
Fig. 1.
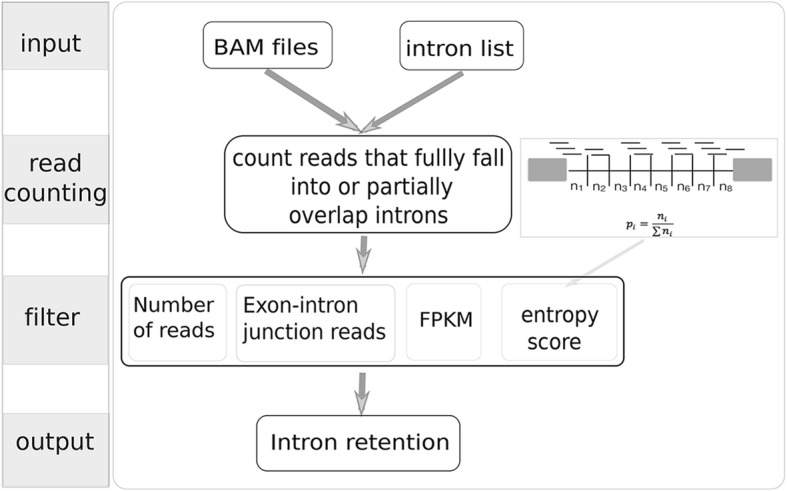
Schematic of the iREAD algorithm. iREAD takes two input files: a BAM file resulting from polyA-enriched RNA-seq read alignment and text file containing a list of independent introns that do not overlap with any exons of any transcripts. iREAD then counts the number of reads (partially) falling into the intron regions. After recording intronic reads, four filters (number of total reads, number of exon-intron junction reads, FPKM and entropy score) are calculated for filtering for high confidence intron retention events