

Abstract
The phenomenon of acquired resistance to chemotherapeutic agents is a long-standing conundrum in cancer treatment. To help delineate drug resistance mechanisms and pave the way for the development of novel strategies, we generated a PC3 prostate cancer cell line resistant to proteasome inhibitor bortezomib for the first time. The resistant cells were found to have an IC50 value of 359.6 nM, whereas the IC50 value of parental cells was 82.6 nM after 24 h of treatment with varying doses of bortezomib. The resistant cells were also partly cross-resistant to the novel proteasome inhibitor carfilzomib; however, they were not resistant to widely used chemotherapeutic agent vincristine sulfate, indicating that enhanced cellular drug efflux via the multidrug resistance (MDR) transporters is not the molecular basis of the resistance. Since both bortezomib and carfilzomib target and inhibit the chymotrypsin-related activity residing in the β5 subunit of the proteasome (PSMB5), we next examined its expression and found surprisingly no significant alteration in the expression profile of the mature form. However, a significant increase in the accumulation of the precursor form of PSMB5 in response to 100 nM bortezomib was observed in the parental cells without a significant accumulation in the resistant cells. The results presented here thus suggest that the molecular mechanisms causing resistance to proteasome inhibitors need to be examined in-depth to overcome the resistance to ubiquitin–proteasome pathway inhibitors in cancer treatment.
Keywords: Bortezomib, Cancer, Metastasis, PC3, Proteasome
Introduction
It is well-known that metastatic disease is responsible for about 90% of cancer-related mortality rates (Guan 2015). Therefore, significant progress in cancer treatment can be achieved if the dissemination of cancer is prevented by targeting the mechanisms of the metastatic process. However, the resistance to conventional chemotherapeutic drugs arises due to primary or acquired resistance mechanisms causing increased drug efflux, decreased drug uptake, alterations in the amount or activity of drug targets, enhanced DNA repair and so on (Zahreddine and Borden 2013). Since its first purification and characterization from rabbit reticulocytes (Hough et al. 1987; Hough and Rechsteiner 1986), the 26S proteasome was shown to play in almost all cellular processes such as protein degradation, cell cycle, apoptosis, DNA repair, and transcription as well as protein quality control and synthesis (Livneh et al. 2016; Yerlikaya and Yontem 2013). A number of studies showed that 26S proteasome is a crucial target in cancer treatment strategies. For instance, Chen and Madura showed that the proteasomal subunits, as well as the activity of the proteasome, were increased by 2–32 fold in more than 90% of primary breast cancer cells as compared to the benign solid tumors (Chen and Madura 2005). Furthermore, numerous studies showed that proteasome inhibitor bortezomib alone has significant growth suppressing and apoptotic activity in many cancer cell lines as well as encouraging antitumor activity in combination with other cytotoxic agents (Adams 2002; Escobar et al. 2011; Okur and Yerlikaya 2019; Yerlikaya and Erin 2008). In fact, the 26S proteasome inhibitor bortezomib and carfilzomib are approved by the Food and Drug Administration (FDA) in 2003 and 2012, respectively, for the treatment of patients with relapsed and refractory multiple myeloma (MM) (Kane et al. 2003; Thompson 2013). However, in a number of studies, it was found that drug resistance develops rapidly in response to the bortezomib treatment. For example, the studies of Oerlemans et al. revealed that bortezomib resistance in human myelomonocytic THP1 cells was associated with (1) an Ala49Thr mutation in the proteasome 5-subunit (PSMB5) protein, and (2) a dramatic overexpression (up to 60 fold) of PSMB5 without any change in other proteasomal subunits (e.g., PSMB6, PSMB7, and PSMA7) (Oerlemans et al. 2008). On the other hand, Wu et al. established two acquired bortezomib-resistant hepatocellular carcinoma (HCC) cell lines, and found that bortezomib-resistant HCC cells had increased expression of β1 and β5 proteasome subunits but no mutation was detected in bortezomib binding pocket in the β5-subunit (Wu et al. 2016). However, to develop novel and more effective cancer treatment strategies, there is a need for an in-depth understanding of the mechanisms underlying the resistance to the proteasome inhibitors. Using PC3 prostate cancer cell line as a model system, here we investigated the mechanisms of resistance to the 26S proteasome inhibitor bortezomib. It was found that the precursor form of PSMB5 was overexpressed in a concentration-dependent manner in the parental cell, while there were no changes in its processing in the resistant cell line. The polyubiquitinated conjugates were also increased to higher levels in the parental cells than those in the resistant cells. In addition, cyclin D1 expression was reduced significantly in the parental cell, but the degree of its change in the resistance cells was not significant.
Materials and methods
Materials
DMEM media, trypsin solution 10X, penicillin–streptomycin, fetal bovine serum, HEPES, sodium pyruvate, sodium bicarbonate, sodium chloride, acrylamide, bis-acrylamide, D-(+)-glucose, sodium dodecyl sulfate, developer and replenisher, fixer and replenisher were purchased from Sigma-Aldrich. PC3 cells (ATCC Cat# CRL-1435, RRID:CVCL_0035) (Bairoch 2018) were provided by Prof. Dr. Serap Kuruca Erdem (İstanbul University, İstanbul, Turkey).
Cell maintenance and development of resistant cells
PC3 prostate cancer cells were cultured in DMEM containing 4.5 g/l glucose, 0.375% sodium bicarbonate, 100 µg/ml streptomycin and 100 U/ml penicillin. The cells were seeded in 35 × 10 mm sterile petri dishes or 96 well multiplate for the experiments as described previously (Aras and Yerlikaya 2016). To obtain resistant PC3 cells, the cells were treated with stepwise increasing concentrations of bortezomib over 6 months. The cells were first treated with 1 nM bortezomib for 3 days until about 70% confluence. Afterwards, the cells were treated with the same concentration during the next two passages. After these passages, the concentration of bortezomib was increased to 2.5 nM for the next three passages. The treatment was similarly continued for 5 nM, 10 nM, 20 nM, 30 nM, 40 nM, 50 nM, 60 nM, 70 nM and 80 nM concentrations. At some points, due to the high cytotoxicity of bortezomib, the cells did not reach about 70% confluency, then the concentration was reduced to a previous concentration and the treatment was continued to allow the cells to regain the rapid proliferation rates. The parental cells were grown and passaged in parallel for subsequent experiments to serve as the best experimental control.
WST-1 assay
WST-1 assay was used to determine the IC50 value of bortezomib in both parental and resistant cells. To determine the IC50 values, the cells (10,000 cells) were seeded in each well on a 96-well plate. When the cells reached about 70% confluency, they were treated with different concentrations of bortezomib (1 nM, 10 nM, 50 nM, 100 nM, 500 nM, 1 µM, 10 µM, 50 µM, 100 µM) for 24 h. Afterwards, the cells were treated for 2 h with RPMI-1640 with 0.5% FBS + 10 mg/ml WST 1. After WST-1 treatment, the absorbances of each well was recorded by RT-2100C microplate reader at 450 nm. The data were analyzed and graphed with GraphPad Prism 5 program. The IC50 values of bortezomib were then obtained by using nonlinear regression to fit the data to the log(inhibitor) versus response-variable slope (Okur and Yerlikaya 2019).
iCELLigence system
To determine the cross-resistance to proteasome inhibitors as well as to the anticancer agents, the iCELLigence system, a real-time, label-free cell analyzer, was used as described before (Aras and Yerlikaya 2016). Briefly, the resistant and parental cells were seeded in E-plate L8 having integrated microelectrode sensors at the bottom of the wells. When the cells start to proliferate and adhere to the microelectrodes, a change in electrical impedance occurs reflecting the biological status of the cells, which is expressed as the cell index (CI), recorded every hour until the end of the experiment (the cell index is a value parallel to the growth and proliferation of the cells). The iCELLigence system thus allows real-time monitoring of time-dependent effects of inhibitors or drugs on cancer cell lines (Aras and Yerlikaya 2016). After adding 150 µl cell culture medium (DMEM with 10% FBS) to each E-Plate L8 well (ACEA Biosciences, San Diego, CA, USA) and incubation at room temperature for 30 min, the background measurement was carried out by following the manufacturer’s protocol (ACEA Biosciences). Subsequently, 12,500 PC3 cells were seeded in each E-Plate L8 well in a final volume of 500 µl. After 24 h of incubation, cells were treated with 100 nM bortezomib, 50 nM carfilzomib, 1 µM carboplatin, 1 µM vincristine sulfate, 10 µM epigallocatechin gallate (EGCG) or 10 µM quercetin.
Western blotting
Western blot analysis was performed as described before (Yerlikaya and Dokudur 2010). In short, parental PC3 or resistant PC3 cells (200,000 per 35 × 10 mm dishes) were grown to the exponential phase of the growth and then treated with an isotonic solution (control), 10 nM bortezomib or 100 nM bortezomib for 24 h. After cell lysis with RIPA buffer containing 1X protease inhibitor cocktail (cat. no. sc-29131, Santa Cruz Biotechnologies Inc.) and protein quantification by the Bradford assay (Bradford 1976), 35 µg protein from each sample was separated on a 12% SDS-PAGE followed by transfer to a PVDF membrane using Bio-Rad Trans-Blot Turbo Transfer system. The membranes were then probed with anti-ubiquitin rabbit polyclonal antibody (1:200, cat. no. sc-9133, Santa Cruz Biotechnologies Inc.), anti-PSMB5 rabbit polyclonal antibody (1:750, cat. no. 12919S, Cell Signalling Technology Inc.) or cyclin D1 rabbit polyclonal antibody (1:750, cat. no. 2922S, Cell Signalling Technology Inc.) in TBS-T for 1 h. To determine equal protein loading, the membranes were also detected with an anti-β-actin rabbit polyclonal antibody (1:3000, cat. no. ab8227, Abcam) in TBS-T for 1 h. To visualize the specific protein bands, Phototope-HRP Western blot detection system was used. After the primary antibody binding, an anti-rabbit HRP-conjugated secondary antibody (1:3000, cat. no. 7074, Cell Signalling Technology Inc.) was added to the membrane for 1 h in TBS-T. Finally, the membranes were incubated with LumiGLO reagent (cat. no. 7072, Cell Signalling Technology Inc.) and the emitted light was captured on an X-ray film in a dark room.
Results
The phenomenon of acquired drug resistance to chemotherapeutic agents is a major obstacle in cancer treatment (Nikolaou et al. 2018). To delineate mechanisms of drug resistance and pave the way for the development of the novel drugs as well as novel combination strategies, overcoming such mechanisms, here we have first developed a PC3 prostate cancer line resistant to 26S proteasome inhibitor bortezomib [a boronic dipeptide which reversibly inhibit the chymotrypsin-like activity residing in the PSMB5 subunit (Lu and Wang 2013)] with stepwise increments in concentrations over 6 months. The parental cells were also grown and passaged in parallel over 6 months to serve as a control for later experiments. Then, the IC50 values of both parental and resistant cells were analyzed to determine the status of the resistance. As can be seen in Fig. 1a and b, the IC50 value of parental PC3 cells (abbreviated as PC3-P) was determined as 82.8 nM after 24 h of treatment with various concentrations of bortezomib; whereas that of the resistant cells (named PC3-R) was 359.6 nM, indicating a 4.3-fold increase in the resistance. Next, we determined whether PC3-R cells were also cross-resistant to carfilzomib, a second-generation proteasome inhibitor, primarily targeting the chymotrypsin-like activity irreversibly (Parlati et al. 2009). Both PC3-P and PC3-R cells were treated with 50 nM carfilzomib or 100 nM bortezomib after 24 h of seeding and then the cell proliferation rates were determined by the real-time cell analyzer iCELLigence as a function of time. As can be seen in Fig. 2, the growth of PC3-P cells was completely inhibited in the presence of 50 nM carfilzomib or 100 nM bortezomib. Interestingly, the resistant cells (PC3-R) showed a reduced proliferation rate as compared to the parental cells (PC3-P). Also, as can be seen in Fig. 2, PC3-R cells were significantly resistant to 100 nM bortezomib after 96 h (p < 0.01) and 120 h (p < 0.001) as compared to PC3-P cells. Similarly, it was noticed that PC3-R cells developed a cross-resistance to carfilzomib (p < 0.01 after 96 h; p < 0.001 after 120 h) as compared to PC3-P cells. To determine whether the resistance phenomenon to bortezomib and carfilzomib by PC3-R cells was due to drug efflux mechanisms, both PC3-P and PC3-R cells were treated with the common chemotherapeutic agent carboplatin (1 µM) and vincristine sulfate (1 µM). As seen in Fig. 3, both cells gave a similar response to both drugs. That is, both PC3-P and PC3-R cells were resistant to carboplatin; whereas, they were highly sensitive to vincristine sulfate under the experimental conditions used in the current study (Fig. 3). Next, the effects of two flavonoids (–)-Epigallocatechin gallate (EGCG) and quercetin, both of which are found to antagonize the antiproliferative and cytotoxic effects of bortezomib (Golden et al. 2009; Yerlikaya et al. 2010), were tested on PC3-P and PC3-R cells using the iCELLigence system. As seen in Fig. 4, both flavonoids did not affect the proliferation rates of PC3-P and PC3-R cells at a relatively high concentration of 10 µM during the treatment periods. As can be seen, the proliferation rates of PC3-P cells in the presence of both flavonoids were again significantly higher than that of PC3-R cells at 96 h and 120 h (Fig. 4).
Fig. 1.
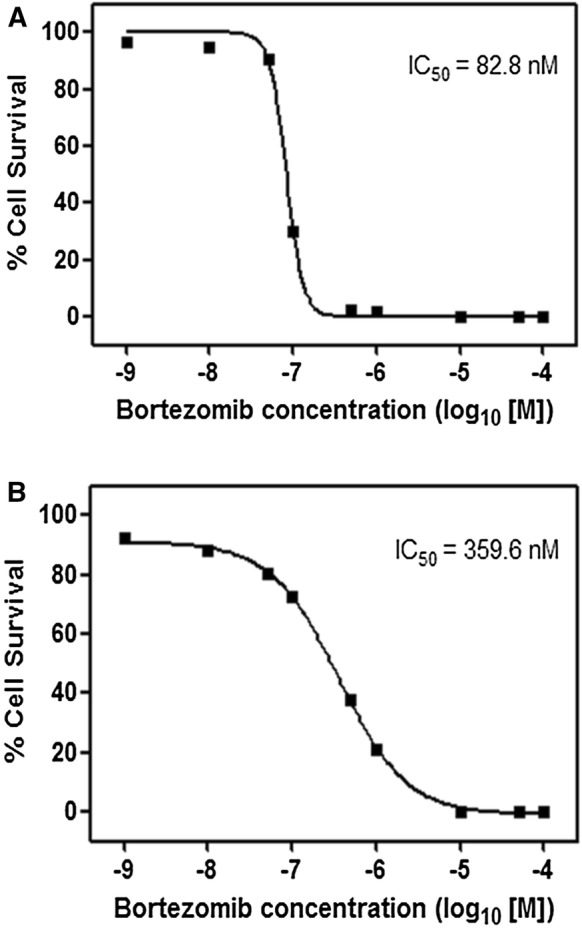
The IC50 values of bortezomib in a parental PC3 and b resistant PC3 cells. The cells were treated with the indicated concentrations of bortezomib in “Materials and methods” section. The cell survival was determined by WST-1 assay (n = 3–5)
Fig. 2.

Effect of proteasome inhibitor carfilzomib (50 nM) and bortezomib (100 nM) on parental PC3 and resistant PC3 cells. a The cell proliferation was determined by the iCELLigence system. b The statistical analysis of cell index values for each treatment. The results are presented as mean ± SD. The groups were compared with two-way ANOVA with Bonferroni multiple comparison post-test. ** represents a p value < 0.01; *** represents a p-value < 0.001
Fig. 3.

Effect of carboplatin (1 µM) and vincristine sulfate (1 µM) on parental PC3 and resistant PC3 cells. a The cell proliferation was determined by the iCELLigence system. b The statistical analysis of cell index values for each treatment. The results are presented as mean ± SD. The groups were compared with two-way ANOVA with Bonferroni multiple comparison post-test. ** represents a p-value < 0.01; *** represents a p-value < 0.001
Fig. 4.

Effect of flavonoids EGCG (10 µM) and quercetin (10 µM) on parental PC3 and resistant PC3 cells. a The cell proliferation was determined by the iCELLigence system. b The statistical analysis of cell index values for each treatment. The results are presented as mean ± SD. The groups were compared with two-way ANOVA with Bonferroni multiple comparison post-test. * represents a p-value < 0.05; ** represents a p-value < 0.01; *** represents a p-value < 0.001
To further delineate the mechanism of resistance, we next examined the accumulation of polyubiquitin conjugates in response to 10 nM and 100 nM bortezomib treatments in both PC3-P and PC3-R cells using Western blot analysis. As can be seen in Fig. 5, polyubiquitin conjugates increased to higher levels in PC3-P cells as compared to the PC3-R cells. Previous studies showed a dramatic increase in the expression of β5 proteasomal subunit (PSMB5) in bortezomib-resistant hepatocellular carcinoma (HCC) cells or human myelomonocytic THP1 cells (Oerlemans et al. 2008; Wu et al. 2016). As seen in Fig. 5, no significant difference in the accumulation of the mature form of PSMB5 was detected between the PC3-P and PC3-R cells; however, to our surprise, there was a significant increase in the precursor form of PSMB5 in PC3-P cells treated with 100 nM bortezomib as compared to PC3-R cells, similarly treated with 100 nM bortezomib. As stated above, the proliferation rate of PC3-R cells was slower than that of the PC3-P cells. Therefore, we examined the expression of cyclin D1 in both cells in response to bortezomib-treatment. As seen in Fig. 5, PC3-R cells (vehicle-treated control sample) had much lower levels of cyclin D1 as compared to the PC3-P cells (vehicle-treated control sample). Quantitation of control band in both cells showed that PC3-P cells (non-treated cells) had a 2.7-fold higher level of cyclin D1 than that in PC3-R cells (non-treated cells). Also, although cyclin D1 expression was reduced in response 10 nM and 100 nM bortezomib treatments in PC3-P cells, no change in cyclin D1 level was observed in PC3-R cells after treatment with 100 nM and 100 nM bortezomib treatments.
Fig. 5.

Western blot analysis of polyubiquitin conjugates, PSMB5 and cyclin D1 in parental and resistant PC3 cells. Cells were treated with 10 nM and 100 nM bortezomib for 24 h. Afterwards, 35 µg protein was separated on 12% SDS-PAGE followed by Western blot analysis as described in detail in “Materials and methods” section. PC3-P, parental PC3 cells; PC3-R, resistant PC3 cells. The result is representative of two experiments, each run in duplicate
Discussion
Drug resistance is a long-standing conundrum in cancer treatment; and therefore, novel treatment strategies targeting and overcoming the resistance mechanisms are urgently needed. In this study, we obtained a bortezomib-resistant PC3 prostate cancer cell line (4.3-fold higher level after 24 h of treatment) for the first time. On the other hand, higher levels of acquired resistance to bortezomib in human myelomonocytic THP1 cells (45- to 129-fold after 72 h of treatment) (Oerlemans et al. 2008), in HepG2 (15-fold after 72 h of treatment) or HuH7 (39-fold after 72 h of treatment) hepatocellular carcinoma cell lines (Wu et al. 2016) were previously obtained. However, Franke et al. obtained a human 8226 myeloma cell, moderately resistant to bortezomib (≤ two fold), using a similar protocol (Franke et al. 2007). These results suggest that the degree of resistance generated is dependent upon the cell type under the study as well as the drug treatment duration (24 h vs 72 h for IC50 determination).
Using the iCELLigence system, PC3-R cells were also found to gain a cross-resistance to carfilzomib, a second-generation, and an irreversible proteasome inhibitor. Franke et al. similarly generated a bortezomib-resistant human monocytic/macrophage THP1 cell line, displaying cross-resistance to carfilzomib (Franke et al. 2012). The resistance of PC3-R cells to bortezomib and carfilzomib was likely not due to the drug efflux mechanisms as both PC3-P and PC3-R cells displayed similar responses to chemotherapeutic agents carboplatin and vincristine sulfate. In accordance with the results presented here, previous findings also support the fact that overexpression of the multidrug resistance (MDR) transporter P-glycoprotein (P-gp) is not the molecular basis of the resistance to bortezomib (Oerlemans et al. 2008; Lu and Wang 2013). Flavonoids EGCG and quercetin are known to inhibit the cytotoxic effects of bortezomib (Golden et al. 2009; Yerlikaya et al. 2010), and especially quercetin has been shown to inhibit the expression of HSP-70 (Hosokawa et al. 1990), which has been implicated in proteasome-resistance (Shringarpure et al. 2006). As presented in Fig. 4, neither EGCG nor quercetin was effectively cytotoxic at a relatively high concentration of 10 µM on PC3-P and PC3-R cells.
A higher accumulation of polyubiquitin conjugates was detected in PC3-P cells as compared to the degree of accumulation in PC3-R cells; this is believed to cause a further decline in ubiquitin–proteasome activity since Bence et al. showed that increased aggregation of misfolded proteins causes ubiquitin–proteasome system (UPS) inhibition (Bence et al. 2001). Although, previous studies showed that bortezomib-resistant HCC or THP1 cells had increased expression of β5 proteasomal subunit (PSMB5) (Oerlemans et al. 2008; Wu et al. 2016), here we have not observed a significant increase in the expression of PSMB5 in PC3-R cells as compared to the PC3-P cells. However, in PC3-P cells, we unexpectedly observed an increase in the expression of the precursor form of PSMB5, which may likely result from inhibition of the processing of the precursor form into a mature form. To our best knowledge, no studies have previously examined or showed the overexpression/processing of the precursor form of PSMB5 in response to bortezomib treatment in wild-type or bortezomib-resistant cells.
During the initial phase of growth, PC3-R cells had a slower growth rate as compared to the parental PC3-P cells. This phenomenon may be attributed to the low level of cyclin D1 (2.7-fold lower in non-treated PC3-R cells). Since overexpression of cyclin D1 is associated with the development as well as the progression of cancer cells (Alao 2007), it was surprising to observe a low level of cyclin D1 in the resistant cells. In addition, it is a short-lived protein and is stabilized by increasing doses bortezomib in HBL100 and T47D cells stably overexpressing cyclin D1 (Ishii et al. 2006). Interesting, we also noticed a decreased expression of endogenous cyclin D1 in response to varying doses of bortezomib in the parental cells. In agreement with our findings, Yu et al. showed that bortezomib alone failed to modify cyclin D1 levels; moreover, combined treatment of cells with bortezomib and histone deacetylase inhibitors (e.g., SAHA) resulted in further down-regulation of cyclin D1 (Yu et al. 2003). Recently, it was shown that DUB3, a deubiquitinating enzyme, regulates and stabilizes cyclin A by removing the polyubiquitin chains built upon cyclin A protein, leading to high levels of cyclin A and progression through the cell cycle (Hu et al. 2019). Chesnel and co-workers also indicated that the degradation of cyclin B through the ubiquitin–proteasome pathway is required for the M-phase exit in Xenopus laevis cell-free extracts (Chesnel et al. 2006). It would be interesting to examine whether the cell proliferation differences between parental and resistant PC3 cells result from the differences in the levels of cyclin A and/or cyclin B protein expressions.
The continued examination of bortezomib-resistant PC3 prostate cancer cells generated here will uncover interesting molecular functions of proteasomal subunits as well as the mechanisms underlying the bortezomib-resistance in cancer cells. In addition, it is believed that our ongoing investigations will undoubtedly help the development of novel strategies targeting cancer cell proliferation.
Acknowledgements
This study is funded by Kütahya Dumlupınar University (Project No 2017-68).
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adams J. The proteasome as a novel target for the treatment of breast cancer. Breast Dis. 2002;15:61–70. doi: 10.3233/BD-2002-15107. [DOI] [PubMed] [Google Scholar]
- Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras B, Yerlikaya A. Bortezomib and etoposide combinations exert synergistic effects on the human prostate cancer cell line PC-3. Oncol Lett. 2016;11:3179–3184. doi: 10.3892/ol.2016.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A. The Cellosaurus, a cell-line knowledge resource. J Biomol Tech. 2018;29:25–38. doi: 10.7171/jbt.18-2902-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chen L, Madura K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005;65:5599–5606. doi: 10.1158/0008-5472.CAN-05-0201. [DOI] [PubMed] [Google Scholar]
- Chesnel F, Bazile F, Pascal A, Kubiak JZ. Cyclin B Dissociation from CDK1 precedes its degradation upon MPF inactivation in mitotic extracts of Xenopus laevis embryos. Cell Cycle. 2006;5:1687–1698. doi: 10.4161/cc.5.15.3123. [DOI] [PubMed] [Google Scholar]
- Escobar M, Velez M, Belalcazar A, Santos ES, Raez LE. The role of proteasome inhibition in nonsmall cell lung cancer. J Biomed Biotechnol. 2011;2011:806506. doi: 10.1155/2011/806506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke NE, Oerlemans R, Zweegman S, van Zantwijk I, Kaspers GJ, Jansen G, Cloos J. Molecular mechanisms of bortezomib resistance in acute lymphoblastic leukemia cells in comparison with multiple myeloma cells. Blood. 2007;110:1016a–1016a. doi: 10.1182/blood.V110.11.1016.1016. [DOI] [Google Scholar]
- Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, Zweegman S, Chan ET, Kirk CJ, Geerke DP, et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26:757–768. doi: 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- Golden EB, Lam PY, Kardosh A, Gaffney KJ, Cadenas E, Louie SG, Petasis NA, Chen TC, Schonthal AH. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113:5927–5937. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015;5:402–418. doi: 10.1016/j.apsb.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hirayoshi K, Nakai A, Hosokawa Y, Marui N, Yoshida M, Sakai T, Nishino H, Aoike A, Kawai K, et al. Flavonoids inhibit the expression of heat shock proteins. Cell Struct Funct. 1990;15:393–401. doi: 10.1247/csf.15.393. [DOI] [PubMed] [Google Scholar]
- Hough R, Rechsteiner M. Ubiquitin-lysozyme conjugates. Purification and susceptibility to proteolysis. J Biol Chem. 1986;261:2391–2399. [PubMed] [Google Scholar]
- Hough R, Pratt G, Rechsteiner M. Purification of two high molecular weight proteases from rabbit reticulocyte lysate. J Biol Chem. 1987;262:8303–8313. [PubMed] [Google Scholar]
- Hu B, Deng T, Ma H, Liu Y, Feng P, Wei D, Ling N, Li L, Qiu S, Zhang L, Peng B, Liu J, Ye M. Deubiquitinase DUB3 regulates cell cycle progression via stabilizing cyclin A for proliferation of non-small cell lung cancer cells. Cells. 2019;8:297. doi: 10.3390/cells8040297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Y, Pirkmaier A, Alvarez JV, Frank DA, Keselman I, Logothetis D, Mandeli J, O’Connell MJ, Waxman S, Germain D. Cyclin D1 overexpression and response to bortezomib treatment in a breast cancer model. J Natl Cancer Inst. 2006;98:1238–1247. doi: 10.1093/jnci/djj334. [DOI] [PubMed] [Google Scholar]
- Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, Ciechanover A. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 2016;26:869–885. doi: 10.1038/cr.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark Res. 2013;1:13. doi: 10.1186/2050-7771-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaou M, Pavlopoulou A, Georgakilas AG, Kyrodimos E. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis. 2018;35:309–318. doi: 10.1007/s10585-018-9903-0. [DOI] [PubMed] [Google Scholar]
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- Okur E, Yerlikaya A. A novel and effective inhibitor combination involving bortezomib and OTSSP167 for breast cancer cells in light of label-free proteomic analysis. Cell Biol Toxicol. 2019;35:33–47. doi: 10.1007/s10565-018-9435-z. [DOI] [PubMed] [Google Scholar]
- Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood. 2009;114:3439–3447. doi: 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- Shringarpure R, Catley L, Bhole D, Burger R, Podar K, Tai YT, Kessler B, Galardy P, Ploegh H, Tassone P, et al. Gene expression analysis of B-lymphoma cells resistant and sensitive to bortezomib. Br J Haematol. 2006;134:145–156. doi: 10.1111/j.1365-2141.2006.06132.x. [DOI] [PubMed] [Google Scholar]
- Thompson JL. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann Pharmacother. 2013;47:56–62. doi: 10.1345/aph.1R561. [DOI] [PubMed] [Google Scholar]
- Wu YX, Yang JH, Saitsu H. Bortezomib-resistance is associated with increased levels of proteasome subunits and apoptosis-avoidance. Oncotarget. 2016;7:77622–77634. doi: 10.18632/oncotarget.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerlikaya A, Erin N. Differential sensitivity of breast cancer and melanoma cells to proteasome inhibitor Velcade. Int J Mol Med. 2008;22:817–823. [PubMed] [Google Scholar]
- Yerlikaya A, Dokudur H. Investigation of the eIF2alpha phosphorylation mechanism in response to proteasome inhibition in melanoma and breast cancer cells. Mol Biol. 2010;44:859–866. doi: 10.1134/S0026893310050122. [DOI] [PubMed] [Google Scholar]
- Yerlikaya A, Yontem M. The significance of ubiquitin proteasome pathway in cancer development. Recent Pat Anticancer Drug Discov. 2013;8:298–309. doi: 10.2174/1574891X113089990033. [DOI] [PubMed] [Google Scholar]
- Yerlikaya A, Okur E, Şeker S, Erin N. Combined effects of the proteasome inhibitor bortezomib and Hsp70 inhibitors on the B16F10 melanoma cell line. Mol Med Rep. 2010;3:333–339. doi: 10.3892/mmr_000000262. [DOI] [PubMed] [Google Scholar]
- Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003;102:3765–3774. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. 2013;4:28. doi: 10.3389/fphar.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]