

Abstract
Objective
Otaplimastat is a neuroprotectant that inhibits matrix metalloprotease pathway, and reduces edema and intracerebral hemorrhage induced by recombinant tissue plasminogen activator (rtPA) in animal stroke models. We aimed to assess the safety and efficacy of otaplimastat in patients receiving rtPA.
Methods
This was a phase 2, 2‐part, multicenter trial in stroke patients (19–80 years old) receiving rtPA. Intravenous otaplimastat was administered <30 minutes after rtPA. Stage 1 was a single‐arm, open‐label safety study in 11 patients. Otaplimastat 80 mg was administered twice daily for 3 days. Stage 2 was a randomized, double‐blind, placebo‐controlled study involving 69 patients, assigned (1:1:1) to otaplimastat 40 mg, otaplimastat 80 mg, or a placebo. The primary endpoint was the occurrence of parenchymal hematoma (PH) on day 1. Secondary endpoints included serious adverse events (SAEs), mortality, and modified Rankin scale (mRS) distribution at 90 days (http://clinicaltrials.gov identifier: NCT02787278).
Results
No safety issues were encountered in stage 1. The incidence of PH during stage 2 was comparable: 0 of 22 with the placebo, 0 of 22 with otaplimastat 40 mg, and 1 of 21 with the 80 mg dose. No differences in SAEs (13%, 17%, 14%) or death (8.3%, 4.2%, 4.8%) were observed among the 3 groups. Three adverse events (chills, muscle rigidity, hepatotoxicity) were judged to be related to otaplimastat.
Interpretation
Intravenous otaplimastat adjunctive therapy in patients receiving rtPA is feasible and generally safe. The functional efficacy of otaplimastat needs to be investigated with further large trials. ANN NEUROL 2020;87:233–245
Recombinant tissue plasminogen activator (rtPA) is the only therapeutic agent approved for patients with acute ischemic stroke (AIS). However, rtPA therapy increases the risk of intracerebral hemorrhage (ICH) or hemorrhagic transformation (HT) through diverse mechanisms.1, 2, 3, 4 It has been shown that rtPA activates matrix metalloproteases (MMPs)5, 6 and aggravates breakdown of the blood–brain barrier, leading to brain edema and HT.7 Thus, there is a need to develop therapeutic strategies to increase the clinical benefit of rtPA in patients with AIS. For this purpose, adjunctive therapies have been developed, some of which have shown promising preclinical results.8, 9, 10, 11 However, clinical trials using such drugs are uncommon; although minocycline,12 uric acid,13 and 3K3A‐APC14 were found to be safe when administered to stroke patients receiving rtPA, their efficacy still remains to be proven.9, 12, 13, 14 Recently, small studies have shown that fingolimod may enhance the efficacy of rtPA administration in patients with AIS receiving rtPA,15 and improve the clinical outcome in patients with a proximal cerebral arterial occlusion in the 4.5‐ to 6‐hour time window.16
Otaplimastat (SP‐8203) is a small molecule with a quinazoline‐2,4‐dione scaffold that improves neurological outcomes through multiple cytoprotective mechanisms in various animal stroke models.17, 18, 19, 20 Notably, in both a standalone treatment and a combined treatment with rtPA, otaplimastat showed significant benefit by reducing infarct volume and edema in embolic stroke models.20 In animal models of stroke, delayed rtPA treatment increased brain ICH, and coadministration of otaplimastat significantly improved neurologic outcome and reduced brain edema and ICH by inhibiting MMP activities through upregulation of tissue inhibitor of metalloproteinase‐1.20
In a phase 1 study, up to 240 mg otaplimastat was well tolerated in 77 healthy volunteers without significant side effects (unpublished data). The purpose of this phase 2a study was to assess the feasibility, safety, and potential efficacy of an intravenous infusion of otaplimastat in patients with AIS treated by rtPA.
Subjects and Methods
Study Design and Participants
This was a 2‐stage phase 2 trial. Stage 1 was an open‐label, unblinded, single‐arm study in which patients were given high‐dose otaplimastat (80 mg twice daily for 3 days) to examine its safety. At the completion of stage 1, a go/no‐go decision was made in an interim meeting by the independent Data and Safety Monitoring Board (DSMB), composed of global stroke experts and a nonvoting statistician. Stage 2 was a randomized, double‐blind, placebo‐controlled study to evaluate the safety and efficacy of otaplimastat 40 mg or 80 mg twice daily for 3 days versus a placebo. The protocol was approved by the local institutional review board, and all patients or legal representatives provided written informed consent. This study was conducted at 8 medical centers in South Korea from June 5, 2016 to August 22, 2017. This study was performed in accordance with International Conference on Harmonization and Good Clinical Practice guidelines.
Patients
AIS patients (19–80 years) who were to receive intravenous rtPA within 4.5 hours were enrolled. Considering that this was the first study that examined the safety of otaplimastat in stroke patients receiving rtPA, patients with a National Institutes of Health Stroke Scale (NIHSS) score of 4 to 10 who were at the relatively lower risk of ICH were enrolled in stage 1. Once the safety of otaplimastat was confirmed in stage 1, we enrolled any patient with an NIHSS score ≥4 in stage 2 to more appropriately reflect routine clinical practice. Inclusion and exclusion criteria are shown in Table 1 (excerpted). Key exclusion criteria included systemic allergic diseases or drug hypersensitivity, abnormal electrocardiogram (ECG) or hematological findings, and a contraindication for the use of rtPA.
Table 1.
Inclusion and Exclusion Criteria for the SAFE‐TPA Trial (Excerpted)
| Inclusion Criteria | Exclusion Criteria |
|---|---|
|
1. Adults aged 19 to 80 years 2. Able to receive rtPA within 4.5 hours after the onset of early symptoms of acute ischemic stroke 3. Available for brain MRI (DWI, GRE/SWI, FLAIR, MRA) scanning 4. Signed informed consent by subject or authorized representative |
1. Systemic allergic diseases or hypersensitivity to specific drugs 2. Patients have condition as follows at screening: (a) Diagnosis with AMI within the past 6 months (b) Arrhythmia causing clinical symptoms such as dyspnea or palpitation within the past 6 months (c) Abnormal ECG findings in stable condition at ER 3. Severe heart failure of NYHA class III or class IV 4. Fever (≥38°C) or infection signs that require antibiotic therapy at screening 5. Pulmonary diseases (asthma, COPD, active tuberculosis, etc) recently treated >1 month at screening 6. Decreased hemoglobin (<10 g/dl), decreased platelet count (<100,000/mm3), or hematocrit of <25% in complete blood count 7. Hemodialysis and/or treatments due to nephropathies, acute or chronic renal failure at screening 8. Diagnosed cancer within 6 months before the screening time, or any treatment for cancer within the previous 6 months, or recurrent/metastatic cancer 9. Pregnancy or breastfeeding 10. Participated in other clinical trials of other drugs within the past 3 months 11. Cannot participate in the trial according to the judgment of investigators 12. Contraindication for the use of rtPA |
|
Stage 1–specific criteria • NIHSS score of 4–10 | |
|
Stage 2–specific criteria • NIHSS score of ≥4 |
AMI = acute myocardial infarction; COPD = chronic obstructive pulmonary disease; DWI = diffusion‐weighted imaging; ECG = electrocardiogram; ER = emergency room; FLAIR = fluid‐attenuated inversion recovery; GRE/SWI = susceptibility‐weighted images generated from gradient‐echo pulse sequences; MRA = magnetic resonance angiography; MRI = magnetic resonance imaging; NIHSS = National Institutes of Health Stroke Scale; NYHA class = New York Heart Association Functional classification for heart failure; rtPA = recombinant tissue plasminogen activator.
Randomization and Masking
All patients received otaplimastat 80 mg during stage 1. Using a computer‐generated allocation sequence, patients were randomly assigned (1:1:1) to placebo, otaplimastat 40 mg, or otaplimastat 80 mg for stage 2. Randomization was stratified by site to guarantee a balanced distribution among the groups and performed by using block randomization with a pregenerated random number list. The patients and investigators were masked to the treatment assignments, with otaplimastat and placebo provided in color‐matched vials (Shin Poong Pharmaceutical, Ansan, Korea). The individual treatment code was stored by the main statistician.
Procedures
Eligibility for rtPA (Actilyse; Boehringer Ingelheim, Biberach, Germany; 0.9 mg/kg) treatment was assessed in the emergency department based on a brain computed tomography (CT) scan. Otaplimastat or placebo was intravenously administered over 30 minutes no later than 30 minutes after starting the rtPA infusion via a different injection route. Study drugs were given twice daily at intervals of 12 hours (±30 minutes) for a total of 6 times over 3 days. Patients underwent magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) within 6 hours after the start of rtPA therapy, and endovascular thrombectomy (EVT) was performed in patients who had an occluded large artery. Afterward, all patients were admitted to the stroke unit. Antiplatelet agents were not used until 1 day after the rtPA infusion. From day 1 to 5, only prespecified doses of aspirin (100 mg/day), clopidogrel (75 mg/day), or both were allowed to patients at the discretion of the attending physician. Afterward, any antiplatelets or anticoagulants were allowed.
All brain imaging was performed using a common protocol and processed by a centralized facility (Central Imaging Core Lab, Asan Medical Center, Seoul, Korea). The standardized MRI protocol consisted of diffusion‐weighted imaging (DWI), susceptibility‐weighted images generated from gradient‐echo pulse sequences (GRE/SWI), and fluid‐attenuated inversion recovery (FLAIR). MRA consisted of time‐of‐flight MRA for the intracranial arteries and contrast‐enhanced MRA for the neck vessels. All CT scans and the initial 4 MRI sequences (DWI/GRE/SWI, FLAIR) were performed as noncontrast. All serial imaging data and additional postprocedural CT images in patients who underwent EVT were retrospectively analyzed to differentiate contrast extravasation from ICH. The imaging equipment and protocol at each trial site were validated with a standardized qualification control and assurance monitoring procedures before and during the study. The image analysis was blinded and assessed by 2 experienced neurologists or by a third investigator to reach a consensus when conflicting judgments occurred.
A brain CT was performed in stage 1 to evaluate the occurrence of parenchymal hematoma (PH) at 24 ± 3 hours after the first administration of otaplimastat. Patients were examined daily until day 5 for vital signs, NIHSS score, and treatment‐emergent adverse events classified using MedDRA version 19.0. On day 5, brain MRI, MRA, ECG, and laboratory tests were done, and the modified Rankin scale (mRS) and Barthel Index (BI) were assessed. All assessments were repeated on day 14. The prespecified criteria for the no‐go decision for stage 2 was the occurrence of 3 or more cases with symptomatic ICH (sICH) causing neurological deterioration (≥4‐point increase in the NIHSS score) or death during the first 5 days. In stage 2, patients received otaplimastat 40 mg, otaplimastat 80 mg, or placebo twice daily for 3 days and followed the same assessment procedures until day 5. The follow‐up visits at day 28 and day 90 included the NIHSS, laboratory tests, chest X‐ray, ECG, and adverse events. The follow‐up at 90 days also included the mRS and BI.
Outcomes
The primary endpoint of both stages was the incidence of PH within 24 hours on a brain CT scan based on post‐thrombolytic ICH being regarded as an early event (<24 hours) in the National Institute of Neurological Disorders and Stroke rtPA trial and all fatal sICH occurring within 24 hours.21 The definition of ICH followed European Cooperative Acute Stroke Study I and II criteria: hemorrhagic infarct (HI) 1, small petechiae along the margins of the infarct; HI 2, confluent petechiae within the infarcted area but without space‐occupying effect; PH 1, a clot not exceeding 30% of the infarcted area with some mild space‐occupying effect; and PH 2, dense blood clot(s) exceeding 30% of the infarct volume with significant space‐occupying effect.22, 23 Secondary safety endpoints were the incidence rate of sICH within 5 days (ie, any ICH confirmed by brain imaging, worsening of ≥2 points on the NIHSS, neurological deterioration persisting >24 hours); incidence of major systemic bleeding according to the International Society on Thrombosis and Hemostasis definition24; and incidence of serious adverse events, adverse events, and adverse drug reactions or death by any cause. Monitoring of adverse events and safety lasted for 30 days after the last visit. Secondary efficacy endpoints were neurological changes assessed by the NIHSS and clinical outcome evaluated by the mRS and BI at 90 days.
Exploratory endpoints were based on MRI findings on day 5: incidence, size, and number of any ICH by GRE, and the rate of cerebral infarct growth and infarct recurrence by DWI.
Statistical Analyses
In stage 1, a safety assessment of 10 subjects was judged to be appropriate for making a relevant decision in the DSMB and 11 patients were enrolled assuming a 10% dropout rate. In stage 2, for the primary endpoint of the incidence of CT‐identified ICH, sample size was calculated based on the simulation results with binomial random variables for the incidence and probability of ≥90% to securely detect the occurrence of at least >1 ICH or sICH events. Assuming an estimated incidence of ICH events of 12.4% in all stroke patients receiving rtPA (19.8% in high‐risk patients), sICH events of 6.9% in all stroke patients receiving rtPA (11.2% in high‐risk patients),25 and a dropout rate of 10%, 69 patients (23 subjects per group) were needed for the study.
The safety population was defined as patients who had received any study treatment at least once, including cases of incomplete infusion. The modified intention‐to‐treat (mITT) population included all randomized patients who had primary endpoint results without major inclusion/exclusion criteria violations. All endpoints in stage 1 were evaluated using the safety population. In stage 2, the primary endpoints and the proportional differences between groups were estimated with the 90% 2‐sided exact confidence interval (CI) in the mITT population.
All secondary safety outcomes and the proportional difference between groups were estimated at each time point with a 2‐sided 90% CI. Secondary efficacy endpoints and exploratory endpoints were evaluated with the Wilcoxon rank‐sum test or Fisher exact test in the mITT population, with Holm–Bonferroni correction for multiplicity. If a patient died before a prespecified visit, missing mRS scores were imputed to the worst possible score (6). The mRS distribution was further analyzed using ordinal logistic regression analysis including adjustment of variables associated with outcomes. Non‐normality and overdispersion that violated the proportional hazard assumption were noted. The effects of treatment in multivariate ordinal logistic regression were assessed after adjusting for the effects of age, sex, baseline NIHSS, tPA treatment time after stroke onset, and the use of endovascular surgery. The categorical group variable of 3 levels was included for analyzing the effect of treatment in the model, and the placebo group was considered as the reference group. We used the van Elteren test to assess temporal changes on the NIHSS score from baseline to day 90, using the cross‐tabulation with 2 rows of each group and the columns of modified ridit scores for NIHSS score changes stratified with time.
The statistical analysis was conducted using SPSS Statistics version 22.0 (IBM, Armonk, NY) and SAS version 9.4 software (SAS Institute, Cary, NC).
Results
Eighty patients were enrolled between June 5, 2016 and August 22, 2018 from 8 South Korean centers. Eleven patients were included in the safety and mITT populations for stage 1 (Fig 1), and their characteristics are shown in Table 2. In stage 2, 69 patients were randomly assigned to 3 groups: 24 to placebo, 24 to otaplimastat 40 mg, and 21 to otaplimastat 80 mg. The stage 2 mITT population included 22 patients in the placebo group, 22 in the otaplimastat 40 mg group, and 21 in the otaplimastat 80 mg group (see Fig 1). The baseline characteristics for stage 2 were balanced among the 3 treatment groups (see Table 2). The median age was 64 years (range = 33–80) and the median NIHSS score was 9 (range = 4–21) on admission. rtPA was administered at a median time of 1.6 hours (range = 0.5–4.2) after stroke onset, and 19 patients (29%, 19/66) underwent EVT. In stage 1, all patients in the safety population (n = 11) received all 6 doses of the study medication. In stage 2, a similar proportion of patients in each group received all 6 doses of the study medication; 88% (21/24) received the placebo, 88% (21/24) received otaplimastat 40 mg, and 91% (19/21) received otaplimastat 80 mg.
Figure 1.
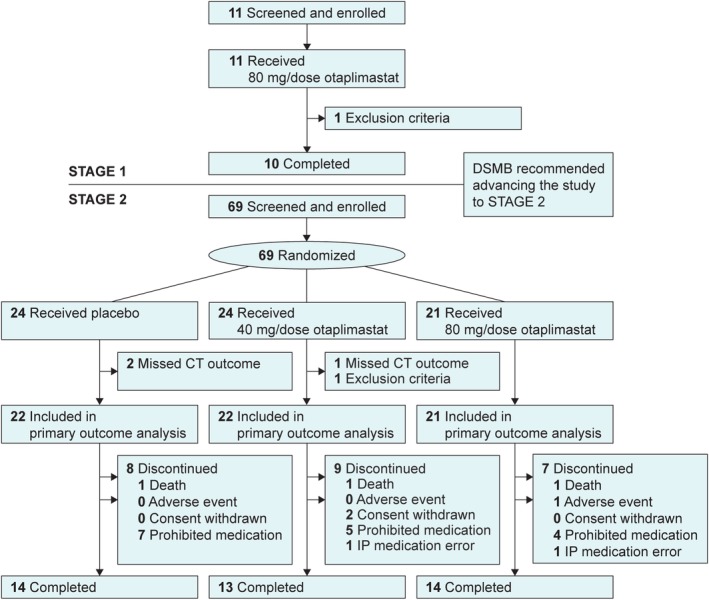
Trial profile. In stage 1, 1 patient was excluded because of low (10 g/dl) hemoglobin (exclusion criterion). In stage 2, 1 patient in the otaplimastat 40 mg group received warfarin (international normalized ratio <1.3, a major exclusion criteria violation) and was excluded. One patient with transient ischemic attack enrolled in stage 2 (the placebo group) was excluded in primary outcome analysis because of missing computed tomography (CT) outcome. Prohibited medication was defined as the use of antiplatelet agents or anticoagulants other than the prespecified dose of aspirin or clopidogrel from days 1 to 5. DSMB = Data and Safety Monitoring Board; IP = Investigational Product.
Table 2.
Baseline Characteristics of the Primary Analysis Population for Stages 1 and 2
| Characteristic | Stage 1, Safety Population | Stage 2, Modified Intention‐to‐Treat Population | ||
|---|---|---|---|---|
| Otaplimastat 80 mg, n = 11 | Placebo, n = 22 | Otaplimastat 40 mg, n = 22 | Otaplimastat 80 mg, n = 21 | |
| Demographics | ||||
| Median age, yr [min−max] | 72 [38–79] | 59 [49–77] | 63.5 [35–80] | 66 [33–79] |
| Female, n (%) | 5 (45%) | 7 (32%) | 5 (23%) | 9 (43%) |
| Risk factors, n (%) | ||||
| Hypertension | 9 (82%) | 13 (59%) | 15 (68%) | 12 (57%) |
| Diabetes | 6 (55%) | 6 (28%) | 6 (28%) | 7 (33%) |
| Hyperlipidemia | 1 (9%) | 4 (1%) | 4 (18%) | 6 (29%) |
| Atrial fibrillation | 0 | 3 (14%) | 5 (23%) | 2 (10%) |
| Smoking, current | 4 (36%) | 8 (37%) | 5 (23%) | 4 (19%) |
| Coronary artery disease | 2 (18%) | 2 (9%) | 0 | 3 (14%) |
| Previous history of stroke | 0 | 3 (14%) | 5 (23%) | 4 (19%) |
| TOAST classification | ||||
| Large artery atherosclerosis | 6 (55%) | 3 (14%) | 6 (27%) | 7 (33%) |
| Cardioembolism | 2 (18%) | 5 (23%) | 10 (45%) | 5 (24%) |
| Small vessel occlusion | 0 | 8 (36%) | 3 (14%) | 4 (19%) |
| Other determined etiology | 0 | 2 (9%) | 1 (5%) | 0 |
| Undetermined etiology | 3 (27%) | 4 (18%) | 2 (9%) | 5 (24%) |
| NIHSS, median [min−max] | 7 [4–10] | 8 [4–19] | 11 [4–21] | 9 [4–19] |
| Endovascular therapy, n (%) | 1 (9%) | 5 (24%) | 7 (32%) | 7 (33%) |
| Time interval, median [min–max] | ||||
| Symptom onset to intravenous rtPA, h | 2.3 [0.7–4.3] | 1.8 [0.9–4.2] | 1.4 [0.5–4.0] | 1.5 [0.8–3.6] |
| Symptom onset to study drug infusion, h | 2.7 [1.2–4.6] | 2.0 [1.3–4.7] | 1.7 [0.9–4.0] | 1.8 [0.8–4.1] |
NIHSS = National Institutes of Health Stroke Scale; rtPA = recombinant tissue plasminogen activator; TOAST = Trial of Org 10172 in Acute Stroke Treatment.
In stage 1, there were no cases of PH identified on brain CT at 24 hours. In addition, no subject experienced sICH or major systemic bleeding. Treatment‐emergent adverse events were noted in all 11 patients (100%), including nausea, dysuria, and pyrexia, each occurring in 27% of patients (3/11). Of the 39 adverse events recorded, 74% (29/39) were of mild severity and 26% (10/39) were of moderate severity. No serious adverse events or deaths were encountered. None of the adverse events was considered drug related. Therefore, this study met the DSMB criteria for advancement to stage 2.
In stage 2, for the primary outcome in the mITT population, PH was absent in the placebo (0/22) and otaplimastat 40 mg groups (0/22). One case of PH was detected in a patient who underwent EVT in the otaplimastat 80 mg group (ie, 1/21, 4.8%, 90% CI = 0.2–20.7), which was not significantly different from the placebo (treatment difference = 4.8%, 90% CI = −20.2 to 29.1). No differences were observed between otaplimastat 40 mg or 80 mg versus placebo in the secondary safety outcomes (Table 3). None of the patients developed sICH or major systemic bleeding in any of the 3 study groups. Treatment‐emergent adverse events occurred in 92% (22/24) of patients receiving placebo, 83% (20/24) taking otaplimastat 40 mg, and 95% (20/21) taking otaplimastat 80 mg (see Table 3). The majority were of mild–moderate severity (Table 4). The incidence of serious adverse events was similar between the treatment groups: 13% (3/24) for placebo (cerebral infarction, acute myocardial infarction, and lung adenocarcinoma), 17% (4/24) for otaplimastat 40 mg (3 cases of cerebral infarction and 1 with stress cardiomyopathy, chills, and muscle rigidity), and 14% (3/21) for the otaplimastat 80 mg group (cerebral infarction, toxic hepatitis, and kidney infection). Of the serious adverse events, chills, muscle rigidity, and toxic hepatitis were considered drug related and eventually resolved. Adverse events led to the withdrawal of 2 subjects in the placebo group (acute myocardial infarction and stroke in evolution) and 2 in the otaplimastat 80 mg group (toxic hepatitis and stroke in evolution). Four patients died, 2 in the placebo group (acute myocardial infarction and stroke in evolution) and 1 in each of the otaplimastat groups (both stroke in evolution). None was considered to be related to the study drug.
Table 3.
Stage 2 Secondary Safety Outcomes
| Outcome | Placebo, n = 24 | Otaplimastat 40 mg, n = 24 | Otaplimastat 80 mg, n = 21 |
|---|---|---|---|
| sICH within 5 days, n [90% CI] | 0 [0.0, 11.7] | 0 [0.0, 11.7] | 0 [0.0, 13.3] |
| Treatment difference, % [90% CI] | 0 [NC] | 0 [NC] | |
| Major systemic bleeding, ISTH, n [90% CI] | 0 [0.0, 11.7] | 0 [0.0, 11.7] | 0 [0.0, 13.3] |
| Treatment difference, % [90% CI] | 0 [NC] | 0 [NC] | |
| Treatment‐emergent adverse events, n (%) [90% CI] | 22 (91.7) [76.0, 98.5] | 20 (83.3) [65.8, 94.1] | 20 (95.2) [79.3, 99.8] |
| Treatment difference, % [90% CI] | −8.3 [−33.1, 17.3] | 3.6 [−21.0, 28.2] | |
| Serious adverse events, n (%) [90% CI] | 3 (12.5) [3.5, 29.2] | 4 (16.7) [5.9, 34.2] | 3 (14.3) [4.0, 32.9] |
| Treatment difference, % [90% CI] | 4.2 [−21.3, 29.2] | 1.8 [−23.2, 26.5] | |
| Adverse drug reactions, n (%) [90% CI] | 0 [0.0, 11.7] | 2 (8.3) [1.5, 24.0] | 1 (4.8) [0.2, 20.7] |
| Treatment difference, % [90% CI] | 8.3 [−17.3, 33.1] | 4.8 [−20.1, 28.9] | |
| Deaths, n (%) [90% CI] | 2 (8.3) [1.5, 24.0] | 1 (4.2) [0.2, 18.3] | 1 (4.8) [0.2, 20.7] |
| Treatment difference, % [90% CI] | −4.2 [−29.2, 21.3] | −3.6 [−28.2, 21.0] |
CI = confidence interval; ISTH = International Society on Thrombosis and Hemostasis; NC = not calculated; sICH = symptomatic intracranial hemorrhage.
Table 4.
Most Common Treatment‐Emergent Adverse Events (Safety Population)
| Adverse Event | Incidence: Patients, n (%) [events, n] | ||
|---|---|---|---|
| Placebo, n = 24 | Otaplimastat 40 mg, n = 24 | Otaplimastat 80 mg, n = 21 | |
| Any adverse event | |||
| Total | 22 (92%) [136] | 20 (83%) [115] | 20 (95%) [109] |
| Mild | 108 (79%) | 86 (75%) | 80 (74%) |
| Moderate | 26 (19%) | 25 (22%) | 26 (24%) |
| Severea | 2 (2%) | 4 (4%) | 3 (3%) |
| Preferred termb | |||
| Headache | 8 (33%) [9] | 6 (25%) [8] | 4 (19%) [4] |
| Pyrexia | 6 (25%) [6] | 4 (17%) [4] | 6 (29%) [6] |
| Productive cough | 3 (13%) [3] | 4 (17%) [4] | 2 (10%) [2] |
| Cough | 2 (8%) [2] | 4 (17%) [4] | 0 |
| Constipation | 6 (25%) [7] | 3 (13%) [3] | 6 (29%) [9] |
| Hiccups | 2 (8%) [2] | 3 (13%) [3] | 1 (5%) [1] |
| Nausea | 2 (8%) [2] | 3 (13%) [3] | 0 |
| Cerebral infarction | 1 (4%) [1] | 3 (13%) [3] | 1 (5%) [1] |
| Diabetes mellitus | 1 (4%) [1] | 3 (12%) [3] | 3 (14%) [3] |
| Hypokalemia | 0 | 3 (13%) [3] | 2 (10%) [2] |
| Urine output decreased | 1 (4%) [1] | 3 (13%) [3] | 2 (10%) [2] |
| Atrial fibrillation | 1 (4%) [1] | 3 (13%) [3] | 2 (10%) [2] |
| Diarrhea | 3 (13%) [3] | 2 (8%) [3] | 1 (5%) [1] |
| Dyspepsia | 5 (21%) [5] | 2 (8%) [2] | 1 (5%) [1] |
| Vomiting | 3 (13%) [3] | 2 (8%) [2] | 1 (5%) [1] |
| Dizziness | 1 (4%) [1] | 2 (8%) [2] | 1 (5%) [1] |
| Insomnia | 2 (8%) [2] | 2 (8%) [2] | 4 (19%) [5] |
| Stroke in evolution | 1 (4%) [1] | 1 (4%) [1] | 3 (14%) [3] |
| Fatigue | 3 (13%) [3] | 1 (4%) [1] | 1 (5%) [1] |
| Gingival bleeding | 3 (13%) [3] | 0 | 3 (14%) [3] |
| Rash | 0 | 0 | 3 (14%) [3] |
| Depression | 4 (17%) [4] | 0 | 2 (10%) [2] |
| Dysuria | 6 (25%) [6] | 0 | 2 (10%) [2] |
Severe adverse events occurred in 9 patients: 2 with placebo (stroke in progression, ST segment elevation myocardial infarction), 4 with otaplimastat 40 mg (stress cardiomyopathy, stroke in progression, recurrent cerebral infarction, muscle rigidity), and 3 with otaplimastat 80 mg (stroke in progression, acute kidney failure, and toxic hepatitis).
Events occurring in ≥10% of patients in any treatment group. The total incidence of infections and infestations including pneumonia and kidney infection was not significantly different among the treatment groups: 17% (4/24) for placebo, 8% (2/24) for otaplimastat 40 mg, and 10% (2/21) for the otaplimastat 80 mg group.
Secondary efficacy outcomes were derived from the mRS, the NIHSS, and the BI to demonstrate feasibility for future efficacy trials. The evaluation of ordinal data showed significantly different distribution of mRS scores with otaplimastat 40 mg versus placebo at 90 days (p = 0.026, Fisher exact test with Holm–Bonferroni multiplicity adjustment) but not with otaplimastat 80 mg (p = 0.502; Fig 2A, Table 5). However, the detection of true trends toward favorable mRS score distribution in 90‐day mRS was limited by the small sample size (adjusted odds ratio = 3.2, 95% CI = 0.9–10.9, p = 0.068). Figure 2B shows temporal changes in the NIHSS scores from baseline over the study period in 3 treatment groups. Although the improvement in the NIHSS scores was more pronounced in the otaplimastat 40 mg group (p = 0.006), the difference was not significant versus placebo at 28 days (p = 0.234) or 90 days (p = 0.414; see Table 5). The BI at 90 days was not significantly different among the 3 groups.
Figure 2.
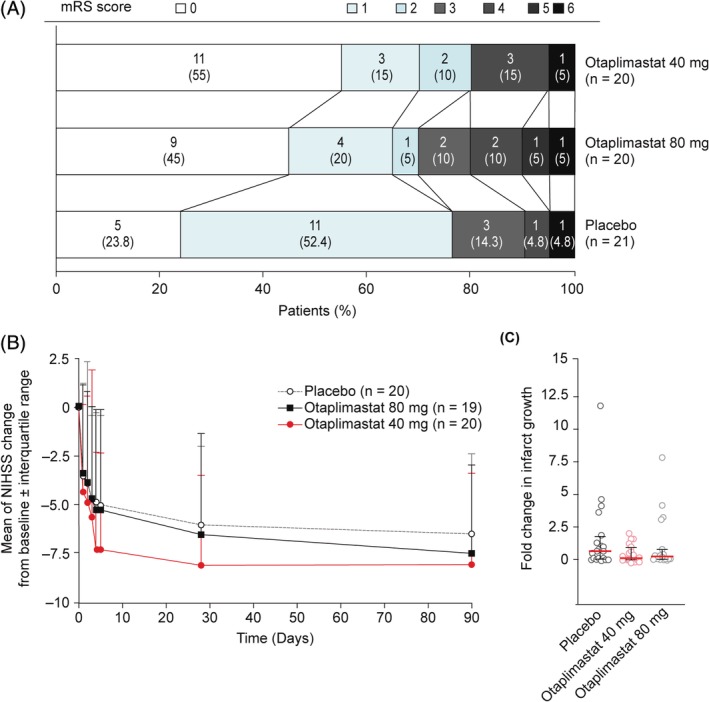
Secondary efficacy outcomes. (A) Distribution of the modified Rankin score (mRS) at 90 days and (B) National Institutes of Health Stroke Scale (NIHSS) score changes from baseline (modified intention‐to‐treat population). mRS distribution at 90 days uses imputed data only for death cases (mRS = 6). Error bars indicate standard deviation values. mRS on day 90: placebo, n = 21; otaplimastat 40 mg, n = 20; 80 mg, n = 20. NIHSS: placebo, n = 22; otaplimastat 40 mg, n = 22; 80 mg, n = 21 on day 0 to day 2. Placebo, n = 20; otaplimastat 40 mg, n = 22; 80 mg, n = 19 on day 3. Placebo, n = 20; otaplimastat 40 mg, n = 20; 80 mg, n = 19 on days 4, 5, and 28. Placebo, n = 19; otaplimastat 40 mg, n = 17; 80 mg, n = 17 on day 90. (C) Fold change of infarct growth was calculated as (individual infarct volume on day 5 − individual infarct volume on day 0)/mean of infarct volumes on day 0. Each point represents an individual patient, with the median (red bar) and interquartile ranges (black bars). Placebo, n = 20; otaplimastat 40 mg, n = 20; 80 mg, n = 19.
Table 5.
Stage 2 Clinical Outcomes (mITT Population)
| Placebo | Otaplimastat 40 mg | Otaplimastat 80 mg | |||||
|---|---|---|---|---|---|---|---|
| Median [IQR] | Median [IQR] | p | OR (95% CI) Adjustedp | Median [IQR] | p | OR (95% CI) Adjusted p | |
| Evaluable, na | 22 | 22 | 21 | ||||
| mRS | |||||||
| mRS at day 0 | 4 [3 to 4] | 4 [3 to 4] | 0.833 | 4 [3 to 4] | 0.192 | ||
| mRS at day 90 | 1.0 [1.0 to 1.0] | 0.0 [0.0 to 2.0] | 0.026 | OR 3.2 (0.9 to 10.9)b p = 0.068b | 1.0 [0.0 to 3.0] | 0.502 | OR 2.0 (0.6 to 6.7)b p = 0.246b |
| NIHSS changes | 0.006c | 0.940c | |||||
| Baseline at day 0 | 8.0 [5.0 to 14.0] | 11.0 [5.0 to 15.0] | >0.999 | 9.0 [5.0 to 13.0] | >0.999 | ||
| Changes at day 5 | −4.0 [−8.0 to −1.5] | −7.0 [−11.0 to −4.5] | 0.387 | −4.0 [−9.0 to −2.0] | 0.866 | ||
| Changes at day 28 | −4.0 [−9.0 to −3.0] | −7.0 [−11.5 to −5.0] | 0.234 | −5.5 [−10.0 to −2.0] | 0.922 | ||
| Changes at day 90 | −5.0 [−10.0 to −4.0] | −8.0 [−11.0 to −5.0] | 0.414 | −7.0 [−10.0 to −5.0] | 0.880 | ||
| Infarct growth, mld | |||||||
| Baseline at day 0 | 4.9 [0.5 to 8.1] | 5.9 [0.9 to 24.0] | 0.579 | 3.2 [0.7 to 15.6] | 0.982 | ||
| Growth at day 5 | 3.2 [0.3 to 7.9] | 1.7 [0.0 to 11.1] | >0.999 | 3.0 [0.5 to 9.0] | 0.866 | ||
| Fold change in 5 days | 0.7 [0.1 to 1.6] | 0.1 [0.0 to 0.9] | 0.303 | 0.3 [0.0 to 0.8] | 0.423 | ||
The mITT population is defined as the population composed of all subjects who belonged to the safety analysis set, fulfilled major inclusion/exclusion criteria, and had at least 1 post‐treatment assessment with primary endpoint. A total of 4 patients missed primary computed tomography outcome analysis and were excluded: 2 in the placebo group (1 transient ischemic attack, 1 death at day 1), and 2 in the 40 mg otaplimastat group (2 withdrew, 1 each at days 0 and 5). Analysis uses observed data. Probability value was obtained by Mann–Whitney test or Fisher exact test at each time point, with multiplicity adjustment by Holm–Bonferroni correction.
mRS at day 90: placebo, n = 21; otaplimastat 40 mg, n = 20; 80 mg, n = 20. NIHSS: placebo, n = 22; otaplimastat 40 mg, n = 22; 80 mg, n = 21 at day 0. Placebo, n = 20; otaplimastat 40 mg, n = 20; 80 mg, n = 19 at day 5 and day 28. Placebo, n = 19; otaplimastat 40 mg, n = 17; 80 mg, n = 17 at day 90. Infarct growth from day 0 to day 5: placebo, n = 20; otaplimastat 40 mg, n = 20; 80 mg, n = 19.
Ordinal logistic regression analysis for mRS distribution (mRS = 0–6) at day 90 uses imputed data for death cases. Unadjusted probability values are: p = 0.257 for placebo vs 40 mg and p = 0.674 for placebo vs 80 mg. Adjusted probability values and ORs show the effect of treatment, adjusted for age, sex, baseline NIHSS, tissue plasminogen activator treatment time after stroke onset, and the use of endovascular surgery.
Changes in NIHSS scores were analyzed with the van Elteren test.
Measurement of infarct growth by diffusion‐weighted imaging on days 0 and 5 (edema unadjusted). Infarct growth on day 5 = individual infarct volume on day 5 − individual infarct volume on day 0. Fold change of infarct growth = infarct growth on day 5 / mean of infarct volumes on day 0.
CI = confidence interval; IQR = interquartile range; mITT = modified intention‐to‐treat; mRS = modified Rankin scale; NIHSS = National Institutes of Health Stroke Scale; OR = odds ratio.
No significant differences were observed among treatment groups in the exploratory endpoints based on the MRI outcomes (Tables 5 and 6). Notably, the DWI‐identified median lesion volume growth (minimal, maximal) was 3.2 (−0.3, 57.2) with placebo, 1.7 (−3.3, 26.4) with otaplimastat 40 mg, and 3.0 (0.0, 89.1) with otaplimastat 80 mg (see Table 5). The incidence of any ICH was comparable at 35% (7/20) with placebo, 35% (7/20) with otaplimastat 40 mg (treatment difference = 0%, 90% CI = −27.8 to 27.8), and 37% (7/19) with otaplimastat 80 mg (treatment difference = 1.8%, 90% CI = −25.7 to 27.6), but a trend toward increased hemorrhage volumes and infarction recurrence in the otaplimastat 80 mg group was shown (see Table 6). Among 69 patients, 54 (78%) were treated with antiplatelet agents and 33% (18/54) of patients developed any ICH. The ICH rate was not significantly different between the patients receiving monotherapy and those receiving dual therapy (data not shown).
Table 6.
Stage 2 Exploratory Magnetic Resonance Imaging Outcomes (Modified Intention‐to‐Treat Population)
| Placebo | Otaplimastat 40 mg | Otaplimastat 80 mg | |||
|---|---|---|---|---|---|
| Value | p | Value | p | ||
| Evaluable, n | 20 | 20 | 19 | ||
| Any ICH at day 5 on GRE | |||||
| Patients, n (%) | 7 (35%) | 7 (35%) | >0.999 | 7 (37%) | >0.999 |
| HI 1 | 5 (25%) | 3 (15%) | >0.999 | 5 (26%) | >0.999 |
| HI 2 | 2 (10%) | 3 (15%) | >0.999 | 1 (5%) | >0.999 |
| PH 1 | 0 | 0 | NA | 1 (5%) | >0.999 |
| PH 2 | 0 | 1 (5%) | >0.999 | 0 | NA |
| Volume, ml, median [IQR] | |||||
| Baseline at day 0 | 0.0 [0.0−0.0] | 0.0 [0.0−0.0] | >0.999 | 0.0 [0.0−0.0] | >0.999 |
| Growth at day 5 | 0.0 [0.0−0.1] | 0.0 [0.0−0.2] | >0.999 | 0.0 [0.0−1.2] | >0.999 |
| Infarction recurrence until day 5 on DWI | |||||
| Patients n (%) | 0 | 1 (5%) | 5 (26%) | ||
| Infarct size, ml, median [IQR] | 0.0 [0.0−0.0] | 0.0 [0.0−0.0] | >0.999 | 0.0 [0.0−0.1] | 0.060 |
Difference between placebo and each treatment group was tested using the Wilcoxon rank sum test, or by Mann–Whitney test or Fisher exact test, with multiplicity adjustment by Holm–Bonferroni correction.
DWI = diffusion‐weighted imaging; GRE = gradient recalled echo; HI = hemorrhagic infarct; ICH = intracerebral hemorrhage; IQR = interquartile range; NA = not applicable; PH = parenchymal hematoma.
Discussion
In this SAFE‐TPA trial, we found no serious adverse events in the stage 1 study. In stage 2, 1 patient developed PH, and 1 had hepatotoxicity in the otaplimastat 80 mg group. In this case, the aspartate aminotransferase level reached up to 759 IU on day 2 and otaplimastat was discontinued (data not shown). This subject was negative for viral hepatitis markers, and the liver function test results returned to the normal level on day 14. Thus, although this potential hepatotoxicity should be investigated further, coadministration of intravenous otaplimastat is feasible and appears to be generally safe in patients with AIS receiving rtPA treatment.
Although rtPA treatment grants better clinical outcome, the risks of ICH and mortality are increased when the treatment is delayed.1, 2, 3, 4 Previous studies have consistently shown that otaplimastat reduces the incidence of ICH in experimental animals with stroke receiving rtPA,15, 16, 17 whereas it did not interfere with the fibrinolytic activity of rtPA (authors’ unpublished data). However, in the current study there were no differences in the incidence of ICH on CT scan on day 1, and on GRE at 5 days between the placebo and otaplimastat groups (see Table 3). The rate of sICH in the present study was low compared with that in previous trials, probably owing to the early thrombolytic therapy (mean tPA treatment delay = 1.78 hours), inclusion of patients with relatively young age (mean = 63 years), less severe strokes (mean baseline NIHSS score = 10), and better thrombolytic recanalization (patient population with nearly complete or complete reperfusion within 5 days with modified Thrombolysis in Cerebral Ischemia [mTICI] 2b–3: >77%).26 Thus, the low occurrence of ICH and the small sample size preclude any conclusion regarding the potential benefit of otaplimastat in preventing ICH in patients with AIS receiving rtPA.
For efficacy outcome, we found that the distribution of the mRS scores was significantly different between the placebo and the otaplimastat 40 mg groups (see Table 5, Fig 2A; p = 0.026), associated with the greater proportion of the patients with good outcome (mRS ≤ 2) in the otaplimastat 40 mg group. The magnitude of the clinical benefit of rtPA treatment itself in our study was larger than that in the recent meta‐analysis with 9 rtPA phase 3 trials (patients with mRS = 0–1 at day 90: 76% vs 31%).3 This very favorable clinical outcome may, at least in part, be attributed to the rapid administration of rtPA (mean = 1.78 hours; see Table 2) after stroke onset in our study, compared with previous rtPA trials (mean = 4.00 hours).3 Considering that the main theoretical neuroprotective mechanism of otaplimastat is to reduce brain edema and ICH after rtPA therapy, especially in delayed therapy,20, 21 the benefit of otaplimastat may have been underappreciated in our patient population.
The 7‐day NIHSS score27 or early decrease in NIHSS score15, 16, 28 has been used as a marker for clinical efficacy in recent exploratory trials in acute stroke. Notably, the NIHSS score tended to decrease more markedly in the otaplimastat 40 mg than that in the placebo group during the acute phase of stroke, although the difference was not statistically significant at each time point (see Table 5, Fig 2B). In addition, the growth of MRI‐identified infarct volume was the lowest in the otaplimastat 40 mg group (see Table 5, Fig 2C). These findings collectively suggest that otaplimastat may exert a beneficial effect during the early phase of stroke in patients receiving rtPA therapy, although further clarification in a larger trial remains necessary.
Recent clinical trials of neuroprotectant as an adjunctive therapy have evaluated several potential clinically effective mechanisms29, 30: reduction of ICH,14, 15, 16 improvement of recanalization and collateral flow,16 and/or suppression of infarct growth.16 Otaplimastat did not significantly decrease the incidence or volume of ICH (see Tables 3 and 6), although this possibility is not entirely excluded, considering the small sample size and early thrombolysis, as discussed earlier. Considering that patients receiving otaplimastat 40 mg had the lowest rate (53% vs 74% placebo) of full recanalization (mTICI score = 3), it is unlikely that this agent improves recanalization. Nevertheless, patients in the otaplimastat 40 mg group exhibited the trend of quickly diminishing neurological deficits. This observation appears to be in line with pleiotropic neuroprotective mechanisms preclinically elucidated that include reduction of inflammatory cell migration, blood–brain barrier stabilization, and enhanced expression of antioxidant enzymes (ie, MnSOD) in the ischemic brain.18, 19
Notably, in patients receiving otaplimastat 80 mg, there was no significant beneficial effect on the mRS distribution. There was no increase in serious adverse effects in this group compared with the otaplimastat 40 mg or placebo groups, except for the one case of hepatotoxicity. Although the otaplimastat 80 mg group had more patients with ICH and recurrent infarction, the clinical outcomes in theses population was not worse than those in other groups with similar initial stroke severity (data not shown). It remains uncertain why the encouraging findings with otaplimastat 40 mg were not mirrored with otaplimastat 80 mg. It was reported that a particular dosage, timing, and duration of MMP inhibition impact the reduction of lesion sizes after ICH, and blood–brain barrier permeability or neurovascular remodeling in the poststroke period.31, 32, 33, 34, 35 For example, an early brief MMP inhibition confers a neuroprotective effect, whereas a prolonged inhibition dysregulates delayed neuroinflammatory responses and hampers recovery.33 Treatment with the MMP inhibitor ilomastat once, but not twice, following focal stroke rescued visual plasticity,34 suggesting the importance of carefully defining dosing regimen to ensure the spatiotemporal inhibition of targeted MMPs and resulting clinical efficacy.
There are limitations in this study. As discussed above, the small number of enrolled patients does not allow us to make a powerful conclusion, especially for the efficacy endpoints. However, the main purpose of this phase 2 study was to examine the feasibility and safety of otaplimastat adjuvant therapy rather than confirming its efficacy. In addition, because we did not administer otaplimastat in patients who did not receive rtPA, it remains unknown whether this agent is feasible and potentially beneficial in patients who are not treated with rtPA. Especially, along with recent positive clinical trials,36, 37, 38 EVT has been increasingly used. Further studies are needed to examine the feasibility, safely, and efficacy of otaplimastat in patients undergoing EVT without intravenous rtPA.
Despite the limitations, we provided evidence that administering otaplimastat to patients with AIS receiving rtPA is feasible and generally safe. There was also an intriguing signal that otaplimastat 40 mg may improve neurological outcomes in these patients. Further clinical studies on otaplimastat or other potentially useful adjunctive therapies for rtPA may help identify novel ways to improve the clinical outcomes of patients with AIS receiving rtPA.39
Author Contributions
J.S.K. formulated the conception and design of this study; J.S.K., K.B.L., J.‐H.P, S.M.S, K.O., E.‐G.K., D.C., Y.‐H.H., and E.‐J.L. contributed to data acquisition; all authors contributed to the analysis and interpretation of the data, and made critical revision and approved for the final version of the manuscript; J.S.K., E.‐J.L., C.J., and B.S.K. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Acknowledgment
This study was supported by a grant from the Korea Health Technology R&D project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (HI15C2796).
We thank the DSMB members, Drs L. B. Goldstein, P. Amarenco, U. Dirnagl, S. Arbe‐Barnes, and Y. J. Lee for their support of the DSMB; K. W. Kim, S. C. Chung, and the Central Imaging Core Lab members at Asan Medical Center, Seoul, Korea for imaging setup and analysis; J. B. Fiebach for independent review of CT/MRI; K.‐S. Yang and J. S. Lee for statistical support for the study; and N. Richardson for editorial support.
References
- 1. National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group . Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 1995;333:1581−1587. [DOI] [PubMed] [Google Scholar]
- 2. Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008;359:1317−1329. [DOI] [PubMed] [Google Scholar]
- 3. Emberson J, Lees KR, Lyden P, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta‐analysis of individual patient data from randomised trials. Lancet 2014;384:1929−1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thomalla G, Simonsen CZ, Boutitie F, et al. WAKE‐UP Investigators. MRI‐guided thrombolysis for stroke with unknown time of onset. N Engl J Med 2018;379:611−622. [DOI] [PubMed] [Google Scholar]
- 5. Wang X, Tsuji K, Lee SR, et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke 2004;35:2726−2730. [DOI] [PubMed] [Google Scholar]
- 6. Lenglet S, Montecucco F, Mach F, et al. Analysis of the expression of nine secreted matrix metalloproteinases and their endogenous inhibitors in the brain of mice subjected to ischaemic stroke. Thromb Haemost 2014;112:363−378. [DOI] [PubMed] [Google Scholar]
- 7. Jickling GC, Liu D, Stamova B, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab 2014;34:185−199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chamorro A, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol 2016;15:869−881. [DOI] [PubMed] [Google Scholar]
- 9. Pena ID, Borlongan C, Shen G, Davis W. Strategies to extend thrombolytic time window for ischemic stroke treatment: an unmet clinical need. J Stroke 2017;19:50−60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanazawa M, Takahashi T, Nishizawa M, Shimohata T. Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. J Atheroscler Thromb 2017;24:240−253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chamorro A. Neuroprotectants in the era of reperfusion therapy. J Stroke 2018;20:197−207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Malhotra K, Chang JJ, Khunger A, et al. Minocycline for acute stroke treatment: a systematic review and meta‐analysis of randomized clinical trials. J Neurol 2018;265:1871−1879. [DOI] [PubMed] [Google Scholar]
- 13. Chamorro A, Amaro S, Castellanos M, et al. Safety and efficacy of uric acid in patients with acute stroke (URICO‐ICTUS): a randomised, double‐blind phase 2b/3 trial. Lancet Neurol 2014;13:453−460. [DOI] [PubMed] [Google Scholar]
- 14. Lyden P, Pryor KE, Coffey CS, et al. Final results of the RHAPSODY trial: a multi‐center, phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3K3A‐APC, a recombinant variant of human activated protein C, in combination with tissue plasminogen activator, mechanical thrombectomy or both in moderate to severe acute ischemic stroke. Ann Neurol 2019;85:125−136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu Z, Fu Y, Tian D, et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation 2015;132:1104−1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian DC, Shi K, Zhu Z, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol 2018;84:717−728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noh SJ, Lee JM, Lee KS, et al. SP‐8203 shows neuroprotective effects and improves cognitive impairment in ischemic brain injury through NMDA receptor. Pharmacol Biochem Behav 2011;100:73−80. [DOI] [PubMed] [Google Scholar]
- 18. Noh SJ, Lee SH, Shin KY, et al. SP‐8203 reduces oxidative stress via SOD activity and behavioral deficit in cerebral ischemia. Pharmacol Biochem Behav 2011;98:150−154. [DOI] [PubMed] [Google Scholar]
- 19. Kim W‐K, Ju C, Jalin AMAA, et al. Therapeutic efficacy and pharmacological mechanisms of SP‐8203 for treatment of cerebral ischemia. Stroke 2015;46:ATP239 (Abstract). [Google Scholar]
- 20. Ju C, Anthony Jalin A, Song HY, et al. Extension of therapeutic time window of tissue plasminogen activator with SP‐8203 combination therapy in rat embolic stroke models. Eur Stroke J 2016;1:581 (Abstract). [Google Scholar]
- 21. National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group . Intracerebral hemorrhage after intravenous t‐PA therapy for ischemic stroke. Stroke 1997;28:2109−2118. [DOI] [PubMed] [Google Scholar]
- 22. Hacke W, Kaste M, Fieschi C, et al. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Cooperative Acute Stroke Study (ECASS). JAMA 1995;274:1017−1025. [PubMed] [Google Scholar]
- 23. Hacke W, Kaste M, Fieschi C, et al. Randomised double‐blind placebo‐controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Second European‐Australasian Acute Stroke Study Investigators. Lancet 1998;352:1245−1251. [DOI] [PubMed] [Google Scholar]
- 24. Schulman S, Kearon C, Subcommittee on Control of Anticoagulation of the Scientific Standardization Committee of the International Society on Thrombosis and Haemostasis . Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non‐surgical patients. J Thromb Haemost 2005;3:692−694. [DOI] [PubMed] [Google Scholar]
- 25. Saposnik G, Fang J, Kapral MK, et al. The iScore predicts effectiveness of thrombolytic therapy for acute ischemic stroke. Stroke 2012;43:1315−1322. [DOI] [PubMed] [Google Scholar]
- 26. Tanne D, Kasner SE, Demchuk AM, et al. Markers of increased risk of intracerebral hemorrhage after intravenous recombinant tissue plasminogen activator therapy for acute ischemic stroke in clinical practice: the Multicenter rt‐PA Stroke Survey. Circulation 2002;105:1679–1685. [DOI] [PubMed] [Google Scholar]
- 27. Kerr DM, Fulton RL, Lees KR, et al. Seven‐day NIHSS is a sensitive outcome measure for exploratory clinical trials in acute stroke: evidence from the Virtual International Stroke Trials Archive. Stroke 2012;43:1401−1403. [DOI] [PubMed] [Google Scholar]
- 28. Broderick JP, Lu M, Kothari R, et al. Finding the most powerful measures of the effectiveness of tissue plasminogen activator in the NINDS tPA Stroke Trial. Stroke 2000;31:2335−2341. [DOI] [PubMed] [Google Scholar]
- 29. Kim JS. tPA helpers in the treatment of acute ischemic stroke: are they ready for clinical use? J Stroke 2019;21:160−174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizuma A, You JS, Yenari MA. Targeting reperfusion injury in the age of mechanical thrombectomy. Stroke 2018;49:1796−1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med 2008;14:497−500. [DOI] [PubMed] [Google Scholar]
- 32. Chang JJ, Emanuel BA, Mack WJ, et al. Matrix metalloproteinase‐9: dual role and temporal profile in intracerebral hemorrhage. J Stroke Cerebrovasc Dis 2014;23:2498−2505. [DOI] [PubMed] [Google Scholar]
- 33. Zhao BQ, Wang S, Kim HY, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med 2006;12:441−445. [DOI] [PubMed] [Google Scholar]
- 34. Pielecka‐Fortuna J, Kalogeraki E, Fortuna MG, et al. Optimal level activity of matrix metalloproteinases is critical for adult visual plasticity in the healthy and stroke‐affected brain. Elife 2015;26:e11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bencsik P, Pálóczi J, Kocsis GF, et al. Moderate inhibition of myocardial matrix metalloproteinase‐2 by ilomastat is cardioprotective. Pharmacol Res 2014;80:36−42. [DOI] [PubMed] [Google Scholar]
- 36. Goyal M, Menon BK, van Zwam WH, et al. Endovascular thrombectomy after large‐vessel ischaemic stroke: a meta‐analysis of individual patient data from five randomised trials. Lancet 2016;387:1723−1731. [DOI] [PubMed] [Google Scholar]
- 37. Nogueira RG, Jadhav AP, Haussen DC, et al; DAWN Trial Investigators . Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med 2018;378:11−21. [DOI] [PubMed] [Google Scholar]
- 38. Albers GW, Marks MP, Kemp S, et al; DEFUSE 3 Investigators . Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med 2018;378:708−718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henninger N, Fisher M. Extending the time window for endovascular and pharmacological reperfusion. Trans Stroke Res 2016;7:284−293. [DOI] [PubMed] [Google Scholar]