

Abstract
Background
Non‐invasive prenatal testing (NIPT) for fetal aneuploidies has rapidly been incorporated into clinical practice. Current NGS‐based methods can reliably detect fetal aneuploidies non‐invasively with fetal fraction of at least 4%. Inaccurate fetal fraction assessment can compromise the accuracy of the test as affected samples with low fetal fraction have an increased risk for misdiagnosis. Using a novel set of fetal‐specific differentially methylated regions (DMRs) and methylation sensitive restriction digestion (MSRD), we developed a multiplex ddPCR assay for accurate detection of fetal fraction in maternal plasma.
Methods
We initially performed MSRD followed by methylation DNA immunoprecipitation (MeDIP) and NGS on fetal and non‐pregnant female tissues to identify fetal‐specific DMRs. DMRs with the highest methylation difference between the two tissues were selected for fetal fraction estimation employing MSRD and multiplex ddPCR. Chromosome Y multiplex ddPCR assay (YMM) was used as a reference standard, to develop our fetal fraction estimation model in male pregnancy samples. Additional 123 samples were tested to examine whether the model is sex dependent and/or ploidy dependent.
Results
In all, 93 DMRs were identified of which seven were selected for fetal fraction estimation. Statistical analysis resulted in the final model which included four DMRs (FFMM). High correlation with YMM‐based fetal fractions was observed using 85 male pregnancies (r = 0.86 95% CI: 0.80–0.91). The model was confirmed using an independent set of 53 male pregnancies.
Conclusion
By employing a set of well‐characterized DMRs, we developed a SNP‐, sex‐ and ploidy‐independent methylation‐based multiplex ddPCR assay for accurate fetal fraction estimation.
Keywords: differentially methylated regions, fetal fraction estimation, multiplex ddPCR, non‐invasive prenatal testing
We have developed a multiplex droplet digital PCR (ddPCR) assay for accurate fetal fraction estimation using methylation sensitive restriction enzymes (MSREs) and a robust set of novel fetal‐specific differentially methylated regions (DMRs). Clinical implementation of our assay as a first‐tier fetal fraction estimation in NIPT can minimize the possibility of incorrect calls due to low fetal fraction estimation.

What is already known about the topic?
Despite the technological advances in NIPT, accurate fetal fraction estimation still presents a challenge.
NGS‐based methods can reliably detect fetal aneuploidies non‐invasively with fetal fraction of at least 4%.
Inaccurate estimation of fetal fraction can potentially compromise the sensitivity of the assay.
What does this study add?
We describe a methylation‐based assay, using a novel set of well‐characterized differentially methylated regions, for accurate estimation of fetal fraction using multiplex ddPCR.
Our assay can rapidly be incorporated into laboratory workflows and can provide accurate fetal fraction information prior to sample library preparation minimizing further any possibility of incorrect calls due to low fetal fraction.
1. INTRODUCTION
Non‐invasive prenatal testing (NIPT) for fetal aneuploidies has rapidly been incorporated into clinical practice. A large number of validation studies have demonstrated high aneuploidy detection rates, leading professional organizations to recommend NIPT as a primary screening in pregnant women regardless of their risk status (Benn et al., 2015; "Committee Opinion No. 640: Cell‐free DNA Screening for Fetal Aneuploidy", 2015; Gregg et al., 2016).
Since the discovery of cell‐free fetal DNA (cffDNA) in the maternal circulation (Lo et al., 1997), several approaches have been developed for the detection of pregnancy‐induced complications (Leung, Zhang, Lau, Chan, & Lo, 2001; Sekizawa et al., 2001) and fetal abnormalities (El Khattabi et al., 2016; Papageorgiou et al., 2011; Tong, Chiu, et al., 2010; Tong et al., 2006; Tong, Jin, et al., 2010; Tsaliki et al., 2012) while the highest impact in the NIPT field was made with the introduction of next‐generation sequencing (Fan, Blumenfeld, Chitkara, Hudgins, & Quake, 2008; Koumbaris et al., 2016; Zhang et al., 2015; Zimmermann et al., 2012); although the limited amount of cffDNA in the maternal circulation still presents a challenge. cffDNA constitutes about 10%–20% of the total cell‐free DNA (Chiu et al., 2011) and can range from less than 4% to more than 30% (Canick, Palomaki, Kloza, Lambert‐Messerlian, & Haddow, 2013; Jiang et al., 2012). Furthermore, it has been shown that NGS‐based methods can reliably detect fetal aneuploidies non‐invasively with fetal fraction of at least 4% (Norton et al., 2012; Palomaki et al., 2011; Samango‐Sprouse et al., 2013). Therefore, inaccurate estimation of fetal fraction can potentially compromise the sensitivity of the assay (Koumbaris et al., 2016; Straver, Oudejans, Sistermans, & Reinders, 2016; Takoudes & Hamar, 2015).
Different approaches have been developed for quantification of fetal fraction in maternal plasma. The most common methods include quantification of Y‐chromosome‐specific sequences using PCR (Lo et al., 1998; Zimmermann, El‐Sheikhah, Nicolaides, Holzgreve, & Hahn, 2005). In order to overcome the limitation of detecting only male‐bearing pregnancies, techniques have been developed to detect paternally inherited polymorphic nucleotides (SNPs) or insertion/deletion polymorphisms as markers for fetal fraction quantification (Barrett et al., 2017; Jiang et al., 2012; Zimmermann et al., 2012). Such approaches can rely heavily on the genotypes of both parents; therefore, their implementation may be limited by the fact that paternal DNA may not be available (Peng & Jiang, 2017). Algorithms such as FetalQuant and FertalQuantSD have been utilized to measure the fetal DNA fraction only from maternal plasma requiring either very high read depth or maternal genotype information, respectively (Jiang et al., 2012, 2016). SNP‐independent algorithms showing good correlation with Y‐chromosome‐based methods have also been developed although it is unclear if their performance is suitable from low fetal fractions. Fragment size differences between fetal and maternal DNA and nucleosome positioning methods showed promising results, but are not accurate enough as a standalone test for direct clinical implementation (Kim et al., 2015; Straver et al., 2016; van Beek et al., 2017). Sex‐ and polymorphism‐independent procedures focusing on the methylation differences between fetal and maternal DNA have also been described. However, the limited number of available fetal‐specific differentially methylated regions in order to account for inter‐individual methylation variability and the high number of positive reactions required for statistical robustness rendered their clinical implementation impractical (Chan et al., 2006; Hindson et al., 2011; Poon, Leung, Lau, Chow, & Lo, 2002; Zejskova, Jancuskova, Kotlabova, Doucha, & Hromadnikova, 2010).
We hereby describe a methylation‐based assay, using a novel set of well‐characterized differentially methylated regions, for accurate estimation of fetal fraction. Taking in consideration the limitations of PCR and methylation‐based methods, we developed a new multiplex SNP‐independent and sex‐independent ddPCR assay for the quantification of fetal fraction. As such, the assay described herein can rapidly be incorporated into laboratory workflows and can provide accurate fetal fraction information prior to sample library preparation minimizing any possibility of incorrect calls due to low fetal fraction, therefore reducing dramatically sample library preparation and sequencing processing cost.
2. METHODS
2.1. Sample collection and preparation
The study was approved by the Cyprus National Bioethics Committee (ΕΕΒΚ/ΕΠ/2013/03, April 2013) and informed consent form was obtained from all participants. Peripheral blood samples of 10 ml were collected anonymously from women with singleton pregnancies of at least 18 years of age and 10th week of gestation into Streck cell‐free DNA BCT tubes. First trimester chrorionic villus samplings (CVS) were collected from collaborating centers.
A mean of 4 ml of plasma was isolated via a double centrifugation protocol of 1,000 g for 10 min, followed by 16,000 g for 10 min and cfDNA isolation from plasma was performed using the Qiasymphony DSP Circulating DNA Kit (Qiagen). DNA isolation from CVS was performed using the QIAmp DNA Mini kit. All samples were stored at −80°C prior to further processing.
2.2. Methylation sensitive restriction digestion
Methylation sensitive restriction digestion was used to enrich fetal‐specific differentially methylated regions for DMR discovery and confirmation of their methylation status on CVS and plasma samples and finally for fetal fraction estimation on pregnancy plasma samples. DNA was simultaneously digested with HhaI and HpaII for 2 hr at 37°C followed by 20 min inactivation at 80°C. Each digestion reaction consisted of 20 U of HhaI, 40 U of HpaII, 20 ul of DNA, 4 ul of 10X CutSmart buffer, and 10 ul HPLC H2O. Two enzymatic reactions were performed for each sample, which were then pooled prior further processing.
2.3. DMR discovery
Extracted DNA from two CVS samples and one female non‐pregnant plasma sample were subjected to blunt ending and sequencing adapter ligation as previously described (Koumbaris et al., 2016). Following methylation sensitive restriction digestion, MeDIP was performed (Borgel, Guibert, & Weber, 2012) and a unique barcode was assigned to all samples. Adaptors from the sequencing reads were removed with cutadapt v1.2 and subsequently aligned using BWA (Li & Durbin, 2009). Duplicate reads were removed from the resulting BAM files using Picard. Following sequencing output normalization, each sample's read depth was calculated for each CpG dinucleotide excluding CpG sites overlapping with repetitive elements and LCRs. The distribution of the normalized read difference between CVS and plasma was then calculated. Sites that lay beyond the 90th quantile of the distribution were used as candidate sites for DMR selection. Subsequently, candidate CpG sites located less than 50bp away were merged to form larger cohesive DMRs. In order to minimize the presence of maternal (background) methylation, only DMRs which did not exhibit any aligned reads in the plasma samples were selected. Final DMR selection criteria included the presence of at least two restriction sites of HpaII and/or HhaI restriction enzymes (NEB) and exclusion of copy number variants (MacDonald, Ziman, Yuen, Feuk, & Scherer, 2014).
2.4. DMR screening
For the purposes of this study, fetal‐specific DMR selection was performed only on chromosome 1. Primers for the selected DMRs were designed with amplicon size ranging from 70 to 110bp. Primer amplification efficiency was tested on control genomic DNA with a pre‐determined concentration (known concentration of 25 ng/ul). Following ddPCR amplification of each primer set, the concentration of the control genomic DNA was calculated. Failure to obtain the expected concentration resulted in the exclusion of the primer from downstream experiments. DMRs with satisfactory amplification efficiency were initially screened using digested pooled DNA obtained from non‐pregnant female plasma samples (Sera Laboratories International). DMRs with less than 3% methylation enrichment in the pool non‐pregnant cfDNA were subsequently confirmed in three normal CVS, three Trisomy 21 CVS, and three individual non‐pregnant female plasma samples following restriction digestion.
Methylation levels for each DMR were calculated in two individual simplex Evagreen‐based ddPCR reactions on QX200 Droplet Digital System (Bio‐Rad) using primers targeting the DMR under investigation and a reference/digestion control region on chromosome 1 (REF1) that did not include restriction sites for HhaI and HpaII. This region was used for calculation of total DNA concentration. DMRs with less than 3% methylation enrichment in non‐pregnant plasma samples and higher than 15% in CVS were selected for multiplexing.
2.5. Multiplex design
2.5.1. Fetal fraction multiplex mix
Selected DMRs were initially multiplexed in a two‐channel octaplex ddPCR reaction using probes labeled with FAM and/or HEX fluorophores (Integrated DNA Technologies, Leuven, Belgium). In total, seven DMRs in addition to the control region (REF1) were combined in the multiplex mix at different ratios of FAM and HEX fluorophores in order to achieve distinct cluster populations for each DMR. Thus, we were able analyze and validate each DMR in the multiplex reaction separately using their amplification efficiency on genomic DNA (0.125 ng/ul; Table 1). Followed by statistical analysis, four out of seven DMRs which showed the best performance for fetal fraction estimation were combined in a multiplex ddPCR assay, termed as fetal fraction multiplex mix (FFMM).
Table 1.
Multiplex mix information designed for fetal fraction estimation using ddPCR followed by methylation sensitive restriction digestion
| Name | Sequence | FAM:HEX ratio | Chromosomal Location | |
|---|---|---|---|---|
| DMR 1a | Forward | CGTTAAGGTAATGAACGGCG | 1:1 | 1p34.2 |
| Reverse | CCAGACCCGCAGAAAGTG | |||
| Probe | TCGAAAGTTCAGCGCCCC | |||
| DMR 2a | Forward | CTTCCAGCCAAGCGCTG | 2:1 | 1p34.1 |
| Reverse | GTGATGCAAATCCGCTCC | |||
| Probe | CCTCGCTTTACGGAAAGAACGC | |||
| DMR 3 | Forward | ACACGTCCCCACCTATTTGG | 1:1 | 1p32.3 |
| Reverse | TGTGGGGAGGAGAAGTGACA | |||
| Probe | TCTTCTCCGGGACCTGAGGT | |||
| DMR 4a | Forward | AGCCTCCCCTTTCCTGTCT | 1:1 | 1p21.2 |
| Reverse | GGTGCGTGTTTTCTGTGATT | |||
| Probe | AGGAGCGTGCACAGGTCCT | |||
| DMR 5a | Forward | GAAGGAAAGGAGCTTAGGCG | FAM | 1p32.1 |
| Reverse | CGCAACCATCGAAGTTCAATC | |||
| Probe | CCTGGACGGAGCTGAGACAAT | |||
| DMR 6 | Forward | AGGAGAACGCTGAGGTCG | FAM | 1p32.2 |
| Reverse | GCGGACTACCTTAGTGGCAC | |||
| Probe | CAACTGCAGCTCGCGCT | |||
| DMR 7 | Forward | GCCGCCTTCAGTAGCACAA | FAM | 1p21.2 |
| Reverse | AGCCCGTGGCCTTAAATAGGA | |||
| Probe | CTCAAACCGCGCATCTCTGGC | |||
| REF 1a | Forward | GTTGTGATGTTCTTAAGGCAGA | HEX | 1p31.1 |
| Reverse | AATTGGGATTTCCACAGGAG | |||
| Probe | TCTTCATAAAAAGGAAAGTAATGGCA |
Markers selected for the final fetal fraction estimation model (FFMM).
2.5.2. Chromosome Y multiplex mix
As reference standard for fetal fraction estimation, a two‐channel chromosome Y multiplex mix (YMM) was developed. Seven primer/probe sets targeting nine regions on chromosome Y and one set of primer/probe targeting chromosome 1 (REF1) were equimolarly added in a single multiplex mix (Table 2).
Table 2.
Multiplex mix information designed for fetal fraction estimation using chromosome Y multiplex mix (YMM)
| Name | Sequence | Location | |
|---|---|---|---|
| CHRY1 | Forward | CAGTGTATTTGTGGAAATGCCT | Yq11.21 |
| Reverse | CTAACTTTTCCAGACAGCAGC | ||
| Probe | ACTGTGTAGTGATAAAGACCTGCT | ||
| CHRY2 | Forward | ACTTCTAGTTTCCTGCTTTAGC | Yp11.31 |
| Reverse | CCAACTGGTTTAATTTGATGGG | ||
| Probe | GTGCAAATTTTATGAAGTCTTGGCA | ||
| CHRY3 | Forward | GACCTGCCCCATCTCCAT | Yq11.221 |
| Reverse | AGGTTGGATGGAAGATGAAGT | ||
| Probe | GTCACAACGACAGTCATCATTTCT | ||
| CHRY4 | Forward | CCTCCTTTGAATACTTATTTACGATT | Yq11.221 |
| Reverse | ACTATGTTTTGCAACCTTTGTT | ||
| Probe | AGTCAAGTTATATGAGTATGTTCAACC | ||
| CHRY5 | Forward | TGTCATCAACATGGGAAGCA | Yq11.221 (two loci) |
| Reverse | TATCTCCCTGAGCAGCAACTA | ||
| Probe | TGG TGA GATCTC TGA GGT CT | ||
| CHRY6 | Forward | CTCATCACCTGAATTTATGTCTATTT | Yq11.222 (two Loci) |
| Reverse | GCTGGGTTTGTCTTTAGGT | ||
| Probe | AAA GAC ACTTGT GGG CCT GT | ||
| CHRY7 | Forward | CGCTTAACATAGCAGAAGCA | Yp11.31 |
| Reverse | AGTTTCGAACTCTGGCACCT | ||
| Probe | TGTCGCACTCTCCTTGTTTTT | ||
| REF1 | Forward | GTTGTGATGTTCTTAAGGCAGA | 1p31.1 |
| Reverse | AATTGGGATTTCCACAGGAG | ||
| Probe | TCTTCATAAAAAGGAAAGTAATGGCA |
2.6. Droplet digital PCR
Each multiplex ddPCR reaction mix consisted of 9.6 ul digested DNA (plasma or CVS), 12 ul of 2X ddPCR Master Mix, 1.2 ul primer/probe mix at final concentrations of 900 nM and 250 nM, respectively, in a final volume of 24 ul. All multiplex ddPCR reactions were performed in triplicate using the following conditions: initial denaturation at 95°C for 10 min, followed by 45 cycles at 95°C for 30 s, 60°C for 60 s and final extension at 98°C for 10 min. The droplet generation was performed on QX200 AutoDG droplet digital system (BioRad), and fluorescent signal was measured and analyzed using the QX200 droplet reader. Replicates with less than 10,000 droplets were excluded from analysis.
2.7. Data analysis
Analysis was performed using R version 3.3.2 on a Linux platform. The training and validation of the suggested fetal fraction quantification model was performed in multiple steps. Initially, fetal fraction estimates obtained from YMM were compared to DYS‐qPCR assay (n = 47), a gold standard assay used for fetal fraction estimation on male pregnancy samples (Kyriakou et al., 2013). Next, YMM was used to train a fetal fraction estimation model using the initial octaplex mix on a training set of male pregnancies (n = 85). A multiple linear regression model was fitted to the data followed by a step‐wise model selection that led to the final model (FFMM). To further test/validate our findings of the estimated model, the fetal fraction was quantified in an independent set of male pregnancies using both YMM and FFMM (n = 53). Furthermore, additional testing of the assay was performed using FFMM on an additional independent set of pregnancy plasma samples, including both male and female pregnancy samples, consisting of both normal (euploid; n = 109) and trisomy 21 (aneuploid) samples (n = 14). Furthermore, our fetal fraction estimation model was tested on non‐pregnant plasma samples.
3. RESULTS
3.1. DMR discovery and screening
Two CVS samples and one non‐pregnant female plasma sample were subjected to methylation sensitive restriction digestion followed by MeDIP and NGS. Reads passing quality filters were aligned to the human reference genome GRCh37/hg19. In total, CVS 433.7 and 416.7 million reads were aligned for CVS samples and 410.2 million reads for the plasma sample. Hundreds of genome‐wide fetal‐specific DMRs were identified between fetal and maternal tissue samples, however for the purposes of this study, we focused on the identification of DMRs on chromosome 1. In total, 93 DMRs were selected, with size ranging from 50 to 759 bp. From these, 43 DMRs, which satisfied all selection criteria were screened using ddPCR (see Section 2.3). Specifically, 151 (n = 151) primer sets targeting the selected DMRs (DMR‐amplicon) were designed and tested on genomic DNA for ddPCR amplification efficiency. Subsequently, the methylation status of each DMR amplicon was investigated on pool plasma obtained from non‐pregnant women using methylation restriction digestion followed by ddPCR. In total, out of the 151 DMR amplicons, 59 and 54 DMR amplicons were excluded from further testing due to inefficient ddPCR amplification and high methylation percentage (>3%) on pool plasma, respectively. The methylation status of the remaining 38 DMR‐amplicons was confirmed on three normal CVS, three trisomy 21 CVS, and three individual non‐pregnant plasma samples. Finally, seven DMRs that showed highest and consistent methylation difference between CVS (normal and trisomy 21) and individual plasma samples were used in a multiplex reaction mix along with a reference marker targeting an unmethylated region (Figures S1 and S2).
3.2. Fetal fraction estimation model
As a first step, fetal fraction estimates obtained from YMM and conventional qPCR‐based DYS assay were obtained and compared in 47 male pregnancy samples. A high degree of correlation was observed between YMM and DYS (r = 0.83 with 95% CI: 0.71–0.9; Figure 1). This allowed us to subsequently use YMM as a reference to train a fetal fraction estimation model using the initial octaplex ddPCR on an independent set of 85 male pregnancy samples. A multiple linear regression model was fitted to the data and a step‐wise model selection was applied resulting in the final model with our covariates/markers (FFMM; Table S1; Venables & Ripley, 2002). All coefficient estimates of the resulting model had p < .024. A high correlation of the model estimates with YMM‐based fetal fractions was observed (r = 0.86; 95% CI: 0.80–0.91; Figure 2a).
Figure 1.
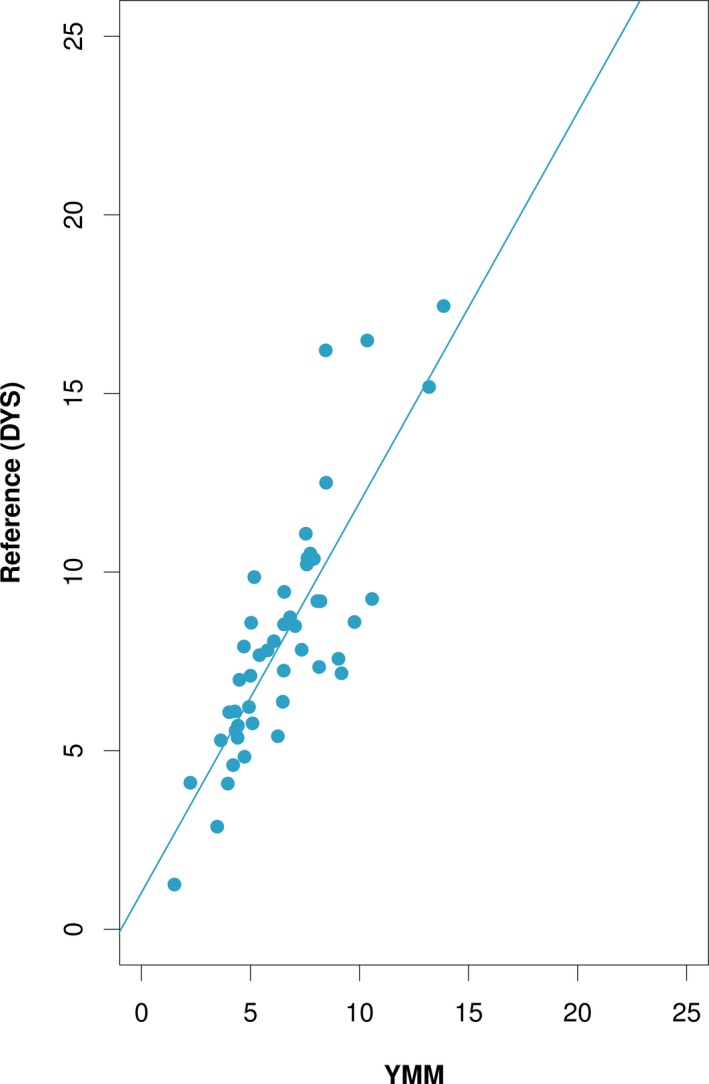
Correlation of fetal fractions on male pregnancies between chr Y ddPCR assay (YMM) and reference standard. Scatter plot analysis of fetal fraction estimates for 47 male samples showed high correlation between YMM and qPCR‐based DYS assay (r = 0.83; 95% CI: 0.71–0.9) that was treated as reference standard. Solid line represents fitted values from linear regression models. These findings allowed us to proceed to the next phase of our analysis, that is, use YMM to train a fetal fraction estimation model using our selected panel of markers
Figure 2.
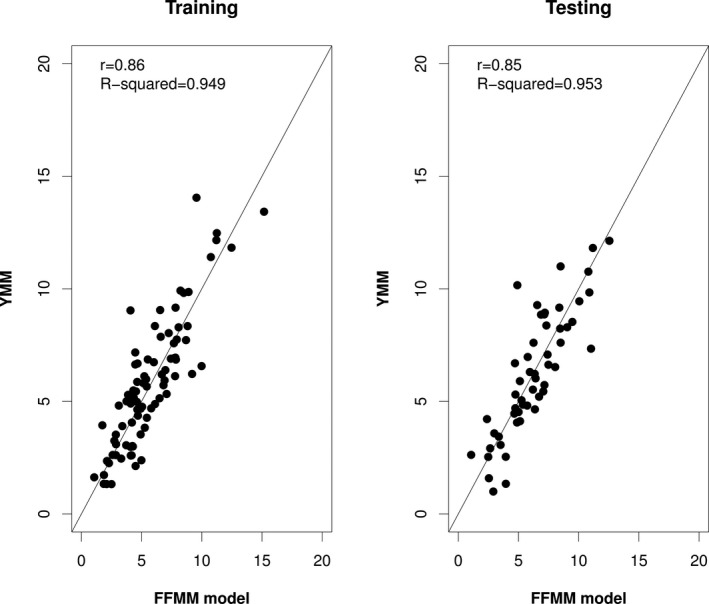
Correlation of fetal fraction estimation between the training/testing model and YMM. Scatter plot analysis of fetal fraction estimates obtained from our model (FFMM) versus YMM on male pregnancy samples. Fetal fraction estimation model using methylation sensitive restriction digestion followed by multiplex ddPCR shows high correlation with YMM (r = 0.86; 95% CI: 0.80–0.91) on a training set of 85 male pregnancy samples (a). Findings were confirmed on additional 53 male pregnancy samples using our estimation model (r = 0.84; 95% CI: 0.74–0.91) (b). The performance of the trained model (n = 85) on an independent set of samples (validation set of size n = 53) is statistically not different (p = .7022). Solid line represents the x = y line
In order to test our findings on an independent dataset, we applied our estimated model on 53 additional male pregnancy samples. A strong correlation of the methylation‐based fetal fraction assay with the corresponding YMM values was confirmed (r = 0.84, 0.74–0.91; Figure 2b). Furthermore, the performance of the trained model (n = 85) was statistically not different from the performance on the testing set (n = 53) (p = 0.7022; Wilcoxon test).
To investigate whether our developed fetal fraction estimation method is gender and/or ploidy independent, additional 109 euploid pregnancies (51 females and 58 males) and 14 trisomy 21 pregnancies (4 females and 10 males) were examined. Since the model is trained and tested on male pregnancy samples, we compared the fetal fraction estimates of the female and abnormal samples with those of the euploid male pregnancy samples. The performance between male (n = 58) and female (n = 51) pregnancy samples did not differ (p = .97; Wilcoxon test; Figure S3). The fetal fraction estimation accuracy was further assessed on 14 trisomy 21 samples showing comparable performance to the euploid male samples (p = .13; Wilcoxon test; Figure S3).
The maternal background (noise) level at very low fetal fractions was assessed using eight non‐pregnant samples. The estimated fetal fractions ranged from 0.2% to 1.09% (Figure S3). The analytical sensitivity to detect pregnancy is estimated to be greater than 99% (97%–99.9%) with 100% analytical specificity (63%–100%). The estimated threshold that discriminates pregnancy from assay noise (non‐pregnant samples) is 1.355%.
4. DISCUSSION
In this study, we present the development and validation of a multiplex assay using methylation sensitive restriction digestion and a robust set of novel fetal‐specific differentially methylated regions for accurate detection of fetal fraction in maternal plasma. For the development of the assay, we employed digital PCR, a powerful a tool used for several clinical applications due to its high sensitivity, simplicity, and precision as it allows quantification from single molecule amplification.
Previous studies have focused on the methylation differences between fetal and maternal DNA for quantification of fetal DNA in the maternal plasma (Chan et al., 2006; Nygren et al., 2010). Inherent biological limitations of methylation‐based assays such as biological methylation stability of the tested markers should be considered prior to their clinical implementation (Nygren et al., 2010). Furthermore, other technologies that is, qPCR‐based assays are characterized by technical limitations such as fluctuations in the amplification efficiency, or the need of different standards for construction of calibration curves (Lun et al., 2008). In addition, ddPCR quantification assays require a high number of positive PCR reactions for statistical robustness for the absolute and accurate quantification of cfDNA due to its limited abundance in maternal plasma (Fan & Quake, 2007). In our efforts to overcome the aforementioned limitations, we initially employed methylation sensitive restriction digestion followed by MeDIP and NGS for the identification of fetal‐specific hypermethylated regions. Hundreds of fetal‐specific DMRs were identified genome‐wide; however, DMR selection process was focused on chromosome 1 as this chromosome has rarely been implicated in first trimester chromosomal abnormalities (excluding 1p36 deletion critical region). Further DMR selection criteria were implemented taking into account the highest methylation difference between fetal (CVS) and maternal tissues (non‐pregnant female plasma) and methylation stability. Finally, seven fetal‐specific DMRs and one reference marker with no restriction sites used as reference/digestion control were combined in a two‐channel fluorophore octaplex reaction for fetal fraction quantification. Multiple linear regression models fitted to the data resulted in a final model consisting of four DMRs in addition to the reference marker. Thus, targeting robust DMRs in a single multiplex reaction, we simultaneously overcame limitations regarding inter‐individual methylation variability and substantially increased the number of positive droplets required for accurate fetal fraction estimation. Additionally, optimal digestion efficiency and removal of maternally derived background were ensured by the incorporation of at least two restriction sites within the tested DMRs.
Our training model was constructed based on the orthogonal assay comparison between Chromosome Y (YMM) and restriction digestion multiplex assay. For both assays, ddPCR was used for quantification with similar starting amount of DNA in order to eliminate possible PCR biases. Strong evidence that YMM was acceptable to be used as a comparison standard for the training model came from the high correlation results between YMM‐ddPCR assay and DYS‐qPCR, a gold standard assay used in the field for male fetus quantification.
The statistical model presented herein attains high correlation with YMM for male pregnancy samples and similar performance in both male and female samples. An examination of 14 trisomy 21 samples also showed evidence that the ploidy status of the sample does not affect our fetal fraction estimates. Furthermore, the results on the performance of the assay on female non‐pregnant samples showed that traces of background methylation were detectable ranging from 0% to 1.1%. DMR selection for the development of the assay was performed based on the highest difference between fetal and maternal methylation as well as methylation stability in CVS and plasma. However, low level methylation (<3%) was also detected on non‐pregnant female plasma samples (Figure S1). This is in agreement with previous studies that showed that, despite the fetal hypermethylation status of well‐characterized DMRs, low maternal methylation was detected (Ioannides et al., 2014; Keravnou et al., 2016). Nevertheless, our fetal fraction estimation model adjusts for the presence of maternal background methylation by regressing methylation levels with YMM. Additional experiments using male, female, euploid, and aneuploid pregnancy samples as well as non‐pregnant controls are required for a more accurate determination of our model's analytical specificity and sensitivity before the clinical implementation of the assay.
In the field of NIPT, fetal fraction estimation is one of the most critical variables which is currently assessed using NGS data. However, on average 2%–6% of NIPT samples is sent for blood redraw at later gestational age due to low fetal fraction estimation. This increases substantially the cost per sample considering the high sequencing consumable and reagent cost. Integration of an accurate, inexpensive, and simple assay, as the one described herein, as a first‐tier screening for fetal fraction estimation prior to sequencing, will reduce dramatically both the sample library preparation and sequencing costs and minimize further the possibility of incorrect calls due to low fetal fraction estimation.
Increased levels of cffDNA in the maternal circulation have also been associated with maternal pathological complications and preterm birth (Monte, 2011; Sekizawa et al., 2001). Studies have shown that prior to the development of clinical symptoms in preeclampsia, one of the most frequent causes of maternal and fetal morbidity and mortality, cffDNA was significantly higher in affected pregnancies than in controls (Leung et al., 2001; Levine et al., 2004; Monte, 2011). Thus, accurate and affordable tests for identifying and monitoring presymptomatic pregnancy complications are in need. Preliminary results presented in this study show potential applicability of our assay in the detection of pregnancy‐associated diseases; nevertheless, its clinical performance should be evaluated in a large cohort of affected and non‐affected pregnancies.
5. CONCLUSION
We developed an NGS, SNP, and sex‐independent assay for the accurate estimation of fetal fraction in maternal plasma. Taking in consideration the high methylation differences between fetal and maternal DNA and methylation stability, we selected and confirmed a set of four DMRs in a multiplex ddPCR assay following methylation sensitive restriction digestion. In this way, we increased the number of positive targets providing statistical robustness to our data overcoming technical and biological limitations. As such, the assay described herein can rapidly be incorporated into laboratory workflows and can provide accurate fetal fraction information prior to sample library preparation minimizing further any possibility of incorrect calls due to low fetal fraction, therefore reducing dramatically sample library preparation and sequencing processing cost.
CONFLICT OF INTEREST
The authors, Marios Ioannides, Achilleas Achilleos, Skevi Kyriakou, Elena Kypri, Charalambos Loizides, Kyriakos Tsangaras, Louiza Constantinou, George Koumbaris, and Philippos C Patsalis are employed by NIPD Genetics.
Supporting information
Ioannides M, Achilleos A, Kyriakou S, et al. Development of a new methylation‐based fetal fraction estimation assay using multiplex ddPCR. Mol Genet Genomic Med. 2020;8:e1094 10.1002/mgg3.1094
Funding information
This work has received funding from the European Research Council (ERC) under 7th Framework Programme under grant agreement No 322953 for the project: A Novel Non‐Invasive Prenatal Diagnosis of Genetic Disorders.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Barrett, A. N. , Xiong, L. I. , Tan, T. Z. , Advani, H. V. , Hua, R. , Laureano‐Asibal, C. , … Choolani, M. (2017). Measurement of fetal fraction in cell‐free DNA from maternal plasma using a panel of insertion/deletion polymorphisms. PLoS ONE, 12(10), e0186771 10.1371/journal.pone.0186771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn, P. , Borrell, A. , Chiu, R. W. K. , Cuckle, H. , Dugoff, L. , Faas, B. , … Yaron, Y. (2015). Position statement from the Chromosome Abnormality Screening Committee on behalf of the Board of the International Society for Prenatal Diagnosis. Prenatal Diagnosis, 35(8), 725–734. 10.1002/pd.4608 [DOI] [PubMed] [Google Scholar]
- Borgel, J. , Guibert, S. , & Weber, M. (2012). Methylated DNA immunoprecipitation (MeDIP) from low amounts of cells. Methods in Molecular Biology, 925, 149–158. 10.1007/978-1-62703-011-3_9 [DOI] [PubMed] [Google Scholar]
- Canick, J. A. , Palomaki, G. E. , Kloza, E. M. , Lambert‐Messerlian, G. M. , & Haddow, J. E. (2013). The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenatal Diagnosis, 33(7), 667–674. 10.1002/pd.4126 [DOI] [PubMed] [Google Scholar]
- Chan, K. A. , Ding, C. , Gerovassili, A. , Yeung, S. W. , Chiu, R. W. , Leung, T. N. , … Lo, Y. D. (2006). Hypermethylated RASSF1A in maternal plasma: A universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clinical Chemistry, 52(12), 2211–2218. 10.1373/clinchem.2006.074997 [DOI] [PubMed] [Google Scholar]
- Chiu, R. W. K. , Akolekar, R. , Zheng, Y. W. L. , Leung, T. Y. , Sun, H. , Chan, K. C. A. , … Lo, Y. M. D. (2011). Non‐invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: Large scale validity study. BMJ, 342, c7401 10.1136/bmj.c7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee Opinion No. 640 . (2015). Cell‐free DNA screening for fetal aneuploidy. Obstetrics and Gynecology, 10.1097/AOG.0000000000001007 [DOI] [PubMed] [Google Scholar]
- El Khattabi, L. A. , Rouillac‐Le Sciellour, C. , Le Tessier, D. , Luscan, A. , Coustier, A. , Porcher, R. , … Dupont, J.‐M. (2016). Could digital PCR be an alternative as a non‐invasive prenatal test for trisomy 21: A proof of concept study. PLoS ONE, 11(5), e0155009 10.1371/journal.pone.0155009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H. C. , Blumenfeld, Y. J. , Chitkara, U. , Hudgins, L. , & Quake, S. R. (2008). Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Academy of Sciences of the United States of America, 105(42), 16266–16271. 10.1073/pnas.0808319105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H. C. , & Quake, S. R. (2007). Detection of aneuploidy with digital polymerase chain reaction. Analytical Chemistry, 79(19), 7576–7579. 10.1021/ac0709394 [DOI] [PubMed] [Google Scholar]
- Gregg, A. R. , Skotko, B. G. , Benkendorf, J. L. , Monaghan, K. G. , Bajaj, K. , Best, R. G. , … Watson, M. S. (2016). Noninvasive prenatal screening for fetal aneuploidy, 2016 update: A position statement of the American College of Medical Genetics and Genomics. Genetics in Medicine, 18(10), 1056–1065. 10.1038/gim.2016.97 [DOI] [PubMed] [Google Scholar]
- Hindson, B. J. , Ness, K. D. , Masquelier, D. A. , Belgrader, P. , Heredia, N. J. , Makarewicz, A. J. , … Colston, B. W. (2011). High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Analytical Chemistry, 83(22), 8604–8610. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannides, M. , Papageorgiou, E. A. , Keravnou, A. , Tsaliki, E. , Spyrou, C. , Hadjidaniel, M. , … Patsalis, P. C. (2014). Inter‐individual methylation variability in differentially methylated regions between maternal whole blood and first trimester CVS. Molecular Cytogenetics, 7(1), 73 10.1186/s13039-014-0073-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, P. , Chan, K. C. A. , Liao, G. J. W. , Zheng, Y. W. L. , Leung, T. Y. , Chiu, R. W. K. , … Sun, H. (2012). FetalQuant: Deducing fractional fetal DNA concentration from massively parallel sequencing of DNA in maternal plasma. Bioinformatics, 28(22), 2883–2890. 10.1093/bioinformatics/bts549 [DOI] [PubMed] [Google Scholar]
- Jiang, P. , Peng, X. , Su, X. , Sun, K. , Yu, S. C. Y. , Chu, W. I. , … Chan, K. C. A. (2016). FetalQuant(SD): Accurate quantification of fetal DNA fraction by shallow‐depth sequencing of maternal plasma DNA. Npj Genomic Medicine, 1, 16013 10.1038/npjgenmed.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keravnou, A. , Ioannides, M. , Tsangaras, K. , Loizides, C. , Hadjidaniel, M. D. , Papageorgiou, E. A. , … Patsalis, P. C. (2016). Whole‐genome fetal and maternal DNA methylation analysis using MeDIP‐NGS for the identification of differentially methylated regions. Genetics Research, 98, e15 10.1017/S0016672316000136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. K. , Hannum, G. , Geis, J. , Tynan, J. , Hogg, G. , Zhao, C. , … Deciu, C. (2015). Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenatal Diagnosis, 35(8), 810–815. 10.1002/pd.4615 [DOI] [PubMed] [Google Scholar]
- Koumbaris, G. , Kypri, E. , Tsangaras, K. , Achilleos, A. , Mina, P. , Neofytou, M. , … Patsalis, P. C. (2016). Cell‐Free DNA Analysis of Targeted Genomic Regions in Maternal Plasma for Non‐Invasive Prenatal Testing of Trisomy 21, Trisomy 18, Trisomy 13, and Fetal Sex. Clinical Chemistry, 62(6), 848–855. 10.1373/clinchem.2015.252502 [DOI] [PubMed] [Google Scholar]
- Kyriakou, S. , Kypri, E. , Spyrou, C. , Tsaliki, E. , Velissariou, V. , Papageorgiou, E. A. , & Patsalis, P. C. (2013). Variability of ffDNA in maternal plasma does not prevent correct classification of trisomy 21 using MeDIP‐qPCR methodology. Prenatal Diagnosis, 33(7), 650–655. 10.1002/pd.4140 [DOI] [PubMed] [Google Scholar]
- Leung, T. N. , Zhang, J. , Lau, T. K. , Chan, L. Y. , & Lo, Y. M. (2001). Increased maternal plasma fetal DNA concentrations in women who eventually develop preeclampsia. Clinical Chemistry, 47(1), 137–139. [PubMed] [Google Scholar]
- Levine, R. J. , Qian, C. , LeShane, E. S. , Yu, K. F. , England, L. J. , Schisterman, E. F. , … Bianchi, D. W. (2004). Two‐stage elevation of cell‐free fetal DNA in maternal sera before onset of preeclampsia. American Journal of Obstetrics and Gynecology, 190(3), 707–713. 10.1016/j.ajog.2003.12.019 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, Y. M. , Corbetta, N. , Chamberlain, P. F. , Rai, V. , Sargent, I. L. , Redman, C. W. , & Wainscoat, J. S. (1997). Presence of fetal DNA in maternal plasma and serum. The Lancet, 350(9076), 485–487. 10.1016/S0140-6736(97)02174-0 [DOI] [PubMed] [Google Scholar]
- Lo, Y. M. D. , Tein, M. S. C. , Lau, T. K. , Haines, C. J. , Leung, T. N. , Poon, P. M. K. , … Hjelm, N. M. (1998). Quantitative analysis of fetal DNA in maternal plasma and serum: Implications for noninvasive prenatal diagnosis. American Journal of Human Genetics, 62(4), 768–775. 10.1086/301800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun, F. M. , Chiu, R. W. , Chan, K. C. , Leung, T. Y. , Lau, T. K. , & Lo, Y. M. (2008). Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clinical Chemistry, 54(10), 1664–1672. 10.1373/clinchem.2008.111385 [DOI] [PubMed] [Google Scholar]
- MacDonald, J. R. , Ziman, R. , Yuen, R. K. , Feuk, L. , & Scherer, S. W. (2014). The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Research, 42(D1), D986–992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monte, S. (2011). Biochemical markers for prediction of preclampsia: Review of the literature. Journal of Prenatal Medicine, 5(3), 69–77. [PMC free article] [PubMed] [Google Scholar]
- Norton, M. E. , Brar, H. , Weiss, J. , Karimi, A. , Laurent, L. C. , Caughey, A. B. , Song, K. (2012). Non‐Invasive Chromosomal Evaluation (NICE) Study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. American Journal of Obstetrics and Gynecology, 207(2), 137.e1–137.e8. 10.1016/j.ajog.2012.05.021 [DOI] [PubMed] [Google Scholar]
- Nygren, A. O. , Dean, J. , Jensen, T. J. , Kruse, S. , Kwong, W. , van den Boom, D. , & Ehrich, M. (2010). Quantification of fetal DNA by use of methylation‐based DNA discrimination. Clinical Chemistry, 56(10), 1627–1635. 10.1373/clinchem.2010.146290 [DOI] [PubMed] [Google Scholar]
- Palomaki, G. E. , Kloza, E. M. , Lambert‐Messerlian, G. M. , Haddow, J. E. , Neveux, L. M. , Ehrich, M. , … Canick, J. A. (2011). DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genetics in Medicine, 13(11), 913–920. 10.1097/GIM.0b013e3182368a0e [DOI] [PubMed] [Google Scholar]
- Papageorgiou, E. A. , Karagrigoriou, A. , Tsaliki, E. , Velissariou, V. , Carter, N. P. , & Patsalis, P. C. (2011). Fetal‐specific DNA methylation ratio permits noninvasive prenatal diagnosis of trisomy 21. Nature Medicine, 17(4), 510–513. 10.1038/nm.2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, X. L. , & Jiang, P. (2017). Bioinformatics approaches for fetal DNA fraction estimation in noninvasive prenatal testing. International Journal of Molecular Sciences, 18(2), 10.3390/ijms18020453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, L. L. , Leung, T. N. , Lau, T. K. , Chow, K. C. , & Lo, Y. M. (2002). Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clinical Chemistry, 48(1), 35–41. [PubMed] [Google Scholar]
- Samango‐Sprouse, C. , Banjevic, M. , Ryan, A. , Sigurjonsson, S. , Zimmermann, B. , Hill, M. , … Rabinowitz, M. (2013). SNP‐based non‐invasive prenatal testing detects sex chromosome aneuploidies with high accuracy. Prenatal Diagnosis, 33(7), 643–649. 10.1002/pd.4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekizawa, A. , Sugito, Y. , Iwasaki, M. , Watanabe, A. , Jimbo, M. , Hoshi, S. , … Okai, T. (2001). Cell‐free fetal DNA is increased in plasma of women with hyperemesis gravidarum. Clinical Chemistry, 47(12), 2164–2165. [PubMed] [Google Scholar]
- Straver, R. , Oudejans, C. B. , Sistermans, E. A. , & Reinders, M. J. (2016). Calculating the fetal fraction for noninvasive prenatal testing based on genome‐wide nucleosome profiles. Prenatal Diagnosis, 36(7), 614–621. 10.1002/pd.4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takoudes, T. , & Hamar, B. (2015). Performance of non‐invasive prenatal testing when fetal cell‐free DNA is absent. Ultrasound in Obstetrics and Gynecology, 45(1), 112 10.1002/uog.14715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, Y. K. , Chiu, R. W. , Akolekar, R. , Leung, T. Y. , Lau, T. K. , Nicolaides, K. H. , & Lo, Y. M. (2010). Epigenetic‐genetic chromosome dosage approach for fetal trisomy 21 detection using an autosomal genetic reference marker. PLoS ONE, 5(12), e15244 10.1371/journal.pone.0015244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, Y. K. , Ding, C. , Chiu, R. W. , Gerovassili, A. , Chim, S. S. , Leung, T. Y. , … Lo, Y. D. (2006). Noninvasive prenatal detection of fetal trisomy 18 by epigenetic allelic ratio analysis in maternal plasma: Theoretical and empirical considerations. Clinical Chemistry, 52(12), 2194–2202. 10.1373/clinchem.2006.076851 [DOI] [PubMed] [Google Scholar]
- Tong, Y. K. , Jin, S. , Chiu, R. W. , Ding, C. , Chan, K. A. , Leung, T. Y. , … Lo, Y. D. (2010). Noninvasive prenatal detection of trisomy 21 by an epigenetic‐genetic chromosome‐dosage approach. Clinical Chemistry, 56(1), 90–98. 10.1373/clinchem.2009.134114 [DOI] [PubMed] [Google Scholar]
- Tsaliki, E. , Papageorgiou, E. A. , Spyrou, C. , Koumbaris, G. , Kypri, E. , Kyriakou, S. , … Patsalis, P. C. (2012). MeDIP real‐time qPCR of maternal peripheral blood reliably identifies trisomy 21. Prenatal Diagnosis, 32(10), 996–1001. 10.1002/pd.3947 [DOI] [PubMed] [Google Scholar]
- van Beek, D. M. , Straver, R. , Weiss, M. M. , Boon, E. M. J. , Huijsdens‐van Amsterdam, K. , Oudejans, C. B. M. , … Sistermans, E. A. (2017). Comparing methods for fetal fraction determination and quality control of NIPT samples. Prenatal Diagnosis, 37(8), 769–773. 10.1002/pd.5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables, W. N. , & Ripley, B. D. (2002). Modern applied statistics with S (4th edn.). New York, NY: Springer. [Google Scholar]
- Zejskova, L. , Jancuskova, T. , Kotlabova, K. , Doucha, J. , & Hromadnikova, I. (2010). Feasibility of fetal‐derived hypermethylated RASSF1A sequence quantification in maternal plasma–next step toward reliable non‐invasive prenatal diagnostics. Experimental and Molecular Pathology, 89(3), 241–247. 10.1016/j.yexmp.2010.09.002 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Gao, Y. , Jiang, F. , Fu, M. , Yuan, Y. , Guo, Y. , … Wang, W. (2015). Non‐invasive prenatal testing for trisomies 21, 18 and 13: Clinical experience from 146,958 pregnancies. Ultrasound in Obstetrics & Gynecology, 45(5), 530–538. 10.1002/uog.14792 [DOI] [PubMed] [Google Scholar]
- Zimmermann, B. , El‐Sheikhah, A. , Nicolaides, K. , Holzgreve, W. , & Hahn, S. (2005). Optimized real‐time quantitative PCR measurement of male fetal DNA in maternal plasma. Clinical Chemistry, 51(9), 1598–1604. 10.1373/clinchem.2005.051235 [DOI] [PubMed] [Google Scholar]
- Zimmermann, B. , Hill, M. , Gemelos, G. , Demko, Z. , Banjevic, M. , Baner, J. , … Rabinowitz, M. (2012). Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenatal Diagnosis, 32(13), 1233–1241. 10.1002/pd.3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.