

Abstract
Purpose of review:
Combination antiretroviral therapy (ART) has enabled tremendous progress in suppressing HIV replication in infected patients. However, ART alone cannot eradicate HIV and its latent, persisting reservoirs. Novel approaches are needed to eradicate the virus or achieve functional cure in the absence of ART.
Recent findings:
Adoptive T-cell therapies were initially tested in HIV-infected individuals with limited efficiency. Benefiting from new and improved methodologies, an increasing array of CAR T-cell therapies has been successfully developed in the cancer immunotherapy field, demonstrating promising new avenues that could be applied to HIV. Numerous studies have characterized various HIV-specific CAR constructs, types of cytolytic effector cells, and CAR-expressing cells’ trafficking to the reservoir compartments, warranting further in vivo efforts. Notably, the ability of CAR cells to persist and function in low-antigen environments in vivo, i.e. in ART-suppressed patients, remains unclear.
Summary:
Despite promising results in pre-clinical studies, only a handful of clinical trials have been initiated worldwide. Several obstacles remain prior to successful application of HIV-specific CAR T-cell therapies in patients. In this review, we survey the current state of the field, and address paths towards realizing the goal of an efficacious HIV CAR T-cell product.
Keywords: HIV-1, chimeric antigen receptor, CD4, broadly neutralizing antibodies, T-cell persistence, gene therapy
1. INTRODUCTION
Antiretroviral therapy (ART) has been a stepping-stone in the treatment of acquired immunodeficiency syndrome (AIDS), but is incapable of eradicating human immunodeficiency virus, type 1 (HIV-1). Instead, latently infected cells persist in reservoir compartments, leading to viral rebound following ART interruption. HIV persistence is also due in part to the unique ability of HIV to evade therapeutics and host immune responses via high mutation rates, further complicating the development of a cure [1]. Alternative treatment options are therefore required to maintain virus remission in the absence of ART. Engineering of T cells with Chimeric Antigen Receptors directed against the envelope of HIV could improve the adaptive immune response against the virus, which is generally observed following HIV-1 infection, but is inefficient at controlling viremia. Despite the recent FDA approval of CAR T cell therapy for B cell leukemia, and the numerous studies in progress for cancer immunotherapies, only two clinical trials have been initiated for anti-HIV CAR T cells (NCT03240328 and NCT01013415). In this review, we will provide an overview of the different CAR T cells strategies that have been attempted, the lessons learned from earlier trials, and challenges that remain to be addressed before bringing CAR T cells to HIV+ patients.
2-. EARLY ADOPTIVE T CELL THERAPY FOR HIV-1 TREATMENT
The basic concept of CAR-based therapies targeted against HIV-1 dates to studies initiated in the late 1980s. For example, soluble CD4 molecules were tested as blocking agents designed to prevent HIV infection and viral replication by interfering with the essential interaction between cell surface CD4 and the viral envelope, but had limited efficiency due to the short serum half-life of soluble CD4, as well as viral resistance in primary HIV-1 isolates [2, 3]. To improve this strategy, cytolytic CD8+ T cells (CTLs) were engineered to express chimeric proteins, including combinations of the extracellular domain of CD4 with the transmembrane and intracellular signaling domains of T cell IgG Fc receptors (CD4ζ-CAR [4]), T cell receptors (TCRs) or the variable regions of isolated monoclonal antibodies [5–7]. By combining an extracellular domain to recognize HIV antigen with an intracellular signaling domain, these constructs efficiently lysed envelope-expressing cells in vitro. Prior to initiation of clinical trials, only one study validated such strategies in vivo, in immunodeficient mice using gene-modified hematopoietic stem and progenitor cells (HSPC) [8]. These studies were reviewed recently [9].
In pioneering clinical trials, adoptive transfer of gene-modified HIV-specific T cells did not significantly impact viremia in vivo due to lack of T cell persistence [10–15]. The subsequent trials built on studies in CMV-infected patients, which demonstrated that the lack of persistence of adoptively transferred CTLs was due to the absence of supporting CD4+ T cells [16]. By using both CD4+ and CD8+ T cells, circulating CD4ζ CAR-modified T-cells persisted for more than 10 years post-infusion in clinical trial patients who participated in long-term follow-up studies, without evidence of toxicities or transformation [11, 17–19] (NCT01013415). Unfortunately, cells persisted at a low frequency (average of 0.01 to 0.1% circulating CD4ζ-CAR cells), and the impact on HIV viremia was low [11, 17–19].
Multiple technical parameters may have limited the efficacy of early CAR trials, but have been addressed more recently in the setting of hematological malignancies. These include low vector transduction efficiencies, the need for CD4 T-cell help, suboptimal CAR constructs, and inadequate ex vivo cell manipulation. Thus, applying these advances to HIV CAR therapies will likely also have a significant impact on the treatment of HIV-infected patients.
3-. DESIGNING THE ANTI-HIV CAR PROTEIN
The “first generation” CAR constructs employed in the early trials described above contained a single intracellular signaling domain derived from the CD3ζ chain of the TCR, fused either to the extracellular region of CD4 (CD4ζ-CAR), or to the variable region of isolated monoclonal antibodies (single chain variable fragment, scFv-CAR; reviewed [20]). These CARs proved to be sensitive to the size of the spacer that separated this domain from the cell surface, impacting not only the conformation and affinity of the chimeric protein, but also its expression and stability [21]. More recent studies employed CD4ζ - or scFv-based chimeric proteins with second or third generation CARs, which contained one or two intracellular costimulatory domains, respectively, and were recently reviewed [9].
The essential interaction between HIV-1 envelope and the CD4 protein has been exploited in the design of the early CARs, ensuring broad targeting of all HIV-1 isolates. Recent studies have validated CD4ζ-CARs in vitro [22, 23] and in vivo in humanized mice and nonhuman primate models infused with HIV-resistant hematopoietic-derived first generation CD4ζ-CAR [24, 25]. These studies showed for the first time the potential of stem cell derived CAR T cells to achieve potent targeting of HIV infected cells in vivo [24, 25], and to target HIV-infected cells as well as reactivated latently infected cell lines [26].
The advantage of antibody-based CARs is the ability to bind specifically to the exogenous viral antigen, and not to uninfected cells. New generations of broadly neutralizing antibodies (bNAbs) were isolated through preferential binding to the trimeric viral envelope, and selected for increased binding potential, specificity and limited off-target epitopes, improving on the previous generation of antibodies [27, 28]. Importantly, bNAb-based CARs achieve potent cytolysis of HIV infected cells and reactivated, latently infected cells in vitro [29–32]. Direct comparison between bNAb- and CD4ζ-based CAR [29, 30] or between bNAbs [31, 32] demonstrated some variations in breadth and potency and suggest that some antibody-derived scFVs might be more adapted than others for CAR T cell applications. Importantly, it remains unclear which assay is the best predictor of in vivo CAR T cells efficiency. Nevertheless, these studies make clear that the bNAb choice is critical to the functionality of HIV-specific CARs, and requires a broad, apples-to-apples comparison.
A combination or “bi-specific” CAR may be required to address the well-characterized capacity of HIV-1 to mutate and escape therapeutic and/or host immune responses, leading to inefficient T cell responses and viral escape, instead of controlling virus replication [15, 33]. Bispecific CARs demonstrated superior efficacy with several HIV-1 primary isolates relative to single CD4ζ CAR [34, 35] and warrant further in vivo investigation.
Comparisons between antibody-based CARs and transgenic TCR-expressing CTLs provided useful insights into the importance of affinities and avidities into CAR T cells activities, as excessively high affinities (e.g. using bNAb-based CARs) might be detrimental to CTL activity [36]. Lower affinities might be optimal for antigen-scFV CAR interactions, which could be explained by serial interactions required for the formation of the immunological synapse mediating cytolysis, as demonstrated in the case of TCR interactions with major histocompatibility complex (MHC) molecules [34, 37, 38]. Low affinity CAR proteins can exhibit improved activity and specificity especially in the presence of low antigen expression [36, 39], as is observed with HIV-1 (Figure 1). These insights into the dynamics of protein interactions involved in CAR efficiency strongly suggest that the affinities of the selected antibodies when designing a CAR construct should be considered. Additionally, in the case of CAR-expressing stem cells undergoing thymic selection (see below), the affinity of the chimeric protein would be of importance, as a strong affinity could lead to negative selection.
Figure 1.
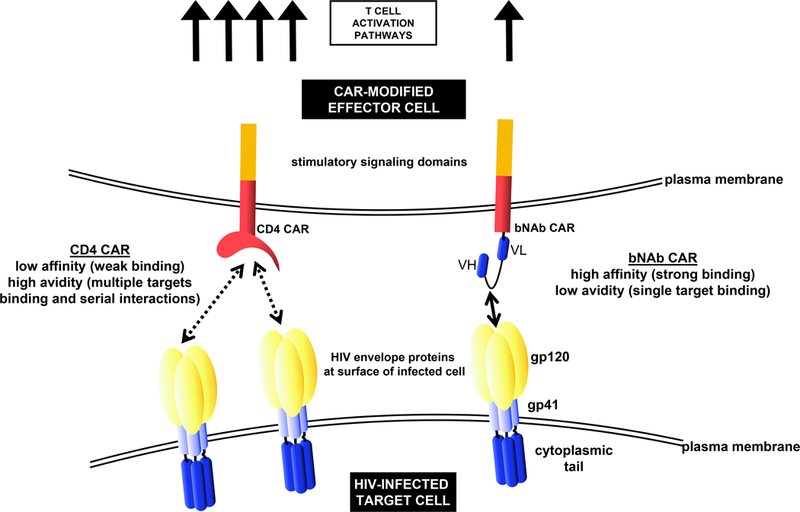
Comparison of CD4ζ - and bNAb-based CARs, and modulation of CAR T cell activation and potency. CD4ζ -based CARs possess lower affinity (strength of the binding between epitope and binding site) but higher avidity (cumulative strength of an interaction, factoring in multiple epitopes/binding sites), potentially leading to more potent T cell activation relative to bNAb-based CARs. The proper combination of affinity, avidity, and accessibility of the targeted epitope(s), among other parameters, are likely key in determining CAR-T cell potency.
4-. CELL TYPES FOR ANTI-HIV CAR
Relative to other cell types in which retroviral vector engineering introduces risks of oncogenic transformation, CAR-modified T-cell products are more stable and low-risk, especially for HIV patients requiring long-term persistence of the treatment [17, 40]. Significant improvements were achieved by incorporating new and improved promoters and costimulatory domain elements, and replacing early gamma retrovirus-based gene therapy vectors with HIV-based lentiviral backbones, favoring integration in open reading frames [30]. Optimized CAR constructs efficiently controlled HIV viremia and viral rebound following ART withdrawal in an NSG mouse model [30], and synergized with latency reversing agents in primary cells isolated from ART-treated individuals [29].
A major obstacle for any therapy against HIV is the ability to demonstrate efficacy at sites of virus persistence in tissues. Despite reports demonstrating the function of CAR T cells against reactivated, latently infected cells in vitro [26] and trafficking of adoptively transferred CTLs to secondary lymphoid tissues [14, 41], few studies have identified anti-HIV CAR T cells in reservoir tissues and/or their ability to target latently infected cells in vivo. Additionally, retention of adoptively transferred anti-HIV TCR T cells in the lungs is a problem that might also affect anti-HIV CAR T cells [42–44]. Addition of chemokine receptors could improve trafficking: for example, co-expression of the CCR7 chemokine receptor and the L-Selectin CD62L in TCR-engineered CD8+ T cells increases trafficking to lymph nodes, while CCR7/CD62L negative cells accumulate in the lungs [44]. CXCR5 similarly improve CAR T cells trafficking to B cell follicles [45]. A glioma-specific CAR demonstrated trafficking to the brain [46]; however, CNS targeting of re-activated, latently infected cells in the context of HIV remains an open question.
Natural killer (NK) cells have great potential as effector cells, due to their potent cytolytic activity [47, 48]. Furthermore, while CAR T cells for malignancies must balance beneficial graft-versus-tumor with toxic graft-versus-host disease (GVHD), NK cells are preferentially associated with graft-versus-leukemia/tumor effects, rather than GVHD [49, 50]. These attributes could enable CAR NK cells’ application in myriad clinical applications. Several studies, including clinical trials, have demonstrated the feasibility and safety of adoptive transfer of NK cells following ex vivo expansion (for a review see [49]). Engineered NK cells bearing CARs are functionally activated in vitro in an HIV-specific manner [51]. While mature NK cells have a limited lifespan from a few days to a few weeks, use of immature NK cells, for example derived from cord blood, induced pluripotent stem cells (iPSCs), or human embryonic stem cells (hESC), could improve their persistence in vivo [49]. In particular, the self-renewal properties of iPSC cells could offer an unlimited source of T or NK cells capable of establishing a memory phenotype [52]. Although first generation CD4ζ-CAR NK cells derived from hESC or iPSC displayed enhanced cytolytic activity against HIV-infected cells relative to unmodified controls in vitro, similar properties were not observed in vivo [22]. Nevertheless, this study provides preliminary data warranting further in vivo investigation of CAR NK cells. Preliminary findings have also been reported using other myeloid cells such as neutrophils or monocytes to target cancer antigens (for a review see [48]). However, more studies assessing in vivo function and toxicity are required, especially for HIV applications.
Delivery of CAR-encoding gene therapy vectors to hematopoietic stem and progenitor cells (HPSC) could combine CAR-T, -NK, and other hematopoietic cells against target antigens (for a review see [20]). This approach could address several challenges encountered by CAR T cells. First, HSPC-derived CAR cells may traffic more effectively to tissues than CAR T cells, for example through differentiation into mature cells that can cross anatomical and physiological barriers. Second, we have shown that HSPC-derived CAR cells persist even in the absence of robust antigen expression, i.e. during ART-suppressed SHIV viremia in nonhuman primates (NHP) [25]. Finally, thymic selection should eliminate self-reacting T cells, improving the safety of this strategy relative to CAR T cells [24].
Our group and Zhen et al. investigated the potential of HSPC-derived CD4ζ-CAR cells in vivo [24, 25]. CAR-encoding HSPCs were capable of multilineage engraftment, giving rise to CAR-expressing monocytes, NK, B and T cells. Mature CAR cells possessed effector function in humanized mice [24] and NHP CAR-expressing cells were found in the spleen, lymph nodes, gut, bone marrow and thymus [25]. In SHIV-infected NHPs, plasma viremia and viral rebound were decreased relative to controls, and no toxicity was detected over 2 years, despite persistent, antigen-dependent expression of CAR+ cells [25]. In this study, the rapid increase in CD4ζ T cells following ART withdrawal suggests the establishment of a memory subset [25]. In summary, autologous transplantation of CD4ζ-CAR-encoding HSPC is safe, feasible, efficient and persistent. Future experiments should focus on the feasibility of this approach with other CAR molecules, including distinct extracellular ligand binding domains and intracellular costimulatory domains.
5-. CONSIDERATIONS FOR ANTI- HIV CAR T CELLS
Enabling life-long CAR T cell therapy
An outstanding question for future clinical trials of HIV-directed CAR cells is the level of HIV antigen needed to observe a CAR-dependent impact. In matching the successes with cancer CARs, HIV-specific CAR T cells might be most active in a high antigen environment, i.e. in unsuppressed patients. Although emerging data suggests that treatment interruption in suppressed HIV+ patients is safe [53], ethical considerations [54] and practical limitations, i.e. risks associated with patients with active plasma viremia, will most likely limit initial HIV CAR trials to stably suppressed, ART-treated patients. CAR T-cells persisted long-term (over a decade) in suppressed patients, possibly due to viral blips in peripheral blood and/or persistent viral antigen in secondary lymphoid tissues. These sources of infrequent, ongoing antigen expression could facilitate restimulation and expansion of CAR-expressing cells; however, the low frequency of CAR T cells and modest effect on viremia suggest that higher antigen expression might be necessary for therapeutically relevant levels of CAR T cell expansion [17]. Although these dynamics may be improved with second and third generation CARs, improving CAR T cell persistence in the absence of antigen expression will be required, especially with the goal of life-long therapy to protect against recrudescent virus that may appear years after remission.
Lack of functional HIV-specific CD4+ T cells also contributes to low CAR T cell persistence [55, 56]. Prior to CAR modification, enrichment/selection for T cells with a memory phenotype and retention of CD4+ T cells in the infused T cell product improve T cell expansion and persistence in vivo (for a review, see [57]). Rational selection of cytokines during T-cell culture, e.g. favoring IL7 and IL15 over IL2, may also aid in the generation of a more robust T-cell product [58, 59]. T-cell exhaustion could also be targeted; similar to cancer patients, HIV infected individuals’ T-cells express higher levels of PD1 [60, 61], Tim3 [62], and other exhaustion markers. Blockade of these pathways could have a binary action by increasing the immune response [63–65] while more effectively targeting latently infected cells [66]. Finally, HSPC-derived CAR cells should also address the problem of T cell persistence, by providing a lifelong source of T-cell progenitors that give rise to functional CAR T-cells in an antigen-dependent manner [25]. Viral escape due to mutations in HIV-1 envelope, and lack of effective T cell stimulation by these mutated epitopes, may also underlie low levels of CAR T cell persistence in future trials [15]. Identifying epitopes expressed in latently infected reservoir cells, ideally those that are essential to virus fitness [67, 68], should minimize selection for potential escape mutations.
Off-target effects, toxicities, and infection-resistant CAR cells
T-cell therapies targeting tumor cells are considered safer when directed against mutated epitopes not expressed on otherwise healthy cells. As such, bNAb-CAR targeting of gp120 might be safer than CD4ζ-CAR, since CD4 is a natural ligand of MHC class II, among others. Notably, no cytolysis of MHC class II-expressing cell lines was observed in CD4ζ-CAR assays [4, 30, 34]. Additionally, the need to control cytolytic activities of cancer CARs prompted the development of non-immunogenic, inducible suicide genes that should be considered for future clinical trials [69–73]. The safety and persistence of CD4ζ-CAR T cells provides strong rationale for further clinical studies with more advanced iterations of this and other HIV-specific CARs [17]. Additionally, while immunogenicity against the bNAb variable region remains possible [35, 74, 75], neither healthy individuals nor HIV-infected patients receiving bNAbs has developed an anti-antibody immune response [33, 76].
A final, but essential question is whether CAR-expressing T cells can be infected by HIV. The env-binding extracellular domain (either CD4 or bNAb) in the CAR could act in place of native CD4 prior to binding CCR5 or CXCR4 coreceptor proteins, enabling infection of CD8+CCR5+ T-cells, for example. Several studies have reported increased HIV infection upon CD4ς-CAR expression [24, 25, 34]. This was not observed with bNAb-based [29] or bispecific CAR [34, 35]. Blockade of CCR5-tropic virus entry into CAR T cells by CCR5 co-receptor disruption [24, 31, 77, 78] or of CXCR4 and CCR5 isolates by co-expression of anti-HIV genes [25, 78] proved efficient. Protecting cells against HIV infection should increase their function and persistence in vivo, and should be strongly considered as a part of any anti-HIV CAR approach.
6-. CONCLUSION
CAR therapies, born in part as a treatment for HIV, represent arguably the most important clinical treatment for cancer developed in decades. Due to a rapidly increasing knowledge base, CARs are poised to make a powerful impact in infectious disease settings such as suppressed HIV infection as well. CAR therapy for HIV+ patients should keep in mind common ground with cancer CARs but also recognize unique aspects. For example, unlike cancer indications, where alternative therapies are frequently unavailable, HIV CAR patients will need to be treated with particular attention to safety, given that these patients can live an almost normal life on ART. Despite the increasing numbers of clinical trials assessing CAR T cells for cancer immunotherapy and recent FDA approval of these treatments, the lessons learnt from these studies must be more aggressively translated to the HIV field, as only two clinical trials are now assessing CAR T cells for HIV-infected patients (NCT03240328 and NCT01013415). Although several obstacles remain to be addressed, new CAR engineering, cellular manufacturing, and subset targeting strategies have the potential to overcome these hurdles, enabling safe, efficient, and specific clearance of HIV+ targets in vivo.
KEY POINTS.
Early attempts using CD4-based chimeric proteins failed to significantly impact HIV viremia in clinical trials, likely due to limited persistence of the modified T cells, non-optimal chimeric proteins, and potential for increased HIV entry upon CD4 paratope expression at the cell surface.
Improved constructs including costimulatory domains, antibody-based or bispecific extracellular domains, suicide genes, and/or HIV resistance strategies have the potential to improve CAR T cell safety and potency for HIV patients.
Future studies should consider new options, such as co-expression of homing receptors or transplantation of CAR-expressing hematopoietic stem and progenitor cells, to allow improved cell trafficking and lifelong persistence of the treatment.
Acknowledgements
We thank Helen Crawford for help in preparing this review.
Financial support and sponsorship
This study was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (U19 AI096111 and UM1 AI126623 to HPK) and National Heart, Lung, and Blood Institute (R01 HL116217 and U19 HL129902 to HPK). HPK is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy.
Footnotes
Conflicts of interest
None.
References and recommended reading
References published within the past 18 months only are bulleted and annotated.
References have one bullet (*) for special interest and two bullets (**) for outstanding interest.
Annotations provide a brief description of the paper’s importance.
- 1.Borrow P, Lewicki H, Wei X, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 1997; 3(2):205–211. [DOI] [PubMed] [Google Scholar]
- 2.Daar ES, Li XL, Moudgil T, Ho DD. High concentrations of recombinant soluble CD4 are required to neutralize primary human immunodeficiency virus type 1 isolates. Proc Natl Acad Sci U S A 1990; 87(17):6574–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore JP, McKeating JA, Huang YX, et al. Virions of primary human immunodeficiency virus type 1 isolates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J Virol 1992; 66(1):235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell 1991; 64(5):1037–1046. [DOI] [PubMed] [Google Scholar]
- 5.Roberts MR, Qin L, Zhang D, et al. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood 1994; 84(9):2878–2889. [PubMed] [Google Scholar]
- 6.Yang OO, Tran AC, Kalams SA, et al. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc Natl Acad Sci U S A 1997; 94(21):11478–11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masiero S, Del Vecchio C, Gavioli R, et al. T-cell engineering by a chimeric T-cell receptor with antibody-type specificity for the HIV-1 gp120. Gene Ther 2005; 12(4):299–310. [DOI] [PubMed] [Google Scholar]
- 8.Hege KM, Cooke KS, Finer MH, et al. Systemic T cell-independent tumor immunity after transplantation of universal receptor-modified bone marrow into SCID mice. J Exp Med 1996; 184(6):2261–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner TA. Quarter Century of Anti-HIV CAR T Cells. Curr HIV/AIDS Rep 2018.* Recent review focusing on clinical trials for HIV using first or second generation CAR T cells.
- 10.Tan R, Xu X, Ogg GS, et al. Rapid death of adoptively transferred T cells in acquired immunodeficiency syndrome. Blood 1999; 93(5):1506–1510. [PubMed] [Google Scholar]
- 11.Walker RE, Bechtel CM, Natarajan V, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood 2000; 96(2):467–474. [PubMed] [Google Scholar]
- 12.Riddell SR, Greenberg PD, Overell RW, et al. Phase I study of cellular adoptive immunotherapy using genetically modified CD8+ HIV-specific T cells for HIV seropositive patients undergoing allogeneic bone marrow transplant. The Fred Hutchinson Cancer Research Center and the University of Washington School of Medicine, Department of Medicine, Division of Oncology. Hum Gene Ther 1992; 3(3):319–338. [DOI] [PubMed] [Google Scholar]
- 13.Riddell SR, Elliott M, Lewinsohn DA, et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med 1996; 2(2):216–223. [DOI] [PubMed] [Google Scholar]
- 14.Brodie SJ, Lewinsohn DA, Patterson BK, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med 1999; 5(1):34–41. [DOI] [PubMed] [Google Scholar]
- 15.Koenig S, Conley AJ, Brewah YA, et al. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat Med 1995; 1(4):330–336. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg PD, Finch RJ, Gavin MA, et al. Genetic modification of T-cell clones for therapy of human viral and malignant diseases. Cancer J Sci Am 1998; 4 Suppl 1:S100–105. [PubMed] [Google Scholar]
- 17.Scholler J, Brady TL, Binder-Scholl G, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 2012; 4(132):132ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitsuyasu RT, Anton PA, Deeks SG, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 2000; 96(3):785–793. [PubMed] [Google Scholar]
- 19.Deeks SG, Wagner B, Anton PA, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Molecular Therapy 2002; 5(6):788–797. [DOI] [PubMed] [Google Scholar]
- 20.Zhen A, Carrillo MA, Kitchen SG. Chimeric antigen receptor engineered stem cells: a novel HIV therapy. Immunotherapy 2017; 9(5):401–410.* Review with a special emphasis on in vivo data for the CD4ζ CAR.
- 21.Patel SD, Moskalenko M, Smith D, et al. Impact of chimeric immune receptor extracellular protein domains on T cell function. Gene Ther 1999; 6(3):412–419. [DOI] [PubMed] [Google Scholar]
- 22.Ni Z, Knorr DA, Bendzick L, et al. Expression of chimeric receptor CD4zeta by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells 2014; 32(4):1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacLean AG, Walker E, Sahu GK, et al. A novel real-time CTL assay to measure designer T-cell function against HIV Env(+) cells. J Med Primatol 2014; 43(5):341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhen A, Kamata M, Rezek V, et al. HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells. Molecular Therapy: the Journal of the American Society of Gene Therapy 2015; 23(8):1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhen A, Peterson CW, Carrillo MA, et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog 2017; 13(12):e1006753.** First study demonstrating the potency of first generation CD4ζ CAR T cells, using human gene modified hematopoieitic stem cells in nonhuman primates.
- 26.Sahu GK, Sango K, Selliah N, et al. Anti-HIV designer T cells progressively eradicate a latently infected cell line by sequentially inducing HIV reactivation then killing the newly gp120-positive cells. Virology 2013; 446(1–2):268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy LE, Burton DR. Identification and specificity of broadly neutralizing antibodies against HIV. Immunol Rev 2017; 275(1):11–20.* Methodological improvements that led to the discovery of a new generation of potent and specific anti-HIV broadly neutralizing antibodies that can be included in CAR constructs.
- 28.Yang G, Holl TM, Liu Y, et al. Identification of autoantigens recognized by the 2F5 and 4E10 broadly neutralizing HIV-1 antibodies. J Exp Med 2013; 210(2):241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu B, Zou F, Lu L, et al. Chimeric antigen receptor T cells guided by the single-chain Fv of a broadly neutralizing antibody specifically and effectively eradicate virus reactivated from latency in CD4+ T lymphocytes isolated from HIV-1-infected individuals receiving suppressive combined antiretroviral therapy. J Virol 2016; 90(21):9712–9724.* Third generation VRC01-based CAR efficiently targets and lyses latently HIV-infected cells reactivated by removal of ART drugs, or by addition of latency reversing agents to cells isolated from ART-treated HIV-infected patients.
- 30.Leibman RS, Richardson MW, Ellebrecht CT, et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog 2017; 13(10):e1006613.* Optimization of a potent anti-HIV CAR by extensive engineering and comparison of a panel of candidate CD4ζ - or antibody-based binding moieties, and promoters. The improved CAR T cell product efficiently killed target cells in vitro and in vivo.
- 31.Hale M, Mesojednik T, Romano Ibarra GS, et al. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther 2017; 25(3):570–579.** Expression of a bNAb-based anti-HIV CAR targted to the gene-edited CCR5 locus is associated with resistance to HIV infection in primary human T cells. Non-random integration of the CAR construct is likely to increase the safety of this therapy.
- 32.Ali A, Kitchen SG, Chen IS, et al. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J Virol 2016; 90(15):6999–7006.* This study compared the in vitro potency of seven different bNAb-based CARs specific for different epitopes on HIV envelope.
- 33.Lynch RM, Boritz E, Coates EE, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med 2015; 7(319):319ra206. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Patel B, Ghanem MH, et al. Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity. J Virol 2015; 89(13):6685–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghanem MH, Bolivar-Wagers S, Dey B, et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy 2018; 20(3):407–419.* Bispecific CAR designed to recognize highly conserved regions on the HIV envelope (CD4 binding site and a dense oligomannose patch). This approach addresses concerns of mutational escape, and showed improved potency relative to a previous VRC01 antibody-based CAR in vitro.
- 36.Oren R, Hod-Marco M, Haus-Cohen M, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol 2014; 193(11):5733–5743. [DOI] [PubMed] [Google Scholar]
- 37.Valitutti S, Muller S, Cella M, et al. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 1995; 375(6527):148–151. [DOI] [PubMed] [Google Scholar]
- 38.Valitutti S The serial engagement model 17 years after: From TCR triggering to immunotherapy. Front Immunol 2012; 3:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turatti F, Figini M, Balladore E, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother 2007; 30(7):684–693. [DOI] [PubMed] [Google Scholar]
- 40.Newrzela S, Cornils K, Li Z, et al. Resistance of mature T cells to oncogene transformation. Blood 2008; 112(6):2278–2286. [DOI] [PubMed] [Google Scholar]
- 41.Tjernlund A, Zhu J, Laing K, et al. In situ detection of Gag-specific CD8+ cells in the GI tract of SIV infected Rhesus macaques. Retrovirology 2010; 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minang JT, Trivett MT, Bolton DL, et al. Distribution, persistence, and efficacy of adoptively transferred central and effector memory-derived autologous simian immunodeficiency virus-specific CD8+ T cell clones in rhesus macaques during acute infection. J Immunol 2010; 184(1):315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolton DL, Minang JT, Trivett MT, et al. Trafficking, persistence, and activation state of adoptively transferred allogeneic and autologous Simian Immunodeficiency Virus-specific CD8(+) T cell clones during acute and chronic infection of rhesus macaques. J Immunol 2010; 184(1):303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ayala VI, Trivett MT, Barsov EV, et al. Adoptive transfer of engineered rhesus simian immunodeficiency virus-specific CD8+ T cells reduces the number of transmitted/founder viruses established in rhesus macaques. J Virol 2016; 90(21):9942–9952.** In this study, adoptive transfer of gene-modified rhesus macaque CD8 T cells directed against SIV decreased the levels of founder viruses. Coexpression of the homing receptors CCR7 and CD62L increased cell trafficking to the lymph nodes while cells that were negative for CCR7 and CD62L were generally localized in the lung.
- 45.Ayala VI, Deleage C, Trivett MT, et al. CXCR5-dependent entry of CD8 T cells into rhesus macaque B-cell follicles achieved through T-cell engineering. J Virol 2017; 91(11).** An interesting approach to improve the trafficking of T cells to B cell follicles in lymphoid tissue reservoir compartments by expression of the CXCR5 homing receptor.
- 46.O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017; 9(399).* In this study, EGFRvIII CAR T cell trafficking to the central nervous system was observed following infusion in glioblastoma patients.
- 47.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005; 106(1):376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrer DC, Dorrie J, Schaft N. Chimeric antigen receptors in different cell types: New vehicles join the race. Hum Gene Ther 2018.* This review outlines CAR therapies using cell types other than T cells, e.g. natural killer and myeloid cells.
- 49.Glienke W, Esser R, Priesner C, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol 2015; 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer dells for the immunotherapy of cancer. Front Immunol 2018; 9:283.** An interesting insight into the advantages, challenges and sources of natural killer cells for CAR therapy applications.
- 51.Tran AC, Zhang D, Byrn R, Roberts MR. Chimeric zeta-receptors direct human natural killer (NK) effector function to permit killing of NK-resistant tumor cells and HIV-infected T lymphocytes. J Immunol 1995; 155(2):1000–1009. [PubMed] [Google Scholar]
- 52.Nishimura T, Kaneko S, Kawana-Tachikawa A, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013; 12(1):114–126. [DOI] [PubMed] [Google Scholar]
- 53.Clarridge KE, Blazkova J, Einkauf K, et al. Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog 2018; 14(1):e1006792.* Comparison of multiple relevant parameters in patients before and after ATI suggests that ATI per se does not negatively impact viral reservoir size, ART resistance, or immune functionality.
- 54.Henderson GE, Peay HL, Kroon E, et al. Ethics of treatment interruption trials in HIV cure research: addressing the conundrum of risk/benefit assessment. J Med Ethics 2018; 44(4):270–276.* Useful case study within the SEARCH cohort, in which patients initiated ART at extremely early time points post-infection, making them prime candidates for subsequent ATI studies. A focus on the viewpoints of patients and other stakeholders should be a primary factor in the design of any ATI-containing clinical trial.
- 55.Kamphorst AO, Ahmed R. CD4 T-cell immunotherapy for chronic viral infections and cancer. Immunotherapy 2013; 5(9):975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wiesel M, Oxenius A. From crucial to negligible: functional CD8(+) T-cell responses and their dependence on CD4(+) T-cell help. Eur J Immunol 2012; 42(5):1080–1088. [DOI] [PubMed] [Google Scholar]
- 57.Patel S, Jones RB, Nixon DF, Bollard CM. T-cell therapies for HIV: Preclinical successes and current clinical strategies. Cytotherapy 2016; 18(8):931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pilipow K, Roberto A, Roederer M, et al. IL15 and T-cell Stemness in T-cell-Based Cancer Immunotherapy. Cancer Res 2015; 75(24):5187–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steele AK, Carrasco-Medina L, Sodora DL, Crawley AM. Increased soluble IL-7 receptor concentrations associate with improved IL-7 therapy outcomes in SIV-infected ART-treated Rhesus macaques. PLoS One 2017; 12(12):e0188427.** Administration of IL7 in SIV-infected, ART-treated rhesus macaques significantly increased CD4 and CD8 T cell proliferation, suggesting a promising role in CAR T cell therapy for HIV-infected patients.
- 60.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006; 443(7109):350–354. [DOI] [PubMed] [Google Scholar]
- 61.D’Souza M, Fontenot AP, Mack DG, et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol 2007; 179(3):1979–1987. [DOI] [PubMed] [Google Scholar]
- 62.Porichis F, Kaufmann DE. HIV-specific CD4 T cells and immune control of viral replication (Review). Current Opinion in HIV & AIDS 2011; 6(3):174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Porichis F, Hart MG, Zupkosky J, et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J Virol 2014; 88(5):2508–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teigler JE, Zelinskyy G, Eller MA, et al. differential inhibitory receptor expression on T cells delineates functional capacities in chronic viral infection. J Virol 2017; 91(23).* A study evaluating expression and blockade of immune inhibitory receptors such as CTLA-4 or PD-1 in both CD4 and CD8 T cell subsets in vitro and in HIV infected patients. Blocking antibodies stimulated cytokine production by CD4 and CD8 T cells. CD4 T cells from progressor patients presented higher levels of the inhibitory receptors than controllers.
- 65.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction [erratum appears in Nat Med. 2006 Nov;12(11):1329]. Nature Medicine 2006; 12(10):1198–1202. [DOI] [PubMed] [Google Scholar]
- 66.Fromentin R, Bakeman W, Lawani MB, et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog 2016; 12(7):e1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng K, Pertea M, Rongvaux A, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 2015; 517(7534):381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rolland M, Manocheewa S, Swain JV, et al. HIV-1 conserved-element vaccines: relationship between sequence conservation and replicative capacity. J Virol 2013; 87(10):5461–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006; 107(6):2294–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barese CN, Felizardo TC, Sellers SE, et al. Regulated apoptosis of genetically modified hematopoietic stem and progenitor cells via an inducible caspase-9 suicide gene in rhesus macaques. Stem Cells 2015; 33(1):91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Resetca D, Neschadim A, Medin JA. Engineering hematopoietic cells for cancer immunotherapy: Strategies to address safety and toxicity concerns. J Immunother 2016; 39(7):249–259. [DOI] [PubMed] [Google Scholar]
- 72.Zhou X, Naik S, Dakhova O, et al. Serial activation of the inducible caspase 9 safety switch after human stem cell transplantation. Mol Ther 2016; 24(4):823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Diaconu I, Ballard B, Zhang M, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther 2017; 25(3):580–592.* Safe, efficient, and dose-dependent elimination of anti-CD19 CAR T cells upon expression of the inducible suicide gene iCasp9 in humanized mice, following administration of the dimerization-inducing small molecule AP20187.
- 74.Lamers CH, Willemsen R, van Elzakker P, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011; 117(1):72–82. [DOI] [PubMed] [Google Scholar]
- 75.Rosenberg Y, Sack M, Montefiori D, et al. Pharmacokinetics and immunogenicity of broadly neutralizing HIV monoclonal antibodies in macaques. PLoS One 2015; 10(3):e0120451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ledgerwood JE, Coates EE, Yamshchikov G, et al. Safety, pharmacokinetics and neutralization of the broadly neutralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin Exp Immunol 2015; 182(3):289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hale M, Lee B, Honaker Y, et al. Homology-directed recombination for enhanced engineering of chimeric antigen receptor T cells. Mol Ther Methods Clin Dev 2017; 4:192–203.** This study improved the safety of CD19-CAR T cells by targeting integration to the CCR5 locus in primary T cells, without loss of function in vitro and in vivo.
- 78.Kamata M, Kim PY, Ng HL, et al. Ectopic expression of anti-HIV-1 shRNAs protects CD8(+) T cells modified with CD4zeta CAR from HIV-1 infection and alleviates impairment of cell proliferation. Biochem Biophys Res Commun 2015; 463(3):216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]