

Abstract
Genome-wide association studies are powerful tools to elucidate the genome-to-phenome relationship. In order to explain most of the observed heritability of a phenotypic trait, a sufficient number of individuals and a large set of genetic variants must be examined. The development of high-throughput technologies and cost-efficient resequencing of complete genomes have enabled the genome-wide identification of genetic variation at large scale. As such, almost all existing genetic variation becomes available, and it is now possible to identify rare genetic variants in a population sample. Rare genetic variants that were usually filtered out in most genetic association studies are the most numerous genetic variations across genomes and hold great potential to explain a significant part of the missing heritability observed in association studies. Rare genetic variants must be identified with high confidence, as they can easily be confounded with sequencing errors. In this study, we used a pre-filtered data set of 1,014 pure Populus trichocarpa entire genomes to identify rare and common small genetic variants across individual genomes. We compared variant calls between Platypus and HaplotypeCaller pipelines, and we further applied strict quality filters for improved genetic variant identification. Finally, we only retained genetic variants that were identified by both variant callers increasing calling confidence. Based on these shared variants and after stringent quality filtering, we found high genomic diversity in P. trichocarpa germplasm, with 7.4 million small genetic variants. Importantly, 377k non-synonymous variants (5% of the total) were uncovered. We highlight the importance of genomic diversity and the potential of rare defective genetic variants in explaining a significant portion of P. trichocarpa's phenotypic variability in association genetics. The ultimate goal is to associate both rare and common alleles with poplar's wood quality traits to support selective breeding for an improved bioenergy feedstock.
Keywords: annotation, genes, genetic architecture, missing heritability, rare defective alleles, small genetic variants, variant calling comparisons
Introduction
In tandem, phenotypic and genomic diversity assessments are key to understand the genetic regulation and architecture of quantitative traits. Genetic association studies in the form of genome-wide association studies (GWAS) have been used extensively to associate genome-wide polymorphisms to phenotypic variation (Visscher et al., 2017). Typical GWAS are only including common genetic variations. Most of these studies, however, failed to explain most of the observed heritability which is coined the missing heritability problem (Manolio et al., 2009; Brachi et al., 2011). It has been suggested that the missing heritability could be found in other forms of hereditary information such as epigenetic factors, epistasis, and rare genetic variation (Maher, 2008). For over a decade, human geneticists have questioned the role of rare genetic variants in complex diseases (Pritchard, 2001; McCarthy et al., 2008; Manolio et al., 2009). Consequently, the first association studies including rare genetic variants and the associated statistical tests originated in the field of human genetics (Cohen et al., 2004; Hoffmann et al., 2010; Wu et al., 2011).
Generally, most genetic polymorphisms in natural population are rare (i.e. found at frequencies lower than 5% in populations). In addition, deleterious variants tend to exist at low frequency in populations because of their negative impact on the phenotype. Non-synonymous genetic variants especially, may have important effects on phenotypes as they alter the amino acid sequence. For instance, a genetic variation leading to a stop codon gain can have drastic impacts on gene products (i.e. RNA and protein). Non-synonymous variants can either be missense or nonsense variants. Missense variants result in a codon change that code for a different amino acid while nonsense variants result in truncated or incomplete gene products. Including rare genetic variants in GWAS along with common genetic variants represents a unique opportunity to explain a significant part of the missing heritability (McClellan and King, 2010). Prior to genetic association studies, however, high confidence identification of the genetic polymorphisms within the studied population is required.
Due to their low frequency, rare genetic variants are challenging to identify. Genetic information for a substantial number of individuals is required to find those genetic variants that are rare in a population. In addition, rare genetic variants can easily be confounded with sequencing errors as high-throughput technologies have sequencing error rates between 0.1 to 1% (Fox et al., 2014). Therefore, rare genetic variants must be identified with high confidence before use in GWAS.
High-throughput sequencing permit the resequencing of large numbers of individuals at reasonable cost. Thanks to this technological advancement, genetic data for model species are now sufficiently large to identify rare genetic variants. Currently, the lack of computing resources remains one of the most important challenges to analyze these overwhelming data sets.
To decrease the confusion of low-frequency genetic variants with sequencing errors, strict quality filters are applied from processing of raw sequencing reads to variant discovery to discard bad quality reads and other chimeras. In addition, comparison between variant calling software resulting in a consensus set of Single Nucleotide Polymorphisms (SNP) lead to increased variant detection accuracy (Baes et al., 2014; Fahrenkrog et al., 2017). This approach minimizes the identification of false genetic variants, even though it will discard true genetic variants that were not identified by all variant callers. Using strict quality filtering and variant caller comparison, it is possible to evaluate both common and rare genetic diversity with high confidence. Sensitivity (the number of true positives) and specificity (the number of false positives) of the data processing and variant calling steps should be optimized according to the objectives of the genomic diversity evaluation.
Some populations are expected to contain a higher number of low-frequency genetic variants than others. Natural, outbreeding, and wide-ranging populations are expected to possess higher heterozygosity and a larger number of low-frequency variants (Petit and Hampe, 2006; Evans et al., 2014). On the contrary, domesticated species typically have reduced genetic diversity because of repeated cycles of artificial selection using a few performant breeders with common genetic backgrounds. Because of this high expected number of low-frequency genetic variants, natural forest tree species represent good candidates for rare variant association studies. In forest trees, rare nonsense variants associated to complex traits have been successfully identified. So far, these variants were found in the following genes and species: a CAD (Cinnamyl alcohol dehydrogenase) in Pinus taeda (MacKay et al., 1997), a CCR (Cinnamoyl-CoA reductase) in two Eucalyptus species (Thumma et al., 2005), an HCT1 (Hydroxycinnamoyl transferase) in Populus nigra (Vanholme et al., 2013), and a KANADI in a P. trichocarpa x P. deltoides pseudo backcross (Muchero et al., 2015). Other studies also highlighted the ubiquity of rare genetic variants and their role in complex trait regulation in poplar species (Evans et al., 2014; Fahrenkrog et al., 2017).
Populus trichocarpa (Torr. & Gray), is a deciduous forest tree species with important ecological and economical aspects. This fast-growing tree mainly ranges along the North American west coast, from Alaska to Baja California Norte (latitude 31°N to 62°N) ( Figure 1 ). The tree is used for pulp and oriented strand board production and represents a good candidate for second-generation biofuel feedstock (Porth and El-Kassaby, 2015). Additionally, P. trichocarpa was the first tree species to have its whole genome sequenced with a genome size close to 500Mbp (Tuskan et al., 2006). Since then, hundreds of whole genome resequencing efforts were conducted (Evans et al., 2014; Muchero et al., 2015; McKown et al., 2017) and numerous phenotypic traits related to phenology and wood properties have been measured in common garden experiments (Porth et al., 2013; Evans et al., 2014; McKown et al., 2014; Muchero et al., 2015).
Figure 1.

The 1,038 P. trichocarpa individuals retrieved from the JGI Genome Portal are represented by red dots across northwestern America. P. trichocarpa natural range is defined in dark grey. P. trichocarpa natural range was drawn from Little (1971).
Contrary to other better-studied model species, forest trees have not been subject to extensive genomic evaluation using whole genome resequencing data. Only a handful of such studies have been performed on economically important trees. Silva-Junior and collaborators used pooled resequencing of 240 Eucalyptus tree genome to develop a SNP chip able to identify 60K SNPs (Silva-Junior et al., 2015). P. trichocarpa is by far the forest tree species with the most available genetic resources. In 2014, Evans and colleagues evaluate the genomic diversity across a data set of 544 WGS of P. trichocarpa individuals identifying 17M variants (Evans et al., 2014). A second data set developed by the US Department Of Energy (DOE) BioEnergy Science Center (BESC) used 882 WGS of P. trichocarpa to identify 28M genetic variants genome-wide (https://bioenergycenter.org/besc/gwas/). To our knowledge, these few studies were the largest genomic evaluation studies performed to date using WGS. It must be noted that large number of individuals have been used in conifers to perform genomic evaluation, but these studies relied on exome or targeted sequencing constrained by the enormous and complex nuclear genome of these species. In 2016, Suren and collaborators used 579 interior spruce samples and 631 lodgepole pine samples to identify 10M SNPs and insertions/deletions (INDELs) in each species using exome capture (Suren et al., 2016).
The goal of the present study was to characterize the genomic diversity of P. trichocarpa individuals across its geographic range. The specific objective was to identifying low frequency genetic variants with high confidence that could be used in GWAS including both common and rare genetic variants. We present here a detailed evaluation of small genetic variants using strict quality filtering and comparison between two variant callers. In addition, we provide functional information obtained from the annotation of the discovered genetic variants. Finally, we performed a Gene Ontology (GO) enrichment of genes in which nonsense variants were found. This is the first study in a plant species aiming at rare allele discovery using a large sampling size from whole genome sequencing (over 1,000 individuals).
Experimental Procedures
P. trichocarpa Sequencing Reads
A total of 1,038 unique P. trichocarpa individuals were sequenced by the US DOE's BESC (Xie et al., 2009; Slavov et al., 2012). These individuals were sampled across most of P. trichocarpa's geographic range in California, Oregon, and Washington, USA, as well as in British Columbia, Canada ( Figure 1 ). These 1,038 P. trichocarpa accessions were retrieved online in fall 2017 from the Joint Genome Institute Genome Portal (https://genome.jgi.doe.gov/portal/) in the form of raw sequencing read files. Whole genome sequencing (WGS) were performed using short paired end reads (100 bp) on an Illumina HiSeq 2000 platform. The sampled individuals were checked for hybrids status after variant discovery based on comparison with closely related species in Principal Component Analysis (PCA; see Results).
Sequencing Reads Quality Filtering
Rare genetic variants and sequencing errors are both found at low frequencies in raw sequencing reads. To differentiate between true genetic variants and sequencing errors, we set stringent quality control on the raw read files. All bioinformatics manipulations were performed on Cedar and Graham computing servers from Compute Canada and on Katak and Manitou computing servers at the Institute of Integrative Biology and Systems, Université Laval (Quebec, Canada). First, we trimmed low-quality reads and sequencing adapters using Trimmomatic (Bolger et al., 2014) ( Figure 2 ). Only bases having a Phred quality score higher than 27 (two chances out of 1,000 that the base is a sequencing error) were kept for further analyses. In addition, reads presenting a mean base quality score below 27 and/or shorter than 50 bases were discarded. The high quality of the cleaned read files was then ensured using FastQC (Andrew, 2010) before the alignment and variant calling steps. The mean number of paired reads per accession was about 66M (range: 24 to 321M) after quality filtering.
Figure 2.
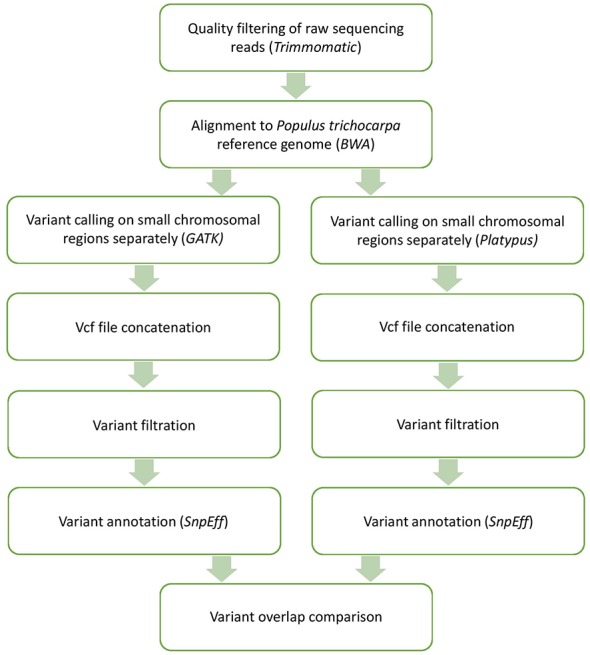
Methodology followed to obtain annotated filtered genetic variants from raw sequencing reads. Software used during each step are in parentheses.
Sequence Alignments
After the quality control steps, each individual accession was align to the reference genome of P. trichocarpa version 3.0 using the Burrow Wheeler Aligner (BWA; Li and Durbin, 2009) ( Figure 2 ). We used the BWA-MEM algorithm that uses seedling alignments with maximal exact matches (MEMs) and then extending seeds with the affine-gap Smith-Waterman algorithm. Twenty-one genomes with average coverage lower than 5X were discarded in order to retain high confidence alignments. Ultimately, 1,017 alignments corresponding to the same number of unique individuals were used in the following analyses.
Variant Calls From Two Different Software Pipelines
In order to obtain high confidence genetic variants, we used two types of variant calling software for result comparison ( Figure 2 ): Platypus version 0.8.1 (Rimmer et al., 2014) and HaplotypeCaller from the Genome Analysis Tool Kit version 3.8 (GATK; DePristo et al., 2011; Poplin et al., 2017). These two variant calling software are widely used for variant discovery therefore facilitating data reproducibility. They also perform well in terms of sensitivity and precision of discovered variants while being computationally efficient thanks to the implementation of multithreading (Sandmann et al., 2017). Platypus enables the user to apply numerous quality filters during variant discovery, while GATK offers a filtering tool for use after variant discovery.
Platypus was used to perform single nucleotide variants (SNV) and INDEL calling on the 1,017 alignment files. As suggested by Platypus default parameter, bases with quality scores below 20 and reads with mapping quality below 20 were ignored during variant calling. The following custom parameters have been used to address rare variant calling: 1) only variants supported by at least 10 reads were considered; 2) reads having less than 40 bases with a quality lower than 20 were discarded; 3); variants where the median minimum quality in a window of 20 nucleotides around the variant fell below 20 were labelled as “bad reads”.
HaplotypeCaller was also used to perform SNV and INDEL calling on the 1,017 alignment files. The filtering tool VariantFiltration from GATK (DePristo et al., 2011) allowed us to apply quality filters to variants discovered by HaplotypeCaller. Parameters for filtering SNPs were set according to GATK recommendations for hard filtering. Variants were filtered out when: 1) their quality divided by nucleotide site depth was lower than 2; 2) they were located on a read with an approximate depth lower than 10; 3) their root mean square mapping quality was lower than 40; 4) their phred-scaled p-value using Fisher's exact test was greater than 60; their symmetric odds ratio of 2x2 contingency table to detect strand bias was greater than 3; their Z-score from Wilcoxon rank sum test of alternative vs. reference read mapping qualities was lower than -8; their z-score from Wilcoxon rank sum test of alternative vs. reference read position bias was lower than -12.5. In addition to the recommended parameters for hard-filtering, variants were filtered out if not supported by at least 10 reads.
Using custom python scripts, we filtered out vcf files obtained by Platypus and HaplotypeCaller. More precisely, variants that were attributed the “bad reads” flag were discarded from the vcf files obtained by the two types of software. Retained variants were therefore validated by each quality criteria settled during the variant calling phase. Additionally, only INDELs smaller than four nucleotides were included.
Parallelization
Given the large size of our data set, we took advantage of task parallelization in order to minimize computation time for the analyses of the two variant calling software ( Figure 3 ). Both Platypus version 0.8.1 and GATK's HaplotypeCaller version 3.8 allow task parallelization within the software, using multiprocessing for Platypus and multithreading for HaplotypeCaller. In addition, we used task parallelization outside the software using a scatter-gathering approach. With this method, large files are divided into smaller regions (scattering) analyzed in parallel, then, the results are collected and merged together (gathering). Both approaches are based on task parallelization, but multiple tasks are run within the software using multiprocessing and multithreading, whereas task parallelization is done by the user and happens outside the software for the scatter-gathering approach. A combination of these two approaches was used in order to minimize both analysis and queue time on calculation servers. Portions of the genome were analyzed in parallel (scatter-gathering) while multiple tasks were also running in parallel on each portion (multiprocessing, multithreading). Using Platypus, we ran the analysis on each chromosome separately, while we had to divide the analyses on smaller chromosomal regions using HaplotypeCaller. The computing resources used for each analysis varied considerably according to the studied chromosomal regions.
Figure 3.

Illustration of different task parallelization approaches. (A) Simplest approach with no task parallelization. (B) Scatter-gathering approach, where task parallelization is done by the user and happens outside the software. (C) Multithreading/multiprocessing approaches, where task parallelization is done by and happens within the software. (D) Combination of multithreading/multiprocessing and scatter-gathering, where task parallelization happens outside and within the software. Yellow backgrounds highlight the multithreading and multiprocessing parallelization. Blue backgrounds highlight the scatter-gathering parallelization.
Variant Annotations
Filtered variants discovered by the two variant callers were annotated using SnpEff (Cingolani et al., 2012). To annotate variants based on the same reference genome used during read alignment we built a custom SnpEff database of the annotated genome of P. trichocarpa version 3.0.
GO Enrichment
Based on variants recovered by the two variant calling software, we performed a gene ontology (GO) enrichment test using PANTHER version 14.1 (Mi et al., 2019). We retrieved the names of P. trichocarpa genes in which stop-gained genetic variants were found. Stop-gained variants can have drastic impact on phenotypes and have been found to affect wood composition in poplars (Muchero et al., 2015). We tested whether stop-gained variants are enriched in specific gene functions with respect to biological processes. PANTHER version 14.1 does not include P. trichocarpa annotations; therefore, we retrieved names of the closest Arabidopsis thaliana genes from P. trichocarpa genes possessing this type of nonsense variants. The closest Arabidopsis genes were determined during the annotation of the P. trichocarpa reference genome v3.1 by aligning A. thaliana TAIR10 proteins to the P. trichocarpa genome (the detailed procedure is available on the P. trichocarpa v3.1 Phytozome page). The closest A. thaliana gene can be found in the gene annotation file of the P. trichocarpa reference genome v3.1 (available on the JGI Genome Portal). This information is available for 84% of the P. trichocarpa genes. We used the PANTHER classification system to perform a statistical overrepresentation test in GO biological processes, using a Fisher's exact test with the names of A. thaliana genes most similar to the targeted P. trichocarpa genes. Fisher's exact test was used rather than the binomial test because the former assumes a hypergeometric distribution, which is more accurate for smaller gene lists. Finally, we applied False Discovery Rate (FDR) correction to the obtained p-values. FDR correction was designed to control the false positive rate in the statistical test results and is generally considered a better choice than Bonferroni correction in enrichment analysis (Mi et al., 2019).
Results
Variant Calling From Platypus and HaplotypeCaller
Before filtering, 31,607,230 genetic variants were identified by Platypus in our data set of 1,017 P. trichocarpa individuals. After filtering by quality and variant size, this number reduced to 15,734,785 variants, no longer than three consecutive nucleotides and distributed across 14,539,625 polymorphic sites. The majority of these variants (64%) showed a frequency in the population lower than 0.05 ( Figure 4 ), i.e. found in less than 51 individuals.
Figure 4.
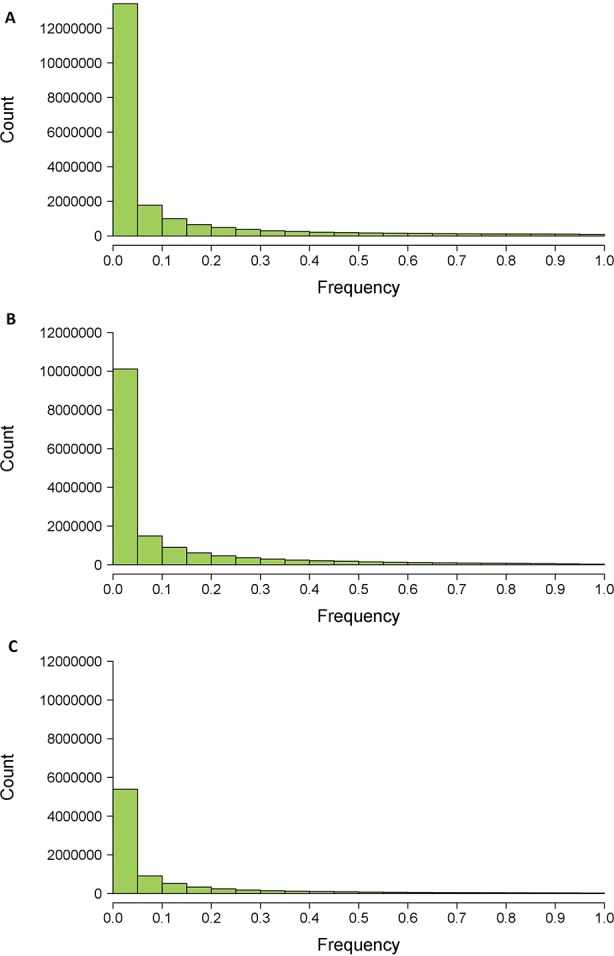
Histograms of variant frequencies after filtering from HaplotypeCaller (A), Platypus (B) and consensus data set (C) between both software.
Before filtering, 35,597,076 genetic variants have been identified by HaplotypeCaller across the 1,017 P. trichocarpa individuals. After filtering by quality and variant size, this number reduced to 19,971,499, no longer than three consecutive nucleotides and distributed across 19,478,954 polymorphic sites. Most of these variants (66%) had a frequency lower than 0.05 in the population ( Figure 4 ).
Variant Annotation
We used SnpEff to annotate genetic variants discovered by Platypus and HaplotypeCaller. Variant annotation uses information from reference genome annotations to describe genetic variants, such as the variants' inter- or intra-genic locations, and for variants located inside gene, the respective gene name and the effect of the variant on the entire nucleotide or gene coding sequence. We must note that the total number of variant annotation greatly exceed the total number of genetic variants. The reason is that some variants belong to more than one gene (i.e. overlapping genes) and here we report annotations for the effect of variants on each gene they belong to because the same variant can have different effects on different genes. On the contrary, we refer to the total number of genetic variants as the total number of nucleotide variation in the genome.
After annotation of variants discovered by Platypus, we found that most of the variants (86%) were located outside of genes, with nearly 11M variants found in intergenic regions and 8.1M and 7.8M variants found in upstream and downstream gene regions, respectively ( Table 1 ). Upstream and downstream regions correspond to 5-kb-long regions around genes in SnpEff default parameter. The remaining variants (about 4.3M) were located in genic regions, with more than 633K non-synonymous variants ( Table 1 ), accounting for 2% of the total.
Table 1.
Annotations obtained from variant calling by Platypus and HaplotypeCaller.
| Annotations | Current study | |||
|---|---|---|---|---|
| 1,014 (1,017) individuals | ||||
| Consensus | Platypus | HaplotypeCaller | ||
| Polymorphic sites | 7,313,551 | (8,368,838) | 14,539,625 | 19,478,954 |
| Total | 7,441,340 | (8,497,509) | 15,734,785 | 19,971,499 |
| intergenic varianta | 5,254,503 | (5,645,996) | 10,886,077 | 15,149,344 |
| downstream gene variantb | 3,955,249 | (4,607,452) | 7,883,178 | 11,059,573 |
| upstream gene variantb | 3,955,094 | (4,478,850) | 8,086,954 | 11,413,237 |
| intron variantc | 1,341,551 | (1,762,003) | 2,427,258 | 3,463,071 |
| missense variantd* | 333,036 | (418,974) | 559,277 | 787,053 |
| 3 prime UTR variante | 269,591 | (345,432) | 484,519 | 672,092 |
| synonymous variantf | 231,894 | (324,970) | 410,776 | 554,853 |
| 5 prime UTR variante | 136,098 | (175,634) | 245,084 | 349,285 |
| splice region variantg | 54,271 | (71,655) | 95,479 | 128,316 |
| 5 prime UTR premature start gainh* | 19,099 | (24,989) | 32,639 | 45,849 |
| frameshift varianti* | 9,766 | (11,103) | 16,937 | 31,172 |
| stop gainedj* | 8,365 | (9,226) | 12,967 | 20,146 |
| splice donor variantk* | 2,694 | (3,208) | 4,387 | 6,237 |
| splice acceptor variantk* | 2,284 | (2,689) | 3,807 | 5,315 |
| stop lostl* | 1,082 | (1,335) | 1,994 | 2,612 |
| start lostm* | 821 | (981) | 1,511 | 2,123 |
| stop retained variantn | 535 | (695) | 925 | 1,246 |
| initiator codon variant° | 115 | (142) | 215 | 280 |
| non_coding_transcript_variantp | 66 | (70) | 368 | 221 |
| intragenic_variantq | 2 | (5) | 13 | 21 |
| exon loss variantr | 2 | (3) | 3 | 3 |
| 5 prime UTR truncations | 2 | (2) | 2 | 2 |
| non canonical start codont | 1 | (1) | 2 | 2 |
| 3 prime UTR truncations | 0 | (1) | 1 | 1 |
For the consensus data set, numbers in brackets indicate the number of variants before suspected hybrids removal, while the number outside the brackets indicates the number of variants after suspected hybrids were already removed.*Non-synonymous variants corresponding to genetic variants inside coding regions altering the amino acid sequence of a protein and identified in both caller analyses. aIntergenic variant: located in intergenic regions and outside upstream and downstream gene regions. bUpstream and downstream variant: located in 5kb regions before and after a gene, respectively. cIntron variant: located in non-translated introns of genes. dMissense variant: located inside coding regions and resulting in an amino acid change. e5 and 3 prime UTR variant: located in 5′ and 3′ untranslated region of a gene, respectively. fSynonymous variant: located inside coding regions and not resulting in an amino acid change. gSplice region variant: located within the region of the splice site. h5 prime UTR premature start gain: resulting in an initiator codon inside the 5′ untranslated region. iFrameshift variant: resulting in a reading frame change, because the number of nucleotides inserted or deleted is not a multiple of three. jStop gained: resulting in a premature stop codon in the coding sequence. kSplice donor and acceptor variant: changing the 2 nucleotide regions at the 5′ and 3′ end of an intron, respectively. lStop lost: resulting in an elongated gene product because of stop codon loss. mStart lost: resulting in initiator codon loss. nStop retained variant: change in one base in the terminator codon, but the terminator remains. °Initiator codon variant: change in at least one base of the first codon of a transcript. pNon-coding transcript variant: located in a non-coding RNA gene. qIntragenic variant: occurs within a gene but falls outside of all transcript features. rExon loss variant: resulting in the loss of an exon from a transcript. s5 and 3 prime UTR truncation: causing the reduction of the 5′ and 3′ untranslated region, respectively. tNon-canonical start codon: a start codon that is not the usual AUG sequence. The total number of variant annotations does not equal the total number of variants. The reason is that some variants are part of several overlapping genes and may have different effect on different genes.
For annotation of variants discovered by HaplotypeCaller, we found that most of the variants were located outside of genes, with 15M variants found in intergenic regions and 11.4M and 11M variants found in upstream and downstream gene regions, respectively ( Table 1 ), accounting for 86.1% of the total. The remaining variants (about 6.1M) were located in genic regions, with nearly 901K non-synonymous variants ( Table 1 ), accounting for 2.1% of the total.
Variant Calling Overlap
In order to add a further quality criterion to the filtering process of genetic variants, we retained only variants recovered by the two variant calling software used in this study (i.e. Platypus and Haplotype Caller). We used the isec command from bcftools to find common variants between the two vcf files leading to a consensus variant set (Li, 2011). As a result, 8.5M genetic variants were recovered by both variant calling software, distributed across 8.4M polymorphic sites ( Figure 5 ).
Figure 5.
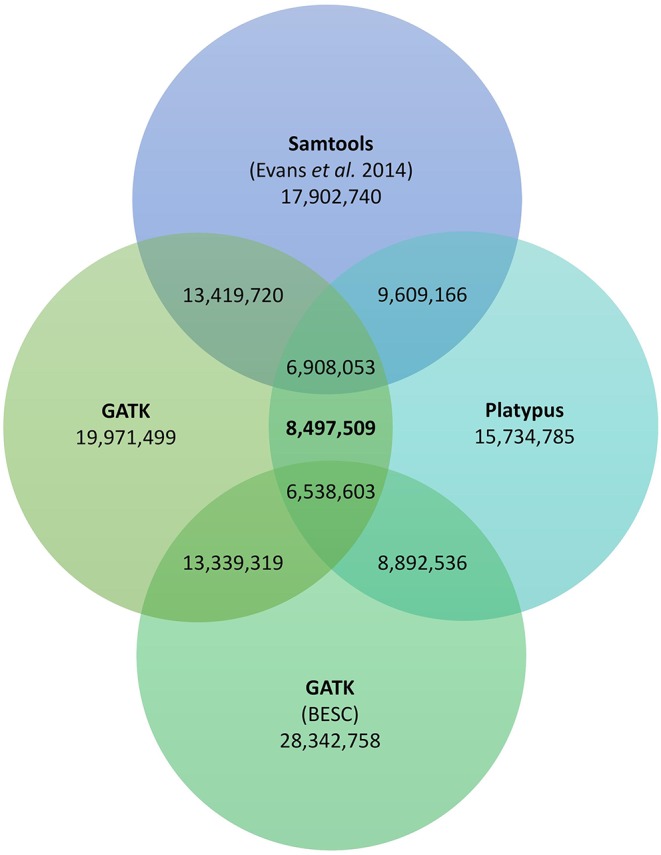
Venn diagram of the number of genetic variants identified in this study in addition to variants identified by Evans and colleagues (Evans et al., 2014) and variants in the BESC data set (https://bioenergycenter.org/besc/gwas/). We used Platypus and GATK's HaplotypeCaller with 1,017 WGS of P. trichocarpa individuals. Evans et al., 2014 used samtools and bcftools conjointly and 544 WGS of P. trichocarpa individuals. The BESC data set was called using multiple tools from GATK and 882 WGS of P. trichocarpa individuals. The number of variants identified by more than one software are indicated where circles overlap. We note here that some of the individuals used in Evans et al. and the BESC data set are the same as the 1,017 used in the present study.
Most of the variants were located outside of genes, with 5.7M variants found in intergenic regions and 4.6M and 4.5M variants found in downstream and upstream gene regions, respectively ( Table 1 ), accounting for 82.4% of the total. The remaining variants (3.2M) were located in genic regions, with nearly 473K non-synonymous variants ( Table 1 ), accounting for 2.6% of the total. Missense variants (419K) accounted for 2.3% of the total and nonsense variants (54K) for 0.3% of the total.
We found 45% more non-synonymous variants compared to synonymous variants. Furthermore, among these non-synonymous variants, missense variants even exceeded synonymous variants by 29%. The total number of genetic variants was only 1.5% higher than the number of polymorphic sites.
To explore result disparities between genomic evaluation studies that used different methods and data set sizes, we also identified the genetic variants commonly found between our study and two other genomic evaluation studies on P. trichocarpa (Evans et al., 2014; https://bioenergycenter.org/besc/gwas/). When comparing our results with the study of Evans and collaborators that used 544 P. trichocarpa individuals and Samtools as a variant caller we found that 81% (6,908,053) of the variants they identified are also present in our consensus data set. When comparing our results with the BESC data set that used 882 P. trichocarpa individuals and tools from GATK we found that 77% (6,538,603) of the variants they identified are also present in our consensus data set ( Figure 5 ). Interestingly, we found less variants in common with the study using 882 individuals comparing to the study using only 544 individuals. Details regarding individual SNP sets from the two variant callers overlap with the SNPs from Evans et al. and the BESC data set and a summary indicating which variants occur within each SNP set are provided in the Supplement ( Tables S1 and S2 ).
Hybrid Identification
In order to identify potential hybrids in our data set, we also identified the genetic variation across two Populus balsamifera, two Populus deltoides, one Populus angustifolia, and one Populus fremontii individuals for comparison. These species are closely related to P. trichocarpa and co-occur naturally in some parts of its natural range (Wang et al., 2019). These four species therefore hybridize naturally with P. trichocarpa. Raw WGS reads were downloaded from the JGI Genome Portal for P. balsamifera and P. deltoides (https://genome.jgi.doe.gov/portal/) and from the Genome Sequence Archive of the BIG Data Center for P. angustifolia and P. fremontii (https://bigd.big.ac.cn/gsa/, accession number CRA001510). Genetic variants from this six genomes were identified with the same bioinformatic pipeline used for P. trichocarpa individuals (see Experimental procedures). We used visual identification from a PCA to identify potential P. trichcocarpa hybrids. To do so, we filtrated the genetic variants identified across the genomes of these four species and the consensus variant set identified in this study using plink (–geno 0.01 –maf 0.1 –hwe 0.01 –LD 50 10 0.1) (Purcell et al., 2007). This filtration step yielded 12,001 variants with which we performed the PCA using plink (–pca 2) ( Figure 6 ).
Figure 6.

Principal component analysis showing the first two principal components of the genetic variation found across 1,017 P. trichocarpa, two P. balsamifera, two P. deltoides, one P. angustifolia, and one P. fremontii individual genomes retrieved from various databases for comparison. Note Nisqually-1 (Tuskan et al., 2006) in the upper right corner used as the overall reference.
The fractions of the genetic variation explained by the two PCAs were 22% and 16%, respectively. Graphical representation of Principal Components (PC) 1 and 2 ( Figure 6 ) clearly separated P. balsamifera, P. deltoides, P. angustifolia and P. fremontii from the 1,017 individuals of our consensus variant set. Every individual from the Tahoe population was slightly separated from the core of P. trichocarpa individuals. The Tahoe population is the southernmost population of our data set, geographically quite distant from the other P. trichcocarpa populations. This suggests that individuals from the Tahoe population differ genetically from other P. trichocarpa populations because of geographic distance and not because of hybridization with other Populus species. One individual each, from the Skagit, Vancouver Island and Willamette populations, respectively, were located halfway between the core of P. trichocarpa individuals and other Populus species, strongly suggesting that these individuals may be hybrids or introgressed. These three individuals were therefore removed from the consensus variant set, hence lowering the number of P. trichocarpa individuals to 1,014.
Biological Pathways Overrepresented Among Functionally Defective Alleles
Among the consensus variants from Platypus and GATK variant calling (based on the finalized 1,014 individuals data set), we found that 8,365 stop-gained variants were distributed in 6,327 P. trichocarpa genes. These genes corresponded to 3,829 synonymous genes for A. thaliana. Analyses of gene function classification in PANTHER show that the set of genes containing stop-gained variants was enriched in 106 GO terms with respect to biological processes. Interestingly, multiple GO terms related to wood properties such as cell wall polysaccharide metabolism, cellulose biosynthesis, phenylpropanoid metabolism and plant-type cell wall biogenesis are enriched in genes possessing stop-gained variants ( Table 2 ).
Table 2.
Results from the Gene ontology (GO) enrichment test performed with PANTHER are presented using a list of A. thaliana genes closest to the P. trichocarpa genes and related to wood formation and in which stop-gained variants where found querying 1,014 black cottonwood individuals.
| GO biological complete | List of genes with stop-gained variants | |||||
|---|---|---|---|---|---|---|
| # | Expected | Enrichment | +/− | raw P-value | FDR | |
| cellulose biosynthetic process | 20 | 6.66 | 3 | + | 1.19E-04 | 8.89E-03 |
| beta-glucan biosynthetic process | 22 | 8.33 | 2.64 | + | 2.69E-04 | 1.82E-02 |
| glucan biosynthetic process | 36 | 15.69 | 2.29 | + | 4.52E-05 | 4.28E-03 |
| cellular polysaccharide biosynthetic process | 49 | 23.6 | 2.08 | + | 2.56E-05 | 2.55E-03 |
| macromolecule metabolic process | 1092 | 792.57 | 1.38 | + | 1.15E-26 | 1.14E-23 |
| organic substance metabolic process | 1556 | 1145.6 | 1.36 | + | 1.82E-39 | 5.41E-36 |
| metabolic process | 1798 | 1334.13 | 1.35 | + | 5.28E-47 | 3.15E-43 |
| organic substance biosynthetic process | 452 | 370.95 | 1.22 | + | 6.11E-05 | 5.13E-03 |
| biosynthetic process | 475 | 393.99 | 1.21 | + | 9.41E-05 | 7.69E-03 |
| cellular biosynthetic process | 441 | 363.73 | 1.21 | + | 1.09E-04 | 8.44E-03 |
| cellular metabolic process | 1487 | 1109.93 | 1.34 | + | 3.52E-34 | 4.20E-31 |
| cellular process | 2021 | 1610.26 | 1.26 | + | 8.40E-36 | 1.67E-32 |
| cellular macromolecule metabolic process | 823 | 606.54 | 1.36 | + | 1.10E-17 | 7.31E-15 |
| cellular polysaccharide metabolic process | 73 | 39.57 | 1.85 | + | 9.51E-06 | 1.07E-03 |
| polysaccharide metabolic process | 93 | 60.67 | 1.53 | + | 2.86E-04 | 1.91E-02 |
| carbohydrate metabolic process | 188 | 138.27 | 1.36 | + | 1.38E-04 | 1.02E-02 |
| primary metabolic process | 1430 | 1049.26 | 1.36 | + | 9.69E-36 | 1.45E-32 |
| cellular carbohydrate metabolic process | 92 | 56.64 | 1.62 | + | 5.83E-05 | 5.04E-03 |
| polysaccharide biosynthetic process | 51 | 28.6 | 1.78 | + | 5.06E-04 | 2.99E-02 |
| cellular carbohydrate biosynthetic process | 53 | 28.74 | 1.84 | + | 1.77E-04 | 1.27E-02 |
| cellular glucan metabolic process | 56 | 29.57 | 1.89 | + | 5.06E-05 | 4.51E-03 |
| glucan metabolic process | 56 | 29.57 | 1.89 | + | 5.06E-05 | 4.44E-03 |
| cell wall polysaccharide metabolic process | 39 | 19.16 | 2.04 | + | 1.99E-04 | 1.38E-02 |
| plant-type cell wall biogenesis | 41 | 20.41 | 2.01 | + | 1.89E-04 | 1.33E-02 |
| cell wall biogenesis | 56 | 31.93 | 1.75 | + | 3.56E-04 | 2.24E-02 |
| cellular component biogenesis | 228 | 171.45 | 1.33 | + | 8.33E-05 | 6.90E-03 |
| cellular component organization or biogenesis | 547 | 426.48 | 1.28 | + | 2.54E-08 | 4.59E-06 |
| phenylpropanoid metabolic process | 34 | 17.08 | 1.99 | + | 8.21E-04 | 4.62E-02 |
| organic cyclic compound metabolic process | 548 | 369.84 | 1.48 | + | 1.69E-17 | 1.01E-14 |
| cellular aromatic compound metabolic process | 518 | 355.81 | 1.46 | + | 2.52E-15 | 1.37E-12 |
Results are sorted hierarchically to better understand the hierarchical relations between over-represented functional classes. We provide for each GO term (up to seven levels): the number of genes present within the analyzed list (#), the expected number of genes under no GO enrichment (Expected), the enrichment value (Enrichment), the sign of the enrichment (+/−), the P-value associated with the enrichment test without multiple testing correction (raw P-value) and multiple testing corrected using False Discovery Rate (FDR).
Discussion
Tool comparison for genomic variant calling has become the standard when using Next Generation Sequencing in clinical diagnostics (Sandmann et al., 2017, e.g.). To our knowledge, this approach has never been used in plant sciences when performing a large scale genomic diversity evaluation using WGS. In our study, we evaluated the genomic diversity across 1,017 individuals of P. trichocarpa in the form of small genetic variation using an existing set of whole genome sequences. Our goal was to identify rare and common genetic variation in the form of SNPs and small INDELs for subsequent use in GWAS. Using stringent filtering steps and variant calling comparison between two software we identified a set of high confidence genetic variants.
Performance Comparison Between Platypus and HaplotypeCaller
Our data set was computationally heavy with more than one thousand P. trichocarpa genomes (~450 Mbp). For this reason, we opted to use variant calling software enabling multithreading to speed up variant identification analyses. HaplotypeCaller from GATK version 3.8 was considerably slower at identifying variants compared to Platypus version 0.8.1. Multithreading for current versions of GATK (version 4) is still under development and not safe for production work, therefore, we used a previous version of GATK (version 3.8). Both software identify variants based on haplotype reconstruction while Platypus also integrates a Bayesian statistical framework for variant discovery. The two software ran on the same data set, but the number of variants identified between each software differed substantially. HaplotypeCaller identified 27% more variants in comparison to Platypus. This discrepancy in the number of variants identified by the two variant calling software highlight the importance of result comparison between variant callers.
Bioinformatic Approaches
We used a scatter-gathering approach coupled to multithreading to perform variant calling on smaller parts of the data separately. We either conducted the variant calling on chromosomes or smaller chromosomal regions separately to reach acceptable running time and computing resource use. This approach allowed us to identify variants across more than one thousand complete genomes of P. trichocarpa within reasonable time. An approach based on a single thread would not have permitted to reach our goal with current computing technologies. The combination of multithreading and scatter-gathering proved very efficient for variant discovery on a large data set.
Consensus Variant Set
Nearly 8.5M genetic variants were identified by the two software and represent high confidence genetic variation. The vast majority of the identified variants had a frequency lower than 0.05 in our data set. Our results are in close agreement with other genetic diversity evaluation studies of P. trichocarpa or closely related species (Evans et al., 2014; Fahrenkrog et al., 2017) and is expected in outcrossing, wide ranging, and undomesticated tree populations (Petit and Hampe, 2006; Fahrenkrog et al., 2017). Most genetic variants are located outside the gene space where nucleotide substitutions are expected to have lower effect on the phenotype and therefore are less subject to purifying selection.
Non-Synonymous/Synonymous Variant Ratio
More surprisingly, inside coding regions, non-synonymous genetic variants were more numerous than synonymous mutations. This pattern has already been observed in a similar study on P. trichocarpa (Evans et al., 2014). Given their higher impact on protein sequence, purifying selection is expected to be stronger on non-synonymous variants compared to synonymous ones. A positive ratio of non-synonymous to synonymous genetic substitutions is associated with positive selection (Yang and Bielawski, 2000). P. trichocarpa is wide-ranging across the west coast of North America and across a large latitudinal gradient from Alaska to southern California. Individuals in our data set were collected across most of P. trichocarpa's range. Consequently, individuals in this study adapted to different environmental conditions and likely exhibit high genetic diversity in response to local adaptation (Evans et al., 2014). Populations genomic studies are needed to evaluate selection pressures and especially adaption acting across P. trichocarpa geographic range.
Comparison With Other Genomic Evaluations on Poplars
Previous studies evaluated the genomic diversity in P. trichocarpa (Evans et al., 2014; BESC SNP data set: https://bioenergycenter.org/besc/gwas/) and Populus deltoides ( Fahrenkrog et al., 2017). Fahrenkrog and colleagues (2017) used targeted resequencing and variant calling overlap between three different software to identified 358K SNPs in 391 unrelated individuals of P. deltoides, which is much lower than the 8.5M variants we found. Their final data set included variants found in a subset of genes, thus reducing the size of the analyzed genome. Intergenic variants were also excluded while most genetic variations are usually found in intergenic regions. Moreover, the variant calling comparison between three different software further decreased the number of identified variants. This approach resulted in a set of high confidence rare and common genetic variants, although less numerous than for studies based on WGS.Using 544 WGS of P. trichocarpa individuals, Evans and colleagues (2014) identified 17M SNPs using one variant caller. This number is more than two times higher than the 8.5M variants we identified using comparison between two variant calling software and stringent quality criteria on two times the number of individuals. Evans and colleagues performed no variant filtration, however, and found that stringent filtering had minimal impact on the sensitivity of known SNP discovery while reducing substantially the number of known SNPs passing the filtering threshold (i.e. specificity). For the targeted identification of rare genetic variants and for sequencing data with low to moderate sequencing depth we believe that variant filtration is highly beneficial. The DOE's BESC also released a SNP data set (a description of how the SNPs were called is available in the method section of the following study: Weighill et al., 2018). This data set included 28M variants identified across 882 WGS of P. trichocarpa. Genetic variants were called using GATK tools. First, variants were called independently for each individual using HaplotypeCaller and merged afterward. Biallellic SNPs were then extracted and filtered using the VariantQualityScoreRecalibration (VQSR) tool. This latter tool uses machine learning to filter variants using a set of known genetic variants (see Weighill et al., 2018 for more information). Similarly, to Evans and colleagues, the BESC data set identified a lot more genetic variants than our study using less individuals. The number of identified variants seems to increase when using only one variant caller. On the contrary, using variant caller comparison the number of individuals scanned does not seem to increase the number of identified variants. Indeed, the number of common variants between our study and the 882 individual data set is slightly lower than the number of common variants between our study and the 544 individuals data set ( Figure 5 ). The number of commonly identified variants can even be greater between two different variant callers than between the same variant caller, i.e. HaplotypeCaller. These observations show that the use of a certain variant caller is not the main factor determining which variants will be identified, instead parameters used during variant discovery and for filtering along with the comparison between variant caller seem to be of considerable importance.
Quality Filtering and Variant Caller Comparison
Application of stringent filtering criteria before and after variant discovery and the result overlap between variant calling software are key factors for genomic diversity evaluation. With current sequencing technologies and variant calling algorithms, a balance must be found between sensitivity and specificity of variant discovery. Increasing severity in quality filters and increasing the number of variant calling software tend to increase the quality of the identified variants while decreasing the total number of variants. Therefore, the goal of genomic diversity evaluation studies must be clearly stated to ensure that optimal parameters for variant identification are used. Common genetic variants can be identified easily with high confidence without using strict quality filters or comparison between variant calling software. On the contrary, rare genetic variants are difficult to identify with high confidence and require strict quality filtering and overlap between results from various variant calling softwares for reliable identification. When identifying both common and rare genetic variants, as in this study, confidence in the identified variants should be prioritized.
Predicting Models for Increased Specificity
When high quality sets of genetic variants are already available as in model species, one can build models to better detect true and false genetic variants using sets of known genetic variants to increase the specificity of variant identification [e.g. VQSR from GATK (McKenna et al., 2010)]. Although these models are very useful for human and some other model species, they do not apply to every study. Large sequence data sets such as WGS and Whole Exome Sequencing (WES) and high quality sets of known genetic variants must be used in order to build accurate predicting models. WGS and WES are now widely used in P. trichocarpa and high quality sets of known common genetic variants are available. Known high quality sets of rare genetic variants, however, are scarce or even non-existent when considering both genic and intergenic regions. Consequently, we did not use such models to increase the specificity of our variant discovery. The consensus set of 8.5M genetic variants, common and rare, identified in this study will be available as a high quality set of known variants to build models aiming at increasing variant specificity in future genomic diversity evaluations of P. trichocarpa and closely related species.
GO Enrichment
We used a GO enrichment test to identify biological pathways overrepresented with genes containing stop-gained genetic variants. A multitude of biological process were overrepresented with genes containing stop-gained variants. Among them, biological processes related to wood properties, and especially secondary cell-wall polysaccharides are of great interest. Previous studies already highlighted the role of functional variants (premature or abolished stop codon, altered start codon, frameshift variant or alternative splice sites) on genes involved in lignin biosynthesis (MacKay et al., 1997; Thumma et al., 2005; Vanholme et al., 2013; Muchero et al., 2015). The lignin and other secondary cell-wall polymers (i.e. cellulose and hemicellulose) biosynthesis pathways may be largely affected by functional variants. Thus, these overrepresented biopathways will help us select candidate genes for further analyses. Recent functional mutations are expected to show greater effects on a phenotype, since such functional allelic variants have not undergone selection to much extent. For example, we detected a stop gain mutation at 2.3% minor allele frequency and with 4.3% carriers in the population for the poplar orthologue of the Arabidopsis irx10 gene (aka PtrGUT2B; Potri.001G068100). Its protein is known to be implicated in xylan backbone formation, and thus a prime target for improving cell wall traits (Porth et al., 2018). Therefore, such variants are important candidates for the purpose of rare variant association studies and ultimately, selective breeding with rare defective alleles (Vanholme et al., 2013; Porth and El-Kassaby, 2015).
Conclusion
We identified 8.5M small genetic variants, common and rare, across more than one thousand P. trichocarpa individuals sampled throughout the species' range. Use of appropriate quality filtering and variant comparison between two variant callers resulted in high-quality sets of genetic variants. With a data set of 1,017 complete genomes, this is the first time that a genomic diversity evaluation of this magnitude has been conducted in P. trichocarpa and, to our knowledge, in any tree species. The high-quality set of known genetic variants identified will be directly available to support other genomic diversity evaluations of P. trichocarpa and other closely related species. Moreover, GWAS including rare and common genetic variants will be conducted using those high-quality variants. Thus, starting out from a wealth of genetic variants uncovered in the present study, we will be able to further narrow down the set of important variants for poplar selective breeding.
Data Availability Statement
Raw sequence data are available under https://phytozome.jgi.doe.gov/pz/portal.html.
Author Contributions
AP obtained data, analyzed all data, and wrote the manuscript. JP supported data analysis. NI, JK, and YE-K provided valuable insights on poplar genomics. JA obtained co-funding. IP designed the study, obtained funding, and helped in drafting the manuscript. All authors read and approved the manuscript.
Conflict of Interest
Author JK was employed by the company New Zealand Forest Research Institute Limited (Scion).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LZ and handling Editor declared their shared affiliation.
Acknowledgments
This research was enabled in part by support provided by Calcul Québec (www.calculquebec.ca) and Compute Canada (www.computecanada.ca). We also acknowledge funding from NSERC Discovery Grants to IP (RGPIN/04748-2017) and JA (RGPIN/05967-2016), respectively, to support this study. WGS data were produced by the US Department of Energy Joint Genome Institute https://www.jgi.doe.gov/in collaboration with the user community.
Support for the poplar GWAS data set was provided by the U.S. Department of Energy, Office of Science Biological and Environmental Research (BER) via the Bioenergy Science Center (BESC) under Contract No. DE-PS02-06ER64304. The poplar GWAS project used resources from the Oak Ridge Leadership Computing Facility and the Compute and Data Environment for Science at Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01384/full#supplementary-material
References
- Andrew (2010). FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. [Google Scholar]
- Baes C. F., Dolezal M. A., Koltes J. E., Bapst B., Fritz-Waters E., Jansen S., et al. (2014). Evaluation of variant identification methods for whole genome sequencing data in dairy cattle. BMC Genomics 15, 948. 10.1186/1471-2164-15-948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachi B., Morris G. P., Borevitz J. O. (2011). Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 12 (10), 232. 10.1186/gb-2011-12-10-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P., Platts A., Wang L. L., Coon M., Nguyen T., Wang L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Aust.) 6, 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. C., Kiss R. S., Pertsemlidis A., Marcel Y. L., McPherson R., Hobbs H. H. (2004). Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305, 869–872. 10.1126/science.1099870 [DOI] [PubMed] [Google Scholar]
- DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans L. M., Slavov G. T., Rodgers-Melnick E., Martin J., Ranjan P., Muchero W., et al. (2014). Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat. Genet. 46, 1089–1096. 10.1038/ng.3075 [DOI] [PubMed] [Google Scholar]
- Fahrenkrog A. M., Neves L. G., Resende M. F. R., Vazquez A. I., de los Campos G., Dervinis C., et al. (2017). Genome-wide association study reveals putative regulators of bioenergy traits in Populus deltoides. New Phytol. 213, 799–811. 10.1111/nph.14154 [DOI] [PubMed] [Google Scholar]
- Fox E. J., Reid-Bayliss K. S., Emond M. J., Loeb L. A. (2014). Accuracy of next generation sequencing platforms. Next Gener. Seq. Appl. 1, 106. 10.4172/jngsa.1000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T. J., Marini N. J., Witte J. S. (2010). Comprehensive approach to analyzing rare genetic variants. PLoS One 5, e13584. 10.1371/journal.pone.0013584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little E. L. (1971). Atlas of United States trees. Volume 1. Conifers and important hardwoods. Misc. Publ. 1146. Washington, DC: U.S. Department of Agriculture, Forest Service; 320 p. [Google Scholar]
- MacKay J. J., O'Malley D. M., Presnell T., Booker F. L., Campbell M. M., Whetten R. W., et al. (1997). Inheritance, gene expression, and lignin characterization in a mutant pine deficient in cinnamyl alcohol dehydrogenase. Proc. Natl. Acad. Sci. 94 (15), 8255–8260. 10.1073/pnas.94.15.8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher B. (2008). Personal genomes: the case of the missing heritability. Nature 456, 18–21. 10.1038/456018a [DOI] [PubMed] [Google Scholar]
- Manolio T. A., Collins F. S., Cox N. J., Goldstein D. B., Hindorff L. A., Hunter D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M. I., Abecasis G. R., Cardon L. R., Goldstein D. B., Little J., Ioannidis J. P. A., et al. (2008). Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369. 10.1038/nrg2344 [DOI] [PubMed] [Google Scholar]
- McClellan J., King M.-C. (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. 10.1016/J.CELL.2010.03.032 [DOI] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., DePristo M. A. (2010). The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Res. 20 (9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKown A. D., Guy R. D., Klápště J., Geraldes A., Friedmann M., Cronk Q. C. B., et al. (2014). Geographical and environmental gradients shape phenotypic trait variation and genetic structure in Populus trichocarpa. New Phytol. 201, 1263–1276. 10.1111/nph.12601 [DOI] [PubMed] [Google Scholar]
- McKown A. D., Klápště J., Guy R. D., Soolanayakanahally R. Y., La Mantia J., Porth I., et al. (2017). Sexual homomorphism in dioecious trees: extensive tests fail to detect sexual dimorphism in Populus. Sci. Rep. 7 (1), 1831. 10.1038/s41598-017-01893-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Huang X., Ebert D., Mills C., Guo X., et al. (2019). Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721. 10.1038/s41596-019-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchero W., Guo J., DiFazio S. P., Chen J.-G., Ranjan P., Slavov G. T., et al. (2015). High-resolution genetic mapping of allelic variants associated with cell wall chemistry in populus. BMC Genomics 16, 24. 10.1186/s12864-015-1215-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit R. J., Hampe A. (2006). Some evolutionary consequences of being a tree. Annu. Rev. Ecol. Evol. Syst. 37, 187–214. 10.1146/annurev.ecolsys.37.091305.110215 [DOI] [Google Scholar]
- Poplin R., Ruano-Rubio V., DePristo M. A., Fennell T. J., Carneiro M. O., Auwera G. A., et al. (2017). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178. 10.1101/201178 [DOI] [Google Scholar]
- Porth I., El-Kassaby Y. A. (2015). Using populus as a lignocellulosic feedstock for bioethanol. Biotechnol. J. 10, 510–524. 10.1002/biot.201400194 [DOI] [PubMed] [Google Scholar]
- Porth I., Klápště J., Skyba O., Hannemann J., McKown A. D., Guy R. D., et al. (2013). Genome-wide association mapping for wood characteristics in Populus identifies an array of candidate single nucleotide polymorphisms. New Phytol. 200, 710–726. 10.1111/nph.12422 [DOI] [PubMed] [Google Scholar]
- Porth I., Maghuly F., El-Kassaby Y. A., Mansfield S. (2018). Localization of gene expression, tissue specificity of Populus xylosyltransferase genes by isolation and functional characterization of their promoters. PCTOC 134, 503–508. 10.1007/s11240-018-1426-5 [DOI] [Google Scholar]
- Pritchard J. K. (2001). Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 69, 124–137. 10.1086/321272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmer A., Phan H., Mathieson I., Iqbal Z., Twigg S. R. F., Wilkie A. O. M., et al. (2014). Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 46, 912–918. 10.1038/ng.3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann S., de Graaf A. O., Karimi M., van der Reijden B. A., Hellström-Lindberg E., Jansen J. H., et al. (2017). Evaluating variant calling tools for non-matched next-generation sequencing data. Sci. Rep. 7, 43169. 10.1038/srep43169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Junior O. B., Faria D. A., Grattapaglia D. (2015). A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 206, 1527–1540. 10.1111/nph.13322 [DOI] [PubMed] [Google Scholar]
- Slavov G. T., Difazio S. P., Martin J., Schackwitz W., Muchero W., Rodgers-Melnick E., et al. (2012). Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol. 196, 713–725. 10.1111/j.1469-8137.2012.04258.x [DOI] [PubMed] [Google Scholar]
- Suren H., Hodgins K. A., Yeaman S., Nurkowski K. A., Smets P., Rieseberg L. H., et al. (2016). Exome capture from the spruce and pine giga-genomes. Mol. Ecol. Resour. 16, 1136–1146. 10.1111/1755-0998.12570 [DOI] [PubMed] [Google Scholar]
- Thumma B. R., Nolan M. F., Evans R., Moran G. F. (2005). Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171, 1257–1265. 10.1534/genetics.105.042028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan G. A., Difazio S., Putnam N., Bhalerao R. R., Bhalerao R. P., Blaudez D., et al. (2006). The genome of black cottonwood, populus trichocarpa (Torr. & Gray). Science 80 313, 1596–1604. 10.2307/20031305 [DOI] [PubMed] [Google Scholar]
- Vanholme B., Cesarino I., Goeminne G., Kim H., Marroni F., Van Acker R., et al. (2013). Breeding with rare defective alleles (BRDA): a natural Populus nigra HCT mutant with modified lignin as a case study. New Phytol. 198, 765–776. 10.1111/nph.12179 [DOI] [PubMed] [Google Scholar]
- Visscher P. M., Wray N. R., Zhang Q., Sklar P., McCarthy M. I., Brown M. A., et al. (2017). 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. 10.1016/J.AJHG.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Zhang L., Zhang Z., Li M., Wang D., Zhang X., et al. (2019). Phylogenomics of the genus populus reveals extensive interspecific gene flow and balancing selection. New Phytol. 225, 1370–1382. 10.1111/nph.16215 [DOI] [PubMed] [Google Scholar]
- Weighill D., Jones P., Shah M., Ranjan P., Muchero W., Schmutz J., et al. (2018). Pleiotropic and epistatic network-based discovery: integrated networks for target gene discovery. Front. Energy Res. 6, 30. 10.3389/fenrg.2018.00030 [DOI] [Google Scholar]
- Wu M. C., Lee S., Cai T., Li Y., Boehnke M., Lin X. (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93. 10.1016/j.ajhg.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C.-Y., Ying C. C., Yanchuk A. D., Holowachuk D. L. (2009). Ecotypic mode of regional differentiation caused by restricted gene migration: a case in black cottonwood (Populus trichocarpa) along the Pacific Northwest coast. Can. J. For. Res. 39, 519–525. 10.1139/X08-190 [DOI] [Google Scholar]
- Yang Z., Bielawski J. P. (2000). Statistical tests of adaptive molecular evolution. Trends Ecol. Evol. 15, 496–502. 10.1016/S0169-5347(00)01994-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data are available under https://phytozome.jgi.doe.gov/pz/portal.html.