accomplish tetrahedral formation is straightforward (Figure 5). Indeed, the puzzling aspect of deacylation is not at all Lys47 as a general base for tetrahedral formation but the energy surface describing the involvement of the Lys47 ammonium in tetrahedral collapse. The expectation that collapse of the tetrahedral species to the product complex, assisted by Lys47 as a general acid, would be exothermic (relative to the tetrahedral) is not realized. Rather, collapse is endothermic. This result suggests that the mechanistic basis for deacylation as the slower step compared to acylation (unusual for an amidase), as is validated by experiment, is an inability of the Lys47 ammonium to optimally engage as a general acid in this tetrahedral collapse.
There is an additional aspect with respect to these energy surfaces that merits comment. The calculated potential-energy barriers at each step are relatively large. There are three possible explanations that may bear on this circumstance. First, and quite simply, PBP 5 is not a robust catalyst, as assessed from the in vitro measurements. The small experimental kcat/KM for the substrate must bring the expectation that computational evaluation of the PBP 5 mechanism will show relatively high energy barriers. Second, the barriers do not reflect the entropic component of the free energy. Favorable entropy, particularly for tetrahedral collapse to the acyl-enzyme and D-Ala, is not captured by a potential-energy surface. Third, the in vitro pH optimum of PBP 5 of 9.5–10.5 does not coincide with the pH optimum for bacterial growth. Since the pH of the E. coli periplasm is in rapid and full equilibrium with the pH of the medium48 it is probable that optimal PBP 5 carboxypeptidase activity involves active-site positioning of residues that may be subtly different from that seen in the (near neutral pH derived) crystal structures. Lastly, the PBP enzymes as a class present the ability to transition from “opened” to “closed” structural states. This is seen experimentally in terms of cosubstrate induced alterations in their protein structure (as observed by the CD spectra of PBPs49 and crystallographically).2,50 We have used a superimposed (combined “apo” and “catalytic”) consensus PBP 5 structure for this computational study to maximize the likelihood that the active site geometry we use is relevant. Nonetheless, the aggregate effect of these factors is to introduce small but measured uncertainty as to the certitude of the computational structures used for the potential-energy surfaces. On this matter we can only emphasize, however, that our conclusion that Lys47 is the catalytic base in both acylation and deacylation is decisive, and is supported by experimental observation from our laboratory.23 No other alternative to Lys47 as the catalytic base for acylation and for deacylation is seen.
This realization refines the key question concerning mechanism. If Lys213 is not a participant in catalysis, what role does this lysine fulfill in this enzyme? The evolutionary conservation of this residue cannot be disputed. The positioning of this residue in the crystal structures of apo PBP 5 (as distinct from its positioning with respect to substrate in our computational structures) is identical to that in the β-lactamase active site, where it has an essential although indirect role in catalysis. The elegant studies of Davies and Nicholas have proven that the catalytic competence of the PBP 5 active site involves an extraordinarily subtle interplay of residue positioning and order.9–11,18 It is possible that a cationic side chain at 198 is necessary to the preservation of active-site integrity. The role of substrate carboxylate positioning and of steric prevention of transpeptidation44 in PBP 5 is fulfilled by Arg198. The role of the general base in both half-reactions of catalysis is fulfilled by Lys47. Lysine213 has no direct involvement beyond the potential for serving as an electrostatic anchor removed from the immediate sphere of the seat of reaction. Lysine213 might have a more direct role in the transpeptidase group of PBPs.
The PBP enzymes accomplish the remarkable biosynthetic assembly of the peptidoglycan polymer that envelops the growing bacterium, and the no less remarkable reassembly of the peptidoglycan during septation. The pivotal enzymatic intermediate at all stages of PBP catalysis is the acyl-enzyme derived from the stem peptide of the glycopeptide biosynthetic intermediate. Whether the acyl-enzyme is fated for transpeptidation, or is fated for hydrolysis, has been decided on the basis of the distinct evolutionary trajectories that have given rise to the various classes of PBPs. In this QM/MM study, the competence for lysine general base catalysis of serine for acylation, and of water for deacylation, is verified. The fate of the PBP 5 acyl-enzyme is decided in favor of hydrolysis, by the entry of a water molecule into contact with this same lysine during the early stages of catalysis. Affirmation of this fate may further involve the active site presence of an arginine residue (a residue not present in the transpeptidase PBPs) both as an electrostatic contact to position the D-Ala terminus of the stem in the active site, and to impede access of the amine of a second stem to the acyl-enzyme. Consistent with the need to preserve the acyl-enzyme for its fate–and consistent with experiment–comparison of the potential-energy surfaces for the acylation and deacylation half-reactions indicates deacylation as the rate limiting step. An outstanding characteristic of PBP 5 catalysis as seen in this study is the utter simplicity of its molecular mechanism: direct interaction with the substrate involves only the catalytic lysine, the catalytic serine, and a water molecule. While it may be conjectured that simplicity has its price–PBP 5 is not a robust catalyst as discerned from in vitro activity, and accordingly the energy surfaces for catalysis show relatively high activation barriers–it cannot be argued that simplicity is inadequate. PBP 5 fulfills its role as an arbiter of the E. coli shape.4 And while it must be expected that the events enabling transpeptidation, as accomplished by other PBP catalysts, are more complex than those seen for PBP 5 amide hydrolysis, it is certainly expected that the core mechanistic simplicity of PBP 5 catalysis (especially for acylation) is preserved by the transpeptidase PBPs.
Supplementary Material
Acknowledgment.
This work was supported by a grant from the National Institutes of Health. Q.S. is a Fellow of the Walther Cancer Research Center.
Footnotes
Supporting Information Available: Complete ref 25, the comparison of the Michaelis complex and tetrahedral intermediate for the cis-and trans-amide conformations, the QM/MM energy calculation for the cis-amide, and the examination of the symmetric assumption for the dual proton transfer in the water-mediated Lys47 protonation pathway are provided. This material is available free of charges via the Internet at http://www.acs.org.
References
- (1).Spratt BG; Cromie KD Rev. Infect. Dis. 1988, 10, 699–711. [DOI] [PubMed] [Google Scholar]; Macheboeuf P; Contreras-Martel C; Job V; Dideberg O; Dessen. A. FEMS Microbiol. Rev. 2006, 30, 673–691. [DOI] [PubMed] [Google Scholar]; Vollmer W; Joris B: Charlier P; Foster S. FEMS Microbiol. Rev. 2008, 32, 259–286. [DOI] [PubMed] [Google Scholar]
- (2).Sauvage E; Kerff F; Terrak M; Ayala JA; Charlier P. FEMS Microbiol. Rev. 2008, 32, 234–258. [DOI] [PubMed] [Google Scholar]
- (3).Norris V; den Blaauwen T; Cabin-Flaman A; Doi RH; Harshey R; Janniere L; Jimenez-Sanchez A; Jin DJ; Levin PA; Mileykovskaya E; Minsky A; Saier MJ; Skarstad K. Microbiol. Mol. Biol. Rev 2007, 71, 230–253. [DOI] [PMC free article] [PubMed] [Google Scholar]; den Blaauwen T; de Pedro MA; Nguyen-Disteche M; Ayala JA FEMS Microbiol. Rev 2008, 32, 321–344. [DOI] [PubMed] [Google Scholar]
- (4).Denome SA; Elf PK; Henderson TA; Nelson DE; Young KD J. Bacteriol 1999,181, 3981–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nelson DE; Young KD J. Bacteriol 2001, 183, 3055–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]; Popham DL; Young KD Curr. Opin. Microbiol 2003, 6, 594–599. [DOI] [PubMed] [Google Scholar]
- (5).Dougherty TJ; Kennedy K; Kessler RE; Pucci MJ J. Bacteriol 1996, 178, 6110–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pratt JM; Holland IB; Spratt BG Nature 1981, 293, 307–309. [DOI] [PubMed] [Google Scholar]; Phoenix DA; Pratt JM FEBS Lett. 1993, 322, 215–218. [DOI] [PubMed] [Google Scholar]; van der Linden MP; de Haan L; Keck W. Biochem. J 1993, 289, 593–598. [PMC free article] [PubMed] [Google Scholar]
- (7).van der Linden MP; de Haan L; Dideberg O; Keck W. Biochem. J 1994, 303, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Davies C; White SW; Nicholas RA J. Biol. Chem 2001, 276 616–623. [DOI] [PubMed] [Google Scholar]
- (9).Nicholas RA; Krings S; Tomberg J; Nicola G; Davies CJ Biol. Chem 2003, 278, 52826–52833. [DOI] [PubMed] [Google Scholar]
- (10).Nicola G; Peddi S; Stefanova M; Nicholas RA; Gutheil WG; Davies C. Biochemistry 2005, 44, 8207–8217. [DOI] [PubMed] [Google Scholar]
- (11).Nicola G; Fedarovich A; Nicholas RA; Davies C. Biochem. J 2005, 392, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Morlot C; Pernot L; Le Gouellec A; Di Guilmi AM; Vernet T; Dideberg O; Dessen AJ Biol. Chem 2005, 280, 15984–15991. [DOI] [PubMed] [Google Scholar]
- (13).Spratt BG Proc. Natl. Acad. Sci. U.S.A 1975, 72, 2999–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Spratt BG J. Bacteriol 1980, 144, 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).de Pedro MA; Young KD; Holtje JV; Schwarz H J. Bacteriol 2003, 185, 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Priyadarshini R; Popham DL; Young KD J. Bacteriol 2006, 188, 5345–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Varma A; de Pedro MA; Young KD J. Bacteriol 2007, 189, 5692–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Spratt BG; Strominger JL J. Bacteriol 1976, 127, 660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Josephine HR; Charlier P; Davies C; Nicholas RA; Pratt RF Biochemistry 2006, 45, 15873–15883. [DOI] [PubMed] [Google Scholar]
- (19).Kumar I; Josephine HR; Pratt RF ACS Chem. Biol 2007, 2, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Malhotra KT; Nicholas RA J. Biol. Chem 1992, 267, 11386–11391. [PubMed] [Google Scholar]
- (21).Korsak D; Vollmer W; Markiewicz Z. FEMS Microbiol. Lett 2005, 251, 281–288. [DOI] [PubMed] [Google Scholar]
- (22).Stefanova ME; Davies C; Nicholas RA; Gutheil WG Biochim. Biophys. Acta 2002, 1597, 292–300. [DOI] [PubMed] [Google Scholar]
- (23).Zhang W; Shi Q; Meroueh SO; Vakulenko SB; Mobashery S. Biochemistry 2007, 46, 10113–10121. [DOI] [PubMed] [Google Scholar]
- (24).Hesek D; Suvorov M; Morio K; Lee M; Brown S; Vakulenko SB; Mobashery SJ Org. Chem 2004, 69, 778–784. [DOI] [PubMed] [Google Scholar]
- (25).Case DA; et al. Amber 8, University of California: San Francisco, CA, 2004. [Google Scholar]
- (26).Sybyl 7.1; Tripos Inc.: St. Louis, MO, 2005.
- (27).Ewing TJA; Kuntz ID J. Comput. Chem 1997, 18, 1175–1189. [Google Scholar]
- (28).Bayly CI; Cieplak P; Cornell WD; Kollman PA J. Phys. Chem 1993, 97, 10269–10280. [Google Scholar]
- (29).Golemi-Kotra D; Meroueh SO; Kim C; Vakulenko SB; Bulychev A; Stemmler AJ; Stemmler TL; Mobashery SJ Biol. Chem 2004, 279, 34665–34673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).MatLab Version 7.4.0 (The Math Works, Inc., Massachusetts), 2007.
- (31).Liu B; Schofield CJ; Wilmouth RC J. Biol. Chem 2006, 281, 24024–24035. Huang DT; Schulman BA Nat. Struct. Mol. Biol 2006, 13, 1045–1047. [Google Scholar]; Shen L; Tatham MH; Dong C; Zagorska A; Naismith JH; Hay RT Nat. Struct. Mol. Biol 2006,13, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lemberg MK; Freeman M. Mol. Cell 2007, 28, 930–940. [DOI] [PubMed] [Google Scholar]
- (32).Vreven T; Morokuma K; Farkas O; Schlegel HB; Frisch MJ J. Comput. Chem 2003, 24, 760–769. [DOI] [PubMed] [Google Scholar]
- (33).Lin H; Truhlar DG J. Phys. Chem. A 2005, 109, 3991–4004. [DOI] [PubMed] [Google Scholar]
- (34).Zhang Y; Kua J; McCammon JA J. Am. Chem. Soc 2002, 124, 10572–10577. [DOI] [PubMed] [Google Scholar]
- (35).Bryant RAR; Hansen DE J. Am. Chem. Soc 1996, 118, 5498–5499. [Google Scholar]; Radzicka A; Wolfenden R. J. Am. Chem. Soc 1996, 118, 6105–6109. [Google Scholar]; Snider MG; Temple BS; Wolfenden R. J. Phys. Org. Chem 2004, 17, 586–591. [Google Scholar]
- (36).Gorb L; Asensio A; Tuñon I; Ruiz-López MF Chem. Eur. J 2007, 11, 6743–6753. [DOI] [PubMed] [Google Scholar]
- (37).Botos I; Wlodawer A. Curr. Opin. Struct. Biol 2007, 17, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Polgar L. Cell. Mol. Life Sci 2005, 62, 2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vineyard D; Zhang X; Lee I. Biochemistry 2006, 45, 11432–11443. Rotanova T; Botos I; Melnikov EE; Rasulova F; Gustchina A; Maurizi MR; Wlodawer A. Protein Sci. 2006, 15, 1815–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lee I; Berdis AJ; Suzuki CK Mol. Biosyst 2006, 2, 477–483. [DOI] [PubMed] [Google Scholar]
- (40).Lodola A; Mor M; Hermann JC; Tarzia G; Piomelli D; Mulholland AJ Chem. Commun 2005, 4399–4401. [DOI] [PubMed] [Google Scholar]; Lodola A; Mor M; Zurek J; Tarzia G; Piomelli D; Harvey JN; Mulholland AJ Biophys. J 2007, 92, L20–L22. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tubert-Brohman I; Acevedo O; Jorgensen WL J. Am. Chem. Soc 2006, 128, 16904–16913. [DOI] [PubMed] [Google Scholar]
- (41).Paetzel M; Strynadka NC; Tschantz WR; Casareno R; Bullinger PR; Dalbey RE J. Biol. Chem 1997, 272, 9994–10003. [DOI] [PubMed] [Google Scholar]; Kim AC; Oliver DC; Paetzel M. J. Mol. Biol 2008, 376, 352–366. [DOI] [PubMed] [Google Scholar]
- (42).Massova I; Mobashery S. Antimicrob. Agents Chemother 1998, 42, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gordon E; Mouz N; Duee E; Dideberg OJ Mol. Biol 2000, 299, 477–485. [DOI] [PubMed] [Google Scholar]; Lim D; Strynadka NC Nat. Struct. Biol 2002, 9, 870–876. [DOI] [PubMed] [Google Scholar]
- (44).Sauvage E; Duez C; Herman R; Kerff F; Petrella S; Anderson JW; Adediran SA; Pratt RF; Frere JM; Charlier P J. Mol. Biol 2007, 371, 528–539. [DOI] [PubMed] [Google Scholar]
- (45).Macheboeuf P; Lemaire D; Dos Santos Martins A; Dideberg O; Jamin M; Dessen AJ Mol. Biol 2008, 376, 405–413. [DOI] [PubMed] [Google Scholar]
- (46).Meroueh SO; Fisher JF; Schlegel HB; Mobashery SJ Am. Chem. Soc 2005, 127, 15397–15407. [DOI] [PubMed] [Google Scholar]
- (47).McDonough MA; Anderson JW; Silvaggi NR; Pratt RF; Knox JR; Kelly JA J. Mol. Biol 2002, 322, 111–122. [DOI] [PubMed] [Google Scholar]
- (48).Wilks JC; Slonczewski JL J. Bacteriol 2007, 189, 5601–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Fuda C; Hesek D; Lee M; Morio K; Nowak T; Mobashery SJ Am. Chem. Soc 2005, 127, 2056–2057. [DOI] [PubMed] [Google Scholar]; Fuda C; Hesek D; Lee M; Heilmayer W; Novak R; Vakulenko SB; Mobashery S. J. Biol. Chem 2006, 281, 10035–10041. [DOI] [PubMed] [Google Scholar]
- (50).Macheboeuf P; Di Guilmi AM; Job V; Vernet T; Dideberg O; Dessen A. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lovering AL; De Castro L; Lim D; Strynadka NC Protein Sci. 2006, 15, 1701–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.