

Abstract
Patients with epidermal growth factor receptor (EGFR)‐mutated non‐small cell lung cancer (NSCLC) harboring BIM deletion polymorphism (BIM deletion) have poor responses to EGFR TKI. Mechanistically, the BIM deletion induces preferential splicing of the non‐functional exon 3‐containing isoform over the functional exon 4‐containing isoform, impairing TKI‐induced, BIM‐dependent apoptosis. Histone deacetylase inhibitor, vorinostat, resensitizes BIM deletion‐containing NSCLC cells to EGFR‐TKI. In the present study, we determined the safety of vorinostat‐gefitinib combination and evaluated pharmacodynamic biomarkers of vorinostat activity. Patients with EGFR‐mutated NSCLC with the BIM deletion, pretreated with EGFR‐TKI and chemotherapy, were recruited. Vorinostat (200, 300, 400 mg) was given daily on days 1‐7, and gefitinib 250 mg was given daily on days 1‐14. Vorinostat doses were escalated based on a conventional 3 + 3 design. Pharmacodynamic markers were measured using PBMC collected at baseline and 4 hours after vorinostat dose on day 2 in cycle 1. No dose‐limiting toxicities (DLT) were observed in 12 patients. We determined 400 mg vorinostat as the recommended phase II dose (RP2D). Median progression‐free survival was 5.2 months (95% CI: 1.4‐15.7). Disease control rate at 6 weeks was 83.3% (10/12). Vorinostat preferentially induced BIM mRNA‐containing exon 4 over mRNA‐containing exon 3, acetylated histone H3 protein, and proapoptotic BIMEL protein in 11/11, 10/11, and 5/11 patients, respectively. These data indicate that RP2D was 400 mg vorinostat combined with gefitinib in BIM deletion/EGFR mutation double‐positive NSCLC. BIM mRNA exon 3/exon 4 ratio in PBMC may be a useful pharmacodynamic marker for treatment.
Keywords: BIM deletion polymorphism, EGFR‐TKI, NSCLC, resistance, vorinostat
Vorinostat, in combination with gefitinib, induced acetylated histone H3 protein expression, as well as a decrease in the BIM mRNA exon 3/exon 4 ratio in PBMC from BIM deletion polymorphism/EGFR mutation double‐positive NSCLC patients. These results provide proof of concept that the combined therapy can mitigate the functional effects of BIM deletion polymorphism.
![]()
1. INTRODUCTION
The majority of patients with non‐small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR)‐activating mutations, such as exon 19 deletion and an L858R point mutation, show marked responses to EGFR‐TKI, such as gefitinib, erlotinib, afatinib, and osimertinib.1, 2, 3, 4, 5 However, 20%‐30% of patients with EGFR‐activating mutations show intrinsic resistance to EGFR‐TKI. Molecular mechanisms of the intrinsic resistance, including a pre‐existing EGFR‐T790M resistance mutation‐positive clone and the activation of alternative pathways, such as hepatocyte growth factor (HGF)/MET, have been discovered.6
Decreased activity of BIM, also known as Bcl‐2‐like protein 11, a proapoptotic molecule that belongs to the Bcl‐2 family, has been recognized as an important mechanism mediating intrinsic resistance to EGFR‐TKI. BIM upregulation is essential for the induction of apoptosis in lung cancer cells with EGFR mutations treated with first‐generation EGFR‐TKI, and low BIM protein levels are associated with resistance to EGFR‐TKI.7, 8 Underlying the importance of BIM in EGFR‐TKI resistance, a functional BIM deletion polymorphism, specifically, a 2903‐bp deletion in intron 2, was discovered in East Asian individuals (13%‐18%) and found to confer inferior responses to EGFR‐TKI.9, 10 Subsequently, the BIM deletion was also found in South American patients with NSCLC (15.7%).11 Mechanistically, the BIM deletion results in the mutually exclusive splicing of exon 3 over the BH3‐encoding (proapoptotic) exon 4 in BIM pre‐mRNA and leads to the production of an inactive BIM protein isoform (BIMγ) lacking the BH3 domain. In turn, this reduces the expression of the proapoptotic BIM protein isoform (BIMEL) in EGFR‐mutant lung cancer cell lines following TKI exposure and is sufficient to confer TKI resistance.9 Several meta‐analyses have reported an association between the BIM deletion polymorphism and shorter progression‐free survival (PFS) in patients with NSCLC harboring EGFR mutations who received either gefitinib or erlotinib treatment.12, 13, 14, 15, 16, 17 Therefore, restoration of BIM activity in patients with the BIM deletion may be an important strategy to overcome intrinsic resistance to EGFR‐TKI in EGFR‐mutated NSCLC.
Vorinostat (suberoylanilide hydroxamic acid [SAHA]), has been approved in 20 countries to date, including Japan, for cutaneous T‐cell lymphoma as monotherapy. It is a small‐molecule inhibitor of histone deacetylase (HDAC) that causes the acetylation of histone proteins, including histone H2, and induces cell differentiation, cell cycle arrest, and apoptosis in several types of tumor cells.18 We previously reported that the combined use of vorinostat and gefitinib was able to preferentially induce transcription of the proapoptotic exon 4‐containing BIM isoform over the inactive exon 3‐containing isoform, thus resensitizing BIM deletion‐containing EGFR‐mutated NSCLC cell lines to TKI in vitro and in vivo.19 Two clinical trials combining TKI and vorinostat have been conducted: a phase I/II study in patients with advanced NSCLC, regardless of the presence/absence of EGFR mutation in Korea,20 and a phase I/II study in patients with advanced EGFR‐mutated NSCLC after EGFR‐TKI progression in Spain.21 However, neither combination regimen showed significant efficacy in these populations, suggesting the need for improved patient selection and the development of pharmacodynamic biomarkers of vorinostat activity.
Based on our preclinical findings, we designed the present phase I study, named VICTORY‐J “Vorinostat‐Iressa Combined Therapy on Resistance by BIM Polymorphism in EGFR Mutant Lung Cancer”, to evaluate the safety of combined therapy with vorinostat and gefitinib, and to determine the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of vorinostat combined with a fixed dose of gefitinib for patients with EGFR‐mutated NSCLC harboring the BIM deletion polymorphism.22 In addition, we conducted pharmacodynamic analyses to identify biomarkers of vorinostat activity.
2. MATERIALS AND METHODS
2.1. Study design
This study was an open‐label, multi‐institutional phase I dose escalation study in patients with EGFR‐mutated (exon 19 deletion and L858R mutation) NSCLC with a BIM deletion polymorphism. Primary endpoint was to determine MTD, which was defined as the highest dose level at which two or fewer of six patients experienced dose‐limiting toxicity (DLT).
Three to six patients were enrolled at each dose level of vorinostat. With a fixed dose of gefitinib, dose escalation of vorinostat was used, in accordance with a conventional 3 + 3 design using an escalation scheme (Figure S1). Initially, three patients were enrolled at the first level. If one or two patients experienced DLT, an additional three patients were enrolled to that level. If three of six patients experienced DLT, the previous dose level was declared the MTD. If two or fewer of the six patients experienced DLT, dose escalation was permitted to continue. After termination of protocol treatment, the patients were allowed any further treatment and followed until death over a period of at least 1 year. This study was conducted in accordance with the International Committee for Harmonization Good Clinical Practice (ICH‐GCP) guidelines and the Declaration of Helsinki. The study protocol was approved by the institutional review boards of all participating institutions. Written informed consent was provided by all patients before registration. This study was registered with UMIN Clinical Trials Registry (UMIN00001519) and ClinicalTrials.gov (NCT02151721).
2.2. Patient eligibility
Prior to enrolment in the study, patients had to fulfil all of the following criteria: histologically or cytologically diagnosed NSCLC (excluding squamous cell carcinoma); NSCLC of clinicopathological stage IIIB or IV for which radical radiation therapy was impractical or there was a recurrence after surgery; EGFR mutations (deletion of exon 19 and L858R mutation of exon 21) for which the clinical benefits of an EGFR‐TKI (gefitinib, erlotinib, or afatinib) were recognized by testing methods that were listed by the national health insurance; history of treatment with an EGFR‐TKI (gefitinib, erlotinib, or afatinib) and a history of pathological deterioration during treatment; history of treatment with cytotoxic anticancer agents (not including pre‐ or postoperative chemotherapy in the previous 1 year or more from the day of final dose); confirmed BIM polymorphism by the PCR fragment analytical method and the sequence method at the central laboratory; a lesion measurable according to the RECIST guidelines version 1.1; 20 years of age and older; ECOG Performance Status (PS) 0 or 1; estimated life expectancy of 12 or more weeks; provision of written informed consent to participate in the present study; and adequate hematological, liver, renal, and respiratory function within 14 days before entry.
Patients were excluded for any of the following reasons: received a cytotoxic anticancer agent within the past 4 weeks, received an EGFR‐TKI within the past 7 days, or surgery or radiotherapy for a primary tumor or mediastinum within the past 6 months; interstitial lung disease or history thereof, radiation pneumonitis treated with corticosteroids or a history thereof; detection of known resistance mutations of EGFR (eg, T790M).
Between March 2014 and February 2017, 527 patients with advanced EGFR‐mutated NSCLC were screened for BIM deletion polymorphism. Among them, 77 patients (14.6%) had BIM deletion polymorphism (75 patients [14.2%] were heterozygous and two patients [0.4%] were homozygous) and a total of 12 patients were enrolled in the present study.
2.3. Treatment and assessment
Vorinostat and gefitinib were purchased from Taiho Pharmaceutical and AstraZeneca, respectively. Vorinostat (level 1: 200 mg, level 2: 300 mg, level 3: 400 mg) was given orally once daily on days 1‐7, and 250 mg gefitinib was given orally once daily in a cycle of 14 days; this continued until the criteria for respite, dosage reduction, or discontinuation of the protocol treatment were met.
Toxicities were graded in accordance with the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. DLT was defined as follows: grade ≥1 intestinal lung disease; grade ≥4 neutropenia lasting 5 days or more; febrile neutropenia; grade ≥3 thrombocytopenia requiring platelet transfusion; grade ≥4 thrombocytopenia; any grade uncontrollable skin toxicity; grade ≥3 nonhematological toxicity. DLT was evaluated during the first two cycles (14 days per cycle) of therapy. Secondary endpoints were the pharmacokinetics and pharmacodynamics of vorinostat and gefitinib, PFS, overall survival (OS), response rate (RR), duration of response and complete response, disease control rate (DCR), and incidence of adverse events defined by CTCAE version 4.0.
We assessed the objective tumor response in accordance with version 1.1 of the revised RECIST guidelines.23 At baseline, we carried out imaging of the chest and abdominal by computed tomography (CT) and brain magnetic resonance imaging (MRI) or CT within 28 days before randomization. After the start of treatment, assessments were carried out at 6‐week intervals during the first 24 weeks and at 12‐week intervals after this period. Brain MRI or CT was required to follow the same schedule, but only for patients with brain metastasis at the time of enrolment (Table S1). Investigators reviewed the images of patients. PFS was defined as the time from the start of protocol treatment to the first occurrence of progression or to death. OS was defined as the time from the start of protocol treatment to death. DCR was defined as the proportion of patients who achieved complete response, partial response, and stable disease for at least 6 weeks.
2.4. Statistical analysis
The population analyzed for the primary endpoint included the enrolled patients with complete safety data on DLT during the first two cycles.
2.5. Genotyping of the BIM deletion polymorphism
Cellular DNAs were extracted from patients’ PBMC using a DNeasy Blood and Tissue Kit (Qiagen). To recognize the presence of the wild‐type and deletion alleles, we conducted PCR as reported previously.19 Primer sequences used were as follows. Forward: 5′‐CCACCAATGGAAAAGGTTCA‐3, reverse: 5′‐CTGTCATTTCTCCCCACCAC‐3′ for detection of wild‐type BIM; and forward: 5′‐ CTGTCATTTCTCCCCACCAC‐3′, reverse: 5′‐ GGCACAGCCTCTATGGAGAA‐3′ for identification of the BIM deletion polymorphism. The primer pairs yielded PCR products of 362 and 284 bp, respectively.
2.6. Pharmacokinetic analysis
Blood sampling for pharmacokinetics was carried out pretreatment and post‐treatment (0.5, 1, 2, 4, 6, 8, 10, 24, 48, and 72 hours after the first dose). Concentration of gefitinib and vorinostat was measured in plasma and serum, respectively.24, 25
2.7. Pharmacodynamic analysis
Patients’ PBMC were sampled at baseline and 4 hours after giving vorinostat on day 2 in cycle 1. Total RNA and proteins were extracted from PBMC using lysis buffer and RNeasy PLUS Mini kit (Qiagen), respectively. Reverse transcription of the collected RNAs was done using SuperScript VILO cDNA synthesis Kit and Master Mix (Invitrogen). Expression of BIM mRNA was quantitatively measured by ViiA 7 Real‐Time PCR System (Applied Biosystems) using the following primers: BIM exon 2A (forward: 5′‐ATGGCAAAGCAACCTTCTGATG‐3′; reverse: 5′‐GGCTCTGTCTGTAGGGAGGT‐3′), BIM exon 3 (forward: 5′‐CAATGGTAGTCATCCTAGAGG‐3′; reverse: 5′‐GACAAAATGCTCAAGGAAGAGG‐3′), BIM exon 4 (forward: 5′‐TTCCATGAGGCAGGCTGAAC‐3′; reverse: 5′‐CCTCCTTGCATAGTAAGCGTT‐3′) and β‐actin (forward: 5′‐GGACTTCGAGCAAGAGATGG‐3′; reverse: 5′‐AGCACTGTGTTGGCGTACAG‐3′).
Proteins harvested from PBMC were separated by SDS‐PAGE. The proteins were transferred onto PVDF membranes (Bio‐Rad); the membranes were immersed in StartingBlock T20 (TBS) Blocking Buffer (ThermoFisher Scientific) for 1 hour at approximately 20°C, and by incubation for longer than 8 hours at 4°C with antibodies against acetylated histone H3 (Lys27), BIM, and β‐actin (Cell Signaling Technology). After three washes in Tris‐buffered saline with polyoxyethylene sorbitan monolaurate (TBST), the membranes were incubated for 1 hour at room temperature with HRP‐conjugated secondary antibodies. Proteins labeled with secondary antibodies were visualized by using SuperSignal West Dura Extended Duration Substrate Enhanced Chemiluminescent Substrate (ThermoFisher Scientific).
3. RESULTS
3.1. Patients and safety
From June 2014 to February 2017, 12 patients were enrolled into this study. Patient characteristics are summarized in Table 1. Previously treated EGFR‐TKI in each patient are shown in Table S2.
Table 1.
Characteristics of patients with EGFR‐mutated NSCLC harboring BIM deletion polymorphism
| Total | Level 1 (200 mg) | Level 2 (300 mg) | Level 3 (400 mg) | |
|---|---|---|---|---|
| Analysis of population | 12 | 3 | 3 | 6 |
| Gender | ||||
| Male/Female | 7/5 | 1/2 | 2/1 | 4/2 |
| Age, y | ||||
| Median (range) | 69.5 (51‐84) | 73.0 (60‐78) | 71.0 (51‐76) | 68.0 (62‐84) |
|
Smoking status Brinkman index | ||||
| Never | 5 | 2 | 2 | 1 |
| 30≤ <1000 | 4 | 0 | 1 | 3 |
| 1000≤ | 3 | 1 | 0 | 2 |
| Performance status | ||||
| 0/1 | 7/5 | 2/1 | 1/2 | 4/2 |
| Histology | ||||
| Adenocarcinoma/Other | 12/0 | 3/0 | 3/0 | 6/0 |
| BIM polymorphism | ||||
| Heterozygote/Homozygote | 11/1 | 3/0 | 3/0 | 5/1 |
| EGFR mutation | ||||
| Exon19 deletion/Exon21 L858R | 8/4 | 2/1 | 2/1 | 4/2 |
|
Duration from previous EGFR‐TKI treatment (days) | ||||
| Median (range) | 58 (8‐574) | 274 (8‐518) | 11 (8‐191) | 58 (14‐574) |
Abbreviations: EGFR, epidermal growth factor receptor; NSCLC, non‐small cell lung cancer.
Planned dose escalation was completed without reaching the MTD. DLT were not observed in any patients. Treatment‐related adverse events are summarized in Table 2. The most common adverse events were diarrhea (92%), anorexia (75%), oral mucositis (58%), rash, weight loss, dysgeusia, nausea (50%), vomiting, and malaise (42%). Treatment‐related grade 3 adverse events included grade 3 hypokalemia (17%), lung infection and thrombocytopenia (8%) (Table 3). No treatment‐related death or grade 4 adverse events were observed.
Table 2.
Number of patients with treatment‐related adverse events
| Total | Level 1 | Level 2 | Level 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Analysis of population | 12 | 3 | 3 | 6 | ||||||
| Grade (CTCAE version 4.0) | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| Diarrhea | 11 (92%) | 3 | 0 | 0 | 2 | 0 | 0 | 3 | 3 | 0 |
| Anorexia | 9 (75%) | 0 | 2 | 0 | 1 | 1 | 0 | 4 | 1 | 0 |
| Oral mucositis | 7 (58%) | 0 | 2 | 0 | 1 | 1 | 0 | 3 | 0 | 0 |
| Rash acneiform | 6 (50%) | 1 | 0 | 0 | 0 | 1 | 0 | 3 | 1 | 0 |
| Weight loss | 6 (50%) | 0 | 2 | 0 | 2 | 0 | 0 | 1 | 1 | 0 |
| Dysgeusia | 6 (50%) | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 2 | 0 |
| Nausea | 6 (50%) | 0 | 1 | 0 | 1 | 0 | 0 | 2 | 1 | 1 |
| Vomiting | 5 (42%) | 2 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 |
| Malaise | 5 (42%) | 1 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 |
| Hypokalemia | 3 (25%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 2 |
| Dry skin | 3 (25%) | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Paronychia | 3 (25%) | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
Table 3.
Number of patients with ≥grade 3 treatment‐related adverse events
| Grade 3 | |
|---|---|
| Nausea | 1 (Level 3) |
| Hypokalemia | 2 (Level 3) |
| Thrombocytopenia | 1 (Level 3) |
| Lung infection | 1 (Level 2) |
| Peripheral neuropathy | 1 (Level 3) |
Adverse events that resulted in drug cessation were observed in four cases: one at level 1 (pneumonia); two at level 2 (diarrhea, vomiting, fever, alanine aminotransferase [ALT] elevation, and loss of appetite); and one at level 3 (neutropenia). One adverse event resulting in vorinostat dose reduction was observed at level 2 (thrombocytopenia).
Based on these data, RP2D was determined as 250 mg gefitinib daily and 400 mg/day vorinostat biweekly.
3.2. Efficacy
Median PFS and OS of 12 patients at levels 1‐3 were 156.5 days (5.2 months; 43‐479 days) and 673 days (22.4 months; 411 days–not reached), respectively (Figure 1). Disease control (stable disease assessed for at least 6 weeks) was achieved in 10 out of 12 (83.3%) patients with a history of progressive disease during EGFR‐TKI treatment (Table 4).
Figure 1.
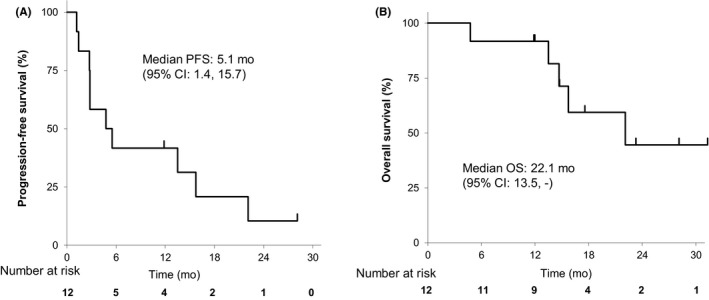
Progression‐free survival (PFS) and overall survival (OS) of patients with BIM deletion‐positive epidermal growth factor receptor‐mutated non‐small cell lung cancer treated with gefitinib combined with vorinostat. Kaplan‐Meier curves for PFS (A) and OS (B) of 12 patients are shown. The 95% confidence intervals were calculated by using the Brookmeyer and Crowley method
Table 4.
Summary of tumor response
| Total | Best overall response by RECIST | SD ≥6 wk | DCR |
DCR 95% CI |
||||
|---|---|---|---|---|---|---|---|---|
| CR | PR | SD | PD | NE | ||||
| 12 | 0 | 0 | 10 | 2 | 0 | 10 | 83.3% | 0.52, 0.98 |
Abbreviations: CI, confidence interval; CR, complete response; DCR, disease‐control rate; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease.
3.3. Pharmacokinetics
Median T max of vorinostat in the 200, 300, and 400 mg groups was 2, 4, and 4 hours, respectively (Table 5, Figure S2); T 1/2 (mean ± SD) was 3.6 ± 3.4, 1.5 ± 0.5, and 2.0 ± 1.1 hours, respectively, and C max (mean ± SD) was 602 ± 166, 661 ± 33, and 1010 ± 335 nmol/L, respectively. AUClast (mean ± SD) was 2340 ± 753, 2430 ± 348, and 4790 ± 2040 nmol·h/L, respectively, and AUCinf (mean ± SD) was 2430 ± 809, 2480 ± 407, and 4810 ± 2040 nmol·h/L, respectively.
Table 5.
Pharmacokinetic parameters
| C max (ng/mL) | AUC last (ng•h/L) | AUC inf (ng•h/L) | T max (h) | t 1/2 (h) | |||
|---|---|---|---|---|---|---|---|
| Gefitinib | 250 mg | No. of patients | 12 | 12 | 11 | 12 | 11 |
| Median (range) | 250 (148‐802) | 3430 (1880‐9850) | 8270 (3190‐23 400) | 4.0 (3.8‐8) | 22.6 (13.3‐42.6) | ||
| Mean (SD) | 294 (181) | 3910 (2140) | 8700 (5430) | 4.6 (1.3) | 23.8 (8.6) |
| C max (nmol/L) | AUC last (nmol•h/L) | AUC inf (nmol•h/L) | T max (h) | t 1/2 (h) | |||
|---|---|---|---|---|---|---|---|
| Vorinostat | Level 1 (200 mg) | No. of patients | 3 | 3 | 3 | 3 | 3 |
| Median (range) | 615 (430‐762) | 2720 (1470‐2830) | 2830 (1500‐2960) | 2.0 (1.1‐4.0) | 1.8 (1.6‐7.5) | ||
| Mean (SD) | 602 (166) | 2340 (753) | 2430 (809) | 2.4 (1.5) | 3.6 (3.4) | ||
| Level 2 (300 mg) | No. of patients | 3 | 3 | 3 | 3 | 3 | |
| Median (range) | 643 (641‐699) | 2250 (2210‐2830) | 2260 (2240‐2950) | 4.0 (1.8‐4.0) | 1.3 (1.1‐2.1) | ||
| Mean (SD) | 661 (33) | 2430 (348) | 2480 (407) | 3.3 (1.3) | 1.5 (0.5) | ||
| Level 3 (400 mg) | No. of patients | 6 | 6 | 6 | 6 | 6 | |
| Median (range) | 944 (695‐1630) | 4250 (2960‐8610) | 4270 (3010‐8650) | 4.0 (0.9‐4.1) | 2.1 (0.8‐3.1) | ||
| Mean (SD) | 1010 (335) | 4790 (2040) | 4810 (2040) | 3.5 (1.3) | 2.0 (1.1) |
Median T max of gefitinib in the 200, 300, and 400 mg groups was 4, 4, and 4.1 hours, respectively (Figure S3); T 1/2 (mean ± SD) was 20.2 ± 7.3, 25.1 ± 5.5, and 25.2 ± 11.4 hours, respectively, and C max (mean ± SD) was 344 ± 44, 240 ± 98, and 295 ± 253 ng/mL, respectively. AUClast (mean ± SD) was 4524 ± 779, 3398 ± 1050, and 3853 ± 3004 ng·h/L, respectively, and AUCinf (mean ± SD) was 8674 ± 3064, 7333 ± 1808, and 9537 ± 8064 ng·h/L, respectively. There was no significant difference between the three groups. These results indicated that vorinostat was unlikely to affect the pharmacokinetics of gefitinib.
3.4. Pharmacodynamics
Pharmacodynamic analyses of PBMC obtained at baseline and 4 hours after giving vorinostat and gefitinib on day 2 in cycle 1 were carried out. As a measure of the effects of vorinostat on the expression and splicing of BIM transcripts, we evaluated, by using real‐time qPCR, BIM exon 2 expression as a surrogate expression marker for all BIM transcripts. In addition, we also separately evaluated the expression of BIM exon 3 and exon 4, which represent the BIM isoform that lacks the proapoptotic BH3 domain and the isoform that has the proapoptotic BH3 domain, respectively. In two cases, (VK‐02) and (VS‐02), we were unable to measure these parameters owing to poor quality of the mRNA and protein, respectively.
We found that the BIM mRNA exon 3/exon 4 ratio was decreased by treatment with gefitinib and vorinostat in all 11 patients assessed (Figure 2A,B). As reported, HDAC inhibitors, alone or in combination with a TKI, may cause the expression of proapoptotic exon 4‐containing BIM transcripts to increase by significantly more than the expression of exon 3‐containing BIM transcripts.19, 26, 27 We also noted that VI‐03, the only patient in this study that was homozygous for the BIM deletion polymorphism, had the highest expression of exon 3‐containing transcripts, as well as the highest exon 3/exon 4 ratio at baseline. This was in line with our previous findings that cells that were homozygous for the BIM deletion polymorphism tended to have higher expression of exon 3‐containing transcripts than of the proapoptotic exon 4‐containing transcripts.9, 26
Figure 2.
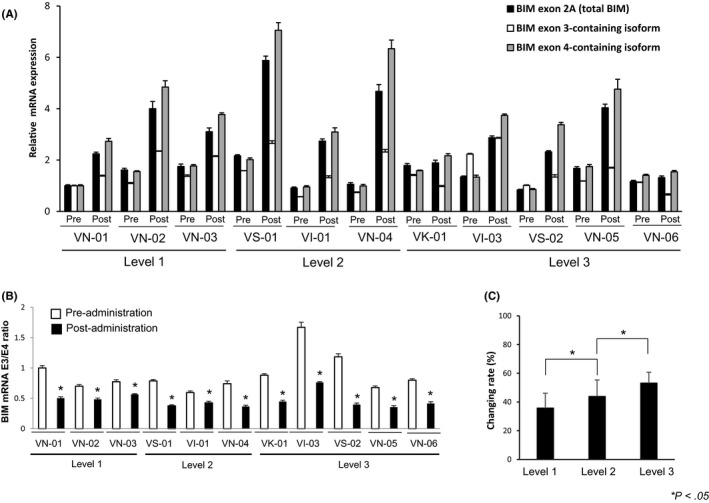
Pharmacodynamic analysis of vorinostat through transcription of BIM mRNA isoforms in PBMC. A, PBMC were harvested at baseline and 4 h after giving gefitinib and 200 mg (n = 3), 300 mg (n = 3), and 400 mg (n = 5) vorinostat on day 2 of cycle 1. RNA was purified from PBMC and mRNA expression of BIM exon 2A (representative of total BIM mRNA expression), exon 3 (representative of the inactive isoform of BIM mRNA), and exon 4 (representative of the proapoptotic isoform of BIM mRNA) is shown. B, BIM mRNA exon 3/exon 4 ratio. *P < .05 vs baseline. C, Change in BIM mRNA exon 3/exon 4 ratio before and 2 d after treatment with vorinostat and gefitinib. Bars indicate mean ± SD
Mean odds ratio of exon 3/exon 4 before and after treatment with gefitinib and vorinostat was 1.9376 (1.5988 at level 1, 1.8436 at level 2, and 2.1974 at level 3), and the results showed a significant upward trend in terms of a vorinostat dose‐dependent increase in proapoptotic exon 4‐containing BIM transcripts (Figure 2C).
In addition, we also assessed the acetylation status of histone H3 as a measure of the inhibitory effect of vorinostat on HDAC activity.28 We also evaluated the protein expression of the pro‐apoptotic BIM isoform, BIMEL, as an indicator of the ability of vorinostat to enhance the proapoptotic isoforms of BIM. As expected, vorinostat markedly increased the protein expression of acetylated histone H3 in 10/11 patients (91%) (3/3 at level 1, 3/3 at level 2, and 4/5 at level 3) (Figure 3). Moreover, vorinostat increased proapoptotic BIM protein (BIMEL) expression in five of 11 patients (45.5%) (1/3 at level 1, 3/3 at level 2, and 1/5 at level 3). Summary of the pharmacokinetic and pharmacodynamic analyses for each patient is shown in Table S3.
Figure 3.
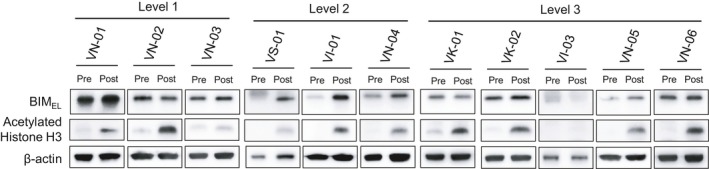
Pharmacodynamic analysis of vorinostat through protein expression of acetylated histone H3 and pro‐apoptotic BIMEL in PBMC. PBMC were harvested at baseline (Pre) and 4 h after dose (Post) of gefitinib and 200 mg (A: n = 3), 300 mg (B: n = 3), and 400 mg (C: n = 5) vorinostat on day 2 of cycle 1. Protein expression in PBMC was determined by western blotting
These results indicated that vorinostat could stimulate histone acetylation through the inhibition of HDAC. Furthermore, vorinostat increased the expression of proapoptotic exon 4‐containing BIM transcript and protein in the majority of patients with EGFR‐mutated NSCLC with BIM deletion polymorphism. This observation was in line with a previous report on the use of HDAC inhibitors alone or in combination with a TKI, which can increase the protein expression of BIM.19, 26, 27 Therefore, these data have shown the technical feasibility of monitoring these parameters as markers of vorinostat activity.
4. DISCUSSION
In the present study, we determined the RP2D of vorinostat, 400 mg/day biweekly, in combination with gefitinib for patients with BIM deletion polymorphism‐positive EGFR‐mutated NSCLC. Two recent clinical trials of vorinostat combined with either gefitinib or erlotinib showed the feasibility of combined therapy in NSCLC. In the Spanish phase I/II trial, vorinostat plus erlotinib (150 mg daily) was evaluated in erlotinib‐refractory EGFR‐mutated NSCLC.21 In the Korean phase I/II trial, vorinostat plus gefitinib (250 mg daily) was evaluated in unselected NSCLC.20 Both trials determined the recommended dose of vorinostat as 400 mg/day biweekly, but drug concentrations in the plasma or serum were not evaluated. Therefore, the pharmacokinetic interactions of vorinostat and EGFR‐TKI have been unknown until now. In our study, we showed that selection of patients with both an EGFR mutation and the BIM deletion polymorphism could be carried out in a clinical setting, and that vorinostat 400 mg/day biweekly was the recommended dose for further studies in BIM deletion polymorphism/EGFR mutation‐positive populations in NSCLC. Furthermore, our pharmacokinetic analyses showed that vorinostat did not affect the dynamics of gefitinib in these populations.
The most important and informative findings in our study are the pharmacodynamic analyses. We found that vorinostat, in combination with gefitinib, induced acetylated histone H3 protein expression, as well as a decrease in the BIM mRNA exon 3/exon 4 ratio in PBMC. These results have provided proof of concept that the combined therapy can mitigate the functional effects of the BIM deletion polymorphism, which, to the best of our knowledge, remains the most common resistance‐conferring germline variant in EGFR‐mutated NSCLC.17 Moreover, we were also able to show increased expression of a proapoptotic BIM protein isoform (BIMEL) in primary patient material. Interestingly, while combined therapy resulted in an increase in protein expression of acetylated histone H3, as well as a decrease in the BIM exon 3/exon 4 ratio, not all individuals who showed these changes had a concomitant increase in BIMEL protein. The reason for this discrepancy is unclear at present, but may encompass both technical and biological explanations. For example, although PBMC samples were harvested and handled according to a common protocol, protein degradation may have occurred between blood sampling and protein purification. For example, in one case (VS‐02), although we detected a decrease in the BIM exon 3/exon 4 ratio, we were unable to detect BIMEL protein. As a result, the BIM mRNA exon 3/exon 4 ratio may be a more robust pharmacodynamic biomarker for vorinostat activity than the measurement of BIMEL protein.
It is well recognized that a small population of cells adapts to initial treatment with EGFR‐TKI as persisters and becomes the base of acquired resistant lesions.29 BIM deletion polymorphism which prevents tumor cell apoptosis is insufficient for tumor cell growth. The persisters need to acquire additional resistance factors which allow tumor cell growth. The persisters need to acquire additional resistance factors which allow tumor cell growth. EGFR‐T790M may be one of such additional resistance factors, because we detected T790M in EGFR‐mutated NSCLC cell line PC‐9, which had been genetically edited to have homozygous BIM deletion polymorphism, after the induction of gefitinib resistance in the in vitro condition.26 Although we did not examine additional resistance mechanisms in specimens from patients enrolled in the VICTROY‐J study, resistant factors other than EGFR‐T790M might also be detectable.
Prognosis of patients with T790M‐negative, EGFR‐mutated NSCLC who are refractory to EGFR‐TKI is reported to be very poor.30, 31, 32, 33 In the present study, we excluded patients with EGFR‐T790M‐positive NSCLC because our previous in vitro results showed that vorinostat did not sensitize T790M‐positive EGFR‐mutated NSCLC cells to gefitinib.19 Therefore, we recruited patients with T790M‐negative EGFR‐mutated NSCLC who were refractory to EGFR‐TKI, and who had been treated with at least one regimen of cytotoxic chemotherapy. As a result, our study comprised heavily pretreated patients. Although the limited number of patients in our study precludes firm conclusions about clinical benefit, the DCR of 83.3% and median PFS of 5.2 months suggests that the combination should be evaluated in phase II studies.
In addition to the gefitinib/vorinostat combination we evaluated, the concept of combining EGFR‐TKI and HDAC inhibitors can be applied by using new‐generation EGFR‐TKI, as well as novel HDAC inhibitors that have recently been described. Recently, the third‐generation EGFR‐TKI, osimertinib, has been recognized as one of the standard drugs for first‐line treatment of EGFR‐mutated NSCLC.5 However, a population of patients show intrinsic resistance, even to osimertinib.5 In these patients, resistance may be mediated by the BIM deletion polymorphism, as we found that BIM deletion polymorphism‐positive EGFR‐mutated NSCLC cell lines (PC‐3) were resistant to osimertinib‐induced apoptosis19, 26; importantly, the addition of vorinostat could resensitize these cells to EGFR‐TKI. Moreover, we reported that HDAC3 was an important regulator of BIM pre‐mRNA splicing and that the activity of vorinostat was likely to require inhibition of HDAC3.26 Therefore, the combination of osimertinib and new‐generation HDAC inhibitors, including HDAC3‐selective inhibitors and others (eg, panobinostat),34 might be a promising first‐line treatment for BIM deletion polymorphism‐positive EGFR‐mutated NSCLC.
In conclusion, we have determined vorinostat at 400 mg/day biweekly combined with gefitinib 250 mg daily as the recommended dose for phase II studies in patients with NSCLC who are double‐positive for BIM deletion polymorphisms and EGFR mutations. Further, it is warranted to evaluate our approach for exploration of the efficacy of combination EGFR‐TKI/HDAC therapy in larger cohorts.
DISCLOSURE
S. Yano obtained grants from Chugai Pharma, Boehringer Ingelheim Japan, Novartis, and received speakers fees from AstraZeneca, Chugai Pharma, Boehringer Ingelheim Japan, Novartis, and Pfizer. T. Hase obtained grants from Boehringer Ingelheim Japan, AstraZeneca, Taiho Pharma, and Novartis, and received speakers fees from Boehringer Ingelheim Japan, Chugai Pharma, AstraZeneca, Ono Pharma, MSD, Bristol‐Myers Squibb, Taiho Pharma, Novartis, and travel support from Taiho Pharma, AstraZeneca, and Novartis. A. Hata obtained speakers fees and research grants from AstraZeneka, Taiho, Boehringer Ingelheim Japan, MSD, and Chugai Pharma. Y. Murakami obtained speakers fees from Taiho Pharma, Merck Sharp & Dohme, AstraZeneca, Chugai Pharma, Lilly Japan, Ono Pharma, Bristol‐Myers Squibb, Pfizer, Novartis, and Boehringer Ingelheim Japan, K. Yoshimura received speakers fees from Chugai Pharma, Astra Zeneca, and Eli Lilly. N. Katakami obtained speakers fees and research grants from AstraZeneka, Taiho, Boehringer Ingelheim Japan, MSD, and Chugai Pharma. T. Takahashi obtained grants from AstraZeneca, Chugai Pharma, Eli Lilly, Ono Pharma, MSD, and Pfizer, and received speakers fees from AstraZeneca, Chugai Pharma, Eli Lilly, from Ono Pharma, MSD, Pfizer, Boehringer Ingelheim Japan, and Roche Diagnostics. Y. Hasegawa obtained grants from AstraZeneca, Eli Lilly, Chugai Pharma, Ono Pharma, Novartis, Bristol‐Myers Squibb, and Taiho Pharma, and received speakers fees from AstraZeneca, Chugai Pharma, Boehringer Ingelheim Japan, MSD. and Pfizer. ST Ong obtained grants from the National Medical Research Council of Singapore (CSASI18may‐0004, NMRC/CSA/0051/2013, and NMRC/GMS/CIRG/1330/2012). The other authors have nothing to disclose.
Supporting information
ACKNOWLEDGMENTS
We thank the patients, their families, and all staff that participated in this study. We are grateful to Dr Hiroyuki Mano, National Cancer Center Research Institute, for encouraging this study. This study was supported by grants from the Japan Agency for Medical Research and Development, AMED, grant numbers 15Aak0101016h0003, 15Ack0106113h0002, and 16ck0106207h0001 (to SY) and Kanazawa University Hospital.
Takeuchi S, Hase T, Shimizu S, et al. Phase I study of vorinostat with gefitinib in BIM deletion polymorphism/epidermal growth factor receptor mutation double‐positive lung cancer. Cancer Sci. 2020;111:561–570. 10.1111/cas.14260
This study was previously presented in part at the ESMO Asia 2017 Congress, Singapore, Nov 17‐19, 2017
This study was registered with UMIN Clinical Trials Registry (UMIN00001519) and ClinicalTrials.gov (NCT02151721).
REFERENCES
- 1. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380‐2388. [DOI] [PubMed] [Google Scholar]
- 2. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121‐128. [DOI] [PubMed] [Google Scholar]
- 3. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13:239‐246. [DOI] [PubMed] [Google Scholar]
- 4. Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327‐3334. [DOI] [PubMed] [Google Scholar]
- 5. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med. 2018;378:113‐125. [DOI] [PubMed] [Google Scholar]
- 6. Yano S, Takeuchi S, Nakagawa T, Yamada T. Ligand‐triggered resistance to molecular targeted drugs in lung cancer: roles of HGF and EGFR ligands. Cancer Sci. 2012;103:1189‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faber AC, Corcoran RB, Ebi H, et al. BIM expression in treatment‐naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Costa C, Molina MA, Drozdowskyj A, et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR‐mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res. 2014;20:2001‐2010. [DOI] [PubMed] [Google Scholar]
- 9. Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 2012;18: 521‐528. [DOI] [PubMed] [Google Scholar]
- 10. Isobe K, Hata Y, Tochigi N, et al. Clinical significance of BIM deletion polymorphism in non‐small‐cell lung cancer with epidermal growth factor receptor mutation. J Thorac Oncol. 2014;9:483‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cardona AF, Rojas L, Wills B, et al. BIM deletion polymorphisms in Hispanic patients with non‐small cell lung cancer carriers of EGFR mutations. Oncotarget. 2016;7:68933‐68942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ying HQ, Chen J, He BS, et al. The effect of BIM deletion polymorphism on intrinsic resistance and clinical outcome of cancer patient with kinase inhibitor therapy. Sci Rep 2015;5:11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma JY, Yan HJ, Gu W Association between BIM deletion polymorphism and clinical outcome of EGFR‐mutated NSCLC patient with EGFR‐TKI therapy: a meta‐analysis. J Cancer Res Ther. 2015;11:397‐402. [DOI] [PubMed] [Google Scholar]
- 14. Huang WF, Liu AH, Zhao HJ, Dong HM, Liu LY, Cai SX. BIM gene polymorphism lowers the efficacy of EGFR‐TKIs in advanced nonsmall cell lung cancer with sensitive EGFR mutations: a systematic review and meta‐analysis. Medicine (Baltimore). 2015;94:e1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nie W, Tao X, Wei H, Chen WS, Li B. The BIM deletion polymorphism is a prognostic biomarker of EGFR‐TKIs response in NSCLC: a systematic review and meta‐analysis. Oncotarget. 2015;6:25696‐25700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou Q, Zhan P, Lv T, Song Y. The relationship between BIM deletion polymorphism and clinical significance of epidermal growth factor receptor‐mutated non‐small cell lung cancer patients with epidermal growth factor receptor‐tyrosine kinase inhibitor therapy: a meta‐analysis. Transl Lung Cancer Res. 2015;4:792‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Su W, Zhang X, Cai X, Peng M, Wang F, Wang Y. BIM deletion polymorphism predicts poor response to EGFR‐TKIs in nonsmall cell lung cancer: an updated meta‐analysis. Medicine (Baltimore). 2019;98:e14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sato A. Vorinostat approved in Japan for treatment of cutaneous T‐cell lymphoma: status and prospects. Onco Targets Ther. 2012;5:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakagawa T, Takeuchi S, Yamada T, et al. EGFR‐TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013;73:2428‐2434. [DOI] [PubMed] [Google Scholar]
- 20. Han JY, Lee SH, Lee GK, et al. Phase I/II study of gefitinib (Iressa(®)) and vorinostat (IVORI) in previously treated patients with advanced non‐small cell lung cancer. Cancer Chemother Pharmacol 2015;75:475‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reguart N, Rosell R, Cardenal F, et al. Phase I/II trial of vorinostat (SAHA) and erlotinib for non‐small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer 2014;84:161‐167. [DOI] [PubMed] [Google Scholar]
- 22. Takeuchi S, Yoshimura K, Fujiwara T, et al. Phase I study of combined therapy with vorinostat and gefitinib to treat BIM deletion polymorphism‐associated resistance in EGFR‐mutant lung cancer (VICTROY‐J): a study protocol. J Med Invest. 2017;64:321‐325. [DOI] [PubMed] [Google Scholar]
- 23. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 24. Ranson M, Hammond LA, Ferry D, et al. ZD1839, a selective oral epidermal growth factor receptor‐tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J Clin Oncol. 2002;20:2240‐2250. [DOI] [PubMed] [Google Scholar]
- 25. Rubin EH, Agrawal NG, Friedman EJ, et al. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. J Clin Cancer Res. 2006;12:7039‐7045. [DOI] [PubMed] [Google Scholar]
- 26. Tanimoto A, Takeuchi S, Arai S, et al. Histone deacetylase 3 inhibition overcomes BIM deletion polymorphism‐mediated osimertinib‐resistance in EGFR‐mutant lung cancer. Clin Cancer Res. 2017;23:3139‐3149. [DOI] [PubMed] [Google Scholar]
- 27. Rauzan M, Chuah CT, Ko TK, Ong ST. The HDAC inhibitor SB939 overcomes resistance to BCR‐ABL kinase Inhibitors conferred by the BIM deletion polymorphism in chronic myeloid leukemia. PLoS ONE. 2017;12:e0174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA. 1998;95:3003‐3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taniguchi H, Yamada T, Wang R, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10(1):259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR‐mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17:1616‐1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hata A, Katakami N, Yoshioka H, et al. Rebiopsy of non‐small cell lung cancer patients with acquired resistance to epidermal growth factor receptor‐tyrosine kinase inhibitor: comparison between T790M mutation‐positive and mutation‐negative populations. Cancer. 2013;119:4325‐4332. [DOI] [PubMed] [Google Scholar]
- 32. Li W, Ren S, Li J, et al. T790M mutation is associated with better efficacy of treatment beyond progression with EGFR‐TKI in advanced NSCLC patients. Lung Cancer. 2014;84:295‐300. [DOI] [PubMed] [Google Scholar]
- 33. Liu Y, Sun L, Xiong ZC, et al. Meta‐analysis of the impact of de novo and acquired EGFR T790M mutations on the prognosis of patients with non‐small cell lung cancer receiving EGFR‐TKIs. Onco Targets Ther. 2017;10:2267‐2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panobinostat approved for multiple myeloma. Cancer Discov. 2015;5:OF4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials