

Abstract
Composites of titanium dioxide (TiO2) and reduced graphene oxide (RGO) have proven to be much more effective photocatalysts than TiO2 alone. However, little attention has been paid so far to the chemical structure of TiO2/RGO interfaces and to the role that the unavoidable residual oxygen functional groups of RGO play in the photocatalytic mechanism. In this work, we develop models of TiO2 rutile (110)/RGO interfaces by including a variety of oxygen functional groups known to be present in RGO. Using hybrid density functional theory calculations, we demonstrate that the presence of oxygen functional groups and the formation of interfacial cross-links (Ti–O–C covalent bonds and strong hydrogen bonds between TiO2 and RGO) have a major effect on the electronic properties of RGO and RGO-based composites. The electronic structure changes from semimetallic to semiconducting with an indirect band gap, with the lowest unoccupied band positioned below the TiO2 conduction band and largely localized on RGO oxygen and carbon orbitals, with some contributions of RGO-bonded Ti atoms. We suggest that this RGO-based lowest unoccupied band acts as a photoelectron trap and the indirect nature of the band gap hinders electron–hole recombination. These results can explain the experimentally observed extended lifetimes of photoexcited charge carriers in TiO2/RGO composites and the enhancement of photocatalytic efficiency of these composites.
Keywords: photocatalysis, titanium dioxide, reduced graphene oxide, electronic structure, charge trapping
Introduction
Photocatalysis has been a subject of intense research over the last few decades: since the first proof-of-concept experiment by Fujishima and Honda,1 there has been an ongoing effort to develop photocatalysts based on inorganic solids.2−6 Such materials have been shown to facilitate a range of solar energy conversion processes, such as the splitting of water into hydrogen and oxygen4−6 and reduction of CO2 for the production of syngas (H2 + CO).2 These processes are important for chemical storage of solar energy, which is necessary for providing us with an alternative to fossil fuels and for production of basic feedstocks for the chemical industry. They are also used for treatment of air and water to remove chemical and biological contaminants, which is a major environmental concern.7
Many successful photocatalysts have been developed based on titanium dioxide (TiO2), chiefly due to its low cost, nontoxicity, chemical stability, and versatility toward chemical modification.3,8−10 The wide band gap of the material (between 3.0 and 3.2 eV depending on the polymorph) enables TiO2 to facilitate a wide range of redox processes, but this wide gap allows absorption of photons in the ultraviolet (UV) range and beyond but not in the visible region of the solar spectrum.3 The photocatalytic performance of TiO2 is also limited by its high charge carrier recombination rate, which is an issue common to most single-component photocatalysts.11,12
A widely used approach for improving TiO2 photocatalysts is to form heterojunctions with other semiconductors.4,9,10 A heterojunction with staggered electronic band positions results in separation of photogenerated charge carriers, thus extending the lifetime of the excited state in the system;11,12 use of semiconductors with narrow optical band gaps (e.g., CdS) also allows TiO2-based heterojunction photocatalysts to make use of the visible region of the solar spectrum.13 However, such heterojunctions often have stability issues since narrow-gap semiconductors such as CdS often suffer from poor stability due to photocorrosion.13
A related and thriving current direction of research is focused on composites of TiO2 with carbon nanostructures.14−17 Following the first photochemical synthesis of a TiO2/graphene composite from graphene oxide by Williams et al.,18 there has been increasing interest in producing composites of TiO2 with graphene materials.16,17 Photocatalytic performance measurements have shown high photocatalytic activities of such composites, containing either the anatase phase of TiO2 or the P25 mixed anatase/rutile phase, under both UV/vis and visible-only irradiation sources.19−23 It has also been shown that the addition of graphene to TiO2 (both the anatase and the rutile phase) greatly extends the lifetime of the excited state relative to pure TiO2.24−26
While there have been many reports demonstrating the enhancement of photocatalytic efficiency, there have been much fewer quantitative studies addressing the mechanisms of this photocatalytic enhancement. For example, transient absorption spectroscopy (TAS) measurements showed that transfer of photoexcited electrons from graphene to TiO2 occurs fast (tens or hundreds of femtoseconds),26,27 faster than charge recombination,27 and that recombination of photoexcited electrons and holes in the composite materials is slower than that in TiO2 alone.24,25 Theoretical calculations confirmed the rapid rate of transfer of photoexcited electrons from graphene to TiO2 (i.e., the role of graphene as a photosensitizer)28 and demonstrated the possibility of charge transfer photoexcitation from graphene to TiO2 induced by visible light.29−31 Our recent calculations also showed that positions of the electronic energy levels of graphene and TiO2 allow transfer of photoexcited electrons from TiO2 to graphene (i.e., the role of graphene as an electron acceptor).32
However, a limitation of all these theoretical studies is that they used the idealized model of pristine graphene to represent the carbon component of the composite.28−32 In practice, the carbon material present in TiO2/carbon composites is typically not pristine graphene but reduced graphene oxide. The commonly used experimental procedure to produce TiO2/graphene composites starts with the production of graphene oxide (GO) from graphite and then photochemical, chemical, or thermal reduction of GO to form reduced graphene oxide (RGO). GO and RGO differ from pristine graphene as they contain certain amounts of oxygen functional groups, such as hydroxyl, epoxide, and carbonyl groups.16,33 Reported carbon/oxygen ratios for GO typically are around 2.6:1–1.7:1 (26–37 at. % oxygen).21,25,34−37 The C/O ratios for RGO range between 14.9:1 and 3.9:1 (6–20 at. %),21,24,25,34−37 depending on the preparation method; in particular, thermal reduction experiments enable tuning of the oxygen content: the higher the temperature, the lower the resulting oxygen content.38 Since these oxygen-containing groups cannot be eliminated in RGO, their effect on photocatalytic properties needs to be understood.
There have been a few experimental and computational studies of isolated GO and RGO, which showed that these materials’ functional groups tend to gather in clusters rather than be evenly distributed34,39−41 and that the ratio of these groups differs between GO and RGO and changes during the reduction process.34,42−45 The functional groups in GO and RGO have a major effect on the electronic properties: they break up the sp2 network of graphene, which leads to both the semiconducting behavior of GO and the reduced conductivity of RGO relative to graphene.33 Computational studies showed that both the type and the concentration of the functional groups strongly affect the band gap40,41 and the work function44 of the material. These studies give insight into the structure–property relationship of GO and RGO and also highlight the complexity of these materials, caused by their amorphous nature and by the great variety of possible arrangements of the functional groups.
In contrast to the multiple studies of isolated RGO, there have been very few studies (either experimental or theoretical) investigating the structure of GO- and RGO-based composites with semiconductor photocatalysts. Notably, several studies, using X-ray photoelectron spectroscopy and infrared absorption spectroscopy, demonstrated the presence of Ti–O–C and Ti–C interfacial bonds in RGO/TiO2 (anatase and P25 phase) composites;19,46−48 these studies suggested that these interfacial bonds could be the reason for the observed improvements of visible-light photocatalytic performance. Insight from theoretical modeling is essential to gain understanding of these structures and their properties; however, very few theoretical studies considered the presence of oxygen functional groups in graphene-based composites. Notably, a recent study by Ferrighi et al.49 has looked at the effect of a bridging oxygen atom (an epoxide group in RGO) at the TiO2/graphene interface and observed stronger electronic hybridization of TiO2 with this RGO compared to pure graphene and a small band gap opening in the RGO and in the TiO2/RGO composite. There are few examples of computational studies of carbon flakes or polyaromatic hydrocarbons anchored on TiO2 by oxygen groups46,50 and small (≤1 nm) TiO2 clusters adsorbed on clean or epoxide-functionalized graphene sheets.51 While all these studies show strong interfacial binding and changes in the electronic structure compared to bare TiO2, these finite systems cannot fully represent the extended structure of graphene and RGO. Since experimentally produced RGO/TiO2 composites contain large (several μm24,26,46) RGO sheets, infinite (2D periodic) RGO models are much closer to experimental systems than small finite flakes. Moreover, theoretical studies mentioned above used very low concentrations of oxygen functional groups (RGO with C/O ratios of 30:149 to 72:1,51 i.e., 1 to 3 at. %), well below the typical oxygen contents of 6–20 at. % in RGO.21,24,25,34−37
Therefore, there is a shortfall in the understanding of the impact of oxygen functional groups and interfacial cross-links on the electronic structure of the TiO2/RGO composites. The reason for this is the complexity of the structure of GO and RGO, which are amorphous and possesses no clearly defined crystal structure.33 Therefore, the choice of a representative structure or, more exactly, a range of representative structures becomes a key challenge. An additional challenge commonly encountered in computational modeling of heterostructures is the incommensurability of the TiO2 and graphene lattices, which requires large composite unit cells.29,32 This greatly increases the computational expense of such calculations, making accurate methods for electronic structure calculations such as the GW method not applicable.52 Therefore, our method of choice is hybrid Hartree–Fock/density functional theory (HF/DFT), which gives a reliable description of the electronic structure of semiconductors such as TiO253,54 but is still computationally demanding. By comparison, pure DFT methods are faster but underestimate band gaps of semiconductors,54 while the DFT + U approach corrects for the band gap underestimation, but there is no universally accepted value of U for TiO2,28−31,51 and variation in U is known to affect not only the band gap but also the localization of excess charges.53
In this study, we construct representative structures for RGO and GO with different oxygen contents and create realistic TiO2/RGO and GO composites and model them using hybrid DFT. We investigate the nature and strength of binding between RGO or GO and the TiO2 rutile (110) surface. Then we analyze the electronic structures of these composite materials. We find notable differences compared to the pure graphene/rutile (110) composite studied in our earlier work,32 both in the nature of interfacial binding and in the electronic structure: in particular, we observe an indirect band gap opening and formation of a distinct RGO-based unoccupied band below the conduction band of TiO2. We propose that this unoccupied band acts as an electron trap state and hinders electron–hole recombination, thereby enhancing the photocatalytic performance of TiO2/RGO composites.
Computational Methods
Geometry Optimizations
All geometry optimizations were done with the Quickstep program,55 part of the CP2K software package (www.cp2k.org). All pure DFT calculations used the PBE56 exchange-correlation functional, and all hybrid HF/DFT calculations used the range-separated HSE0657 functional. All structures were first optimized using the PBE functional and then reoptimized using the HSE06 functional afterward. In all cases, calculations included Grimme’s D2 dispersion correction.58 All optimization calculations utilized double-ζ basis sets with diffuse and polarization functions optimized for use in CP2K (denoted as DZVP-MOLOPT-GTH-qn in the program)59 and Goedecker–Teter–Hutter pseudopotentials.59,60 All hybrid HF/DFT calculations used the auxiliary density matrix method,61 which is implemented in CP2K. In these calculations, Hartree–Fock exchange is computed using a small auxiliary basis set and density matrix, while all non-HF parts of the calculation are computed using the primary basis set and density matrix. Auxiliary basis set cpFIT3 (contracted, 3 Gaussian exponents per valence orbital, includes polarization functions) was used for carbon, oxygen, and hydrogen, while FIT3 (3 Gaussian exponents per valence orbital) was used for all titanium atoms. All optimization calculations were done at the Γ point only. Binding and interaction energy calculations accounted for the basis set superposition error (BSSE) using the counterpoise (CP) method.62
Electronic Structure Calculations
After geometry optimizations were completed, an optimized wave function was produced in a single-point calculation using the CRYSTAL14 software package.63 All system properties and one-electron properties were then obtained from subsequent CRYSTAL14 analyses, with the exception of crystalline orbitals that were calculated using CRYSTAL17.64 The range-separated HSE0657 hybrid HF/DFT functional with Grimme’s D2 dispersion correction was used. All calculations used all-electron triple-ζ basis sets with diffuse and polarization functions devised by Peintinger et al.,65 and a Monkhorst–Pack k-point mesh of 12 × 12 × 1. In order to present absolute orbital energies, each density of states (DoS) and band structure shown is corrected for the energy of the electron in vacuum, which is done by shifting the energies of the plots. The energy of the electron in vacuum is taken as zero energy. The magnitude of the energy shift in the DoS and band structure plots is determined by calculating the electrostatic energy in the vacuum region of the simulation cell sufficiently far away from the atoms (>50 Å along the cell’s z axis).
Unit Cell Construction
The structure of the rutile (110) slab used in this study is the same as the 9 atomic layer slab we used in our previous work.32 Similarly, each RGO and GO structure we investigated was based on the graphene supercell featured in the same work.32 Each RGO and GO/rutile (110) composite therefore used a 2 × 5 extended three unit cell (9 atomic layers) thick slab of rutile with the (110) surface exposed (60 titanium atoms, 120 oxygen atoms) and a 3 × 6 supercell of an orthorhombic graphene unit cell (72 carbon atoms) as a basis. To create GO and RGO models, graphene sheets were functionalized with hydroxyl and epoxide groups. Five structures were created, with the carbon/oxygen ratios based on the experimental reports of oxygen contents in GO and RGO: the C/O ratio in GO was taken to be 2:1 (50% oxygen coverage),24,26 while in RGO they were 6:1 (16% oxygen coverage) and 12:1 (8% oxygen coverage)—the higher and lower ends of the experimentally measured oxygen content in RGO;20,35,38 highly reduced GO with C/O ratios of 18:1 and 36:1 was also modeled for comparison. These structures are henceforth referred to as GO, 6-RGO, 12-RGO, 18-RGO, and 36-RGO, respectively. The hydroxyl and epoxide groups were positioned to create amorphous arrangements, according to the following criteria based on the literature: (i) the functional groups should form clusters rather than be isolated,34,39−41 (ii) the functional groups should be, on average, evenly distributed above and below the graphene plane,39 and (iii) RGO structures should have islands of functional groups surrounded by sp2 regions, while GO structures should have sp2 islands surrounded by functional groups. Color-coded images of the GO, 6-RGO, and 12-RGO ratio structures are shown in Figure 1; the 18-RGO and 36-RGO structures were formed from 12-RGO by consecutively removing hydroxyl and epoxide functional groups. Each system contains different quantities of hydroxyl and epoxide groups: 30 hydroxyl and 10 epoxide groups in the GO system; 6 hydroxyl and 6 epoxide groups in 6-RGO; 4 hydroxyl and 2 epoxide groups in 12-RGO; 2 hydroxyl and 2 epoxide groups in 18-RGO; 2 hydroxyl and 0 epoxide groups in 36-RGO. These ratios of hydroxyl and epoxide functional groups are in line with the ranges reported in earlier experimental and computational studies of GO and RGO.34,42−45 No requirement was made to use constant ratios of these groups because the reported concentrations of hydroxyl, epoxide, and carbonyl groups in GO and RGO vary.34,42−45 More hydroxyl than epoxide groups were added to promote interfacial interactions; the number of hydroxyl groups was kept even to avoid producing a spin-polarized system and to avoid the more costly spin-polarized calculations. Spin-polarized calculations were carried out on one system, the 12H-RGO (NCL) composite described in the next section; both singlet and triplet calculations converged to the singlet state, and the densities of states produced by spin-polarized and spin-averaged calculations were very similar; therefore, only spin-averaged calculations were done for all further systems.
Figure 1.

Images showing the studied structures of the GO and RGO systems with C/O ratios of (a) 2:1 (GO), (b) 6:1 (6-RGO), and (c) 12:1 (12-RGO). Color coding: oxygen (red); hydrogen (white); sp2 carbon (dark grey); sp3 carbon (green).
When the GO cell (optimized cell parameters a = 13.101 Å and b = 15.080 Å) is interfaced with the rutile (110) cell (a = 13.058 Å and b = 14.975 Å), this results in very small compressive strain on GO: −0.33 and −0.70% along the a and b directions, respectively. By comparison, the graphene/TiO2 composite studied in our earlier work32 has small tensile strains on graphene of +2.32 and +1.44% along the a and b directions, respectively (cf. optimized cell parameters of the 3 × 6 graphene supercell a = 12.762 Å and b = 14.760 Å). The slight increase in size of the GO cell compared to graphene is attributed to the greater number of sp3 carbon atoms in the GO system. We expect that the RGO systems with fewer oxygen functional groups would be intermediate in size between graphene and GO, with the difference proportional to the oxygen content. Our tests done elsewhere32 showed that this very small amount of strain on graphene resulted in <0.1 eV changes in graphene band gaps, total energies, and Fermi level positions. We therefore expect that the very small strain on GO and RGO in the composite structures will have a negligible effect on their electronic properties.
Results and Discussion
Design of Composite Unit Cells
Composites of TiO2 with the GO and RGO model structures described in the Computational Methods section were produced by placing the graphene-based structures on top of the rutile (110) slab (Figure 2 and Figure S1 in the Supporting Information). Several structures were considered for RGO-based composites, aiming to achieve either interfacial hydrogen bonding (structures labeled 12H and 6H) or chemisorption (structures 12C and 6C). To achieve chemisorption, a hydrogen atom was removed from a hydroxyl group on the face of RGO facing rutile (110) to facilitate the formation of a Ti–O–C bond to a nearby 5-coordinated Ti atom, and one more hydrogen was removed from a hydroxyl group on the other face of RGO (creating an epoxide group) to avoid spin-unpaired systems.
Figure 2.
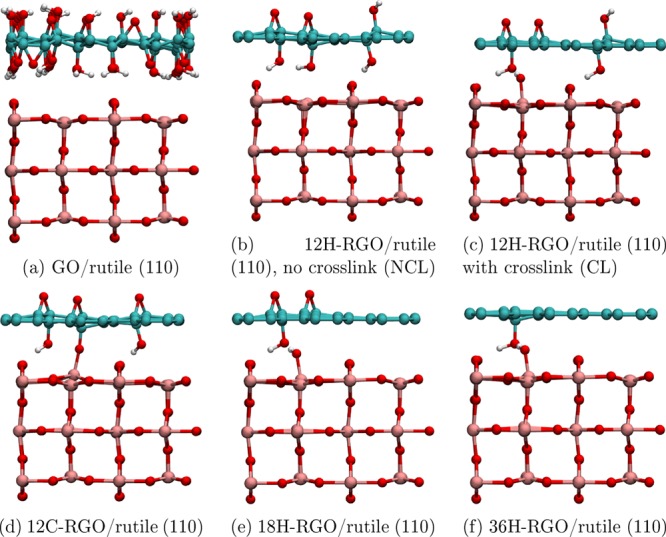
Optimized structures of the GO/ and RGO/rutile (110) composites used in this work: (a) GO/rutile (110); 12:1 RGO/rutile (110): (b) hydrogen-bonded, no cross-link (NCL), (c) hydrogen-bonded with cross-link (CL), (d) chemisorbed; (e) 18:1 RGO/rutile (110); and (f) 36:1 RGO/rutile (110).
The composite structures were optimized first using dispersion-corrected PBE and then HSE06 functional, as described in the Computational Methods section. The exception is the 6H-RGO and 6C-RGO/rutile structures (Figure S1), which were optimized only using PBE + D, as it was found to be too difficult to attain self-consistent field (SCF) convergence for these systems using HSE06. Therefore, only the GO-based and 12-RGO-based composites and the 18- and 36-RGO composites based on them (e.g., all structures shown in Figure 2) were used for the following analysis of the electronic structure.
Notably, several different structures were obtained for composites based on 12-RGO (shown in Figure 2b–d). The initial optimization of the 12H-RGO/rutile composite system using PBE + D resulted in a purely hydrogen-bonded interface structure (Figure 2b), while further optimization with HSE06 + D resulted in a rearrangement where a hydroxyl group from RGO was transferred to the surface of rutile (110). The latter structure is denoted the cross-linked (CL) structure (Figure 2c), and it contains a Ti–O–H···O–C bonding arrangement with a hydroxyl group on RGO (where ··· indicates a strong hydrogen bonding interaction between H and O with a bond length of 1.55 Å). In contrast, the 12C-RGO/rutile (110) composite (Figure 2d) forms a Ti–O–C covalent bond between RGO and the rutile (110) surface. Covalent bond formation was also attempted in the 6C-RGO/rutile (110) system by positioning 6-RGO with one of its oxygen atoms directly above an undercoordinated Ti atom of TiO2; however, this did not result in interfacial covalent bonding between RGO and the rutile (110) surface, but interfacial hydrogen bonds were formed instead. Thus, unlike 12-RGO, which has a low density of functional groups, 6-RGO could not approach the rutile (110) slab close enough to form Ti–O covalent bonds because of short-range repulsion between the hydroxyl groups of RGO and 2-coordinated bridging oxygens of rutile (110). The structures of 18H- and 36H-RGO/rutile (110) (Figure 2e,f, respectively) were created from the cross-linked 12H-RGO/rutile (110) system by consecutively removing oxygen functional groups not involved in the cross-link.
Binding Properties of the TiO2/RGO and GO Interfaces
To investigate the strength of binding of the RGO and GO structures to the rutile (110) surface, interfacial binding (Ebind) and interaction (Eint) energies were calculated as follows
| 1 |
where Etot is the total energy of the composite system, Eru(opt) is the total energy of the optimized rutile (110) slab, Egr(opt) is the total energy of the optimized GO or RGO sheet, and EBSSE is the basis set superposition error correction. This gives the overall energy difference resulting from bringing the two parts of the composite together. This overall interaction energy can be decomposed into the energy changes due to binding the two parts together (Ebind) and the structural deformation (Edef) resulting from the combination of the two parts
| 2 |
| 3 |
where Eru(def) and Egr(def) are the total energies of the rutile and graphene parts of the composite fixed in the geometries that they adopt in the composite system, respectively. These energies, calculated with Quickstep using the PBE functional with the D2 correction, are shown in Table 1. Comparing the results for the GO, RGO, and previously calculated pure graphene based composite32 shows that having a small number of oxygen functional groups (as in the case of RGO) strengthens interfacial binding, while the much greater number of such groups present in the GO system has a detrimental effect on interfacial binding strength.
Table 1. Binding Energies of the GO/ and RGO/Rutile (110) Composite Systems Calculated Using PBE + Da.
| system | Eint (eV) | Ebind (eV) | Edef (eV) | Edef(ru) (eV) | Edef(gr) (eV) |
|---|---|---|---|---|---|
| GO/rutile (110) | –1.12 | –3.02 | 1.91 | 0.76 | 1.15 |
| 6H-RGO/rutile (110) (NCL) | –2.45 | –3.06 | 0.61 | 0.51 | 0.10 |
| 6C-RGO/rutile (110) (NCL) | –2.23 | –3.05 | 0.82 | 0.52 | 0.30 |
| 12H-RGO/rutile (110) (NCL) | –1.74 | –3.66 | 1.92 | 0.77 | 1.15 |
| 12H-RGO/rutile (110) (CL) | –4.21 | –5.66 | 1.45 | 1.11 | 0.34 |
| 12C-RGO/rutile (110) (CL) | –3.44 | –7.39 | 3.95 | 2.49 | 1.46 |
| 18H-RGO/rutile (110) (CL) | –3.97 | –5.14 | 1.17 | 0.95 | 0.22 |
| 36H-RGO/rutile (110) (CL) | –4.10 | –5.45 | 1.35 | 1.16 | 0.18 |
| graphene/rutile (110) | –1.67 | –3.24 | 1.57 | 0.89 | 0.69 |
As seen in Table 1, all cross-linked RGO/rutile composites with low oxygen contents (12C-, 12H-, 18H-, and 36H-RGO (CL) composites) have large interaction energies, much larger than that for the pristine graphene/TiO2 composite, showing a large energy gain due to forming interfacial covalent or strong hydrogen bonds. In particular, the strong interaction observed in the cross-linked 12H-, 18H-, and 36H-RGO/rutile (110) composites involving transfer of a hydroxyl group from RGO to the TiO2 surface can be explained by the large binding energy gain due to forming a new Ti–O bond (bond energy 666.5 ± 5.6 kJ mol–166), which outweighs the cost of breaking a single C–O bond (energy 385 ± 6.3 kJ mol–166) and results in a net decrease of the total energy. In the covalently bonded 12C-RGO/rutile (110) composite, a large energy gain due to the formation of a new interfacial Ti–O bond is accompanied by a large deformation of both the rutile and RGO subsystems. While the energy barrier to formation of the Ti–O–C bond was not calculated in this work, it can be expected based on these deformation energies that this type of cross-link will form mainly at elevated temperatures, such as those used in thermal reduction of GO.20,38
By comparison, weak interfacial hydrogen bonds make only a small contribution to the overall interaction energy: for example, the removal of a hydrogen-bonded hydroxyl group from the 12H-RGO/rutile (CL) composite to form the 18H-RGO composite reduces the interaction energy by only 0.24 eV. The interaction energy of the non-cross-linked 12H-RGO/rutile (NCL) composite (with two interfacial hydrogen bonds) is only 0.07 eV larger than the interaction energy of the pure graphene based composite bound only by dispersion interactions. As seen in Table 1, the binding energy gain from these hydrogen bonds is balanced by an increase in the RGO deformation energy. The two 6-RGO-based composites are also relatively weakly bound because they are bound only by interfacial hydrogen bonds with no interfacial covalent bonds.
Interestingly, even epoxide groups that do not participate in interfacial binding nevertheless affect the binding energies: the removal of two epoxide groups from the 18H-RGO/rutile (CL) composite to form the 36H-RGO composite increases the binding energy by 0.30 eV. This can be attributed to the electron-withdrawing character of epoxide groups, which draw electron density from the neighboring atoms involved in the interfacial binding and thus weaken the binding. This effect will be discussed in more detail below.
The GO/rutile (110) interface, surprisingly, has the smallest interaction energy among the structures considered here, even smaller than pure graphene/rutile (110). A likely explanation is that the GO sheet moves up away from TiO2 to minimize the repulsion between TiO2 oxygens and RGO epoxide and hydroxyl oxygens, and the interfacial dispersion interactions are therefore weakened. Moreover, having many functional groups may lead to intra-GO hydrogen bonding, leaving few functional groups available to form hydrogen bonds with the surface of TiO2.
In order to explain the rather weak interfacial bonding in the GO/rutile (110) composite, the GO/rutile and 12H-RGO/rutile (CL) structures were analyzed for the presence of hydrogen bonding (Figure 3a–c). Hydrogen bonds were visualized using the VMD software package,67 with a maximum bond distance cutoff of 3 Å and O–H···O maximum bond angle deviations of 20 and 50° from the ideal value of 180°. From the images of the GO composite in Figure 3a,b, it can be seen that there are few hydrogen bonds between GO and rutile (110), and such bonds are weak, as the hydrogen bond angles deviate very far (over 20°) from the ideal value of 180°. Instead, multiple hydrogen bonds between functional groups within GO are formed. This contrasts strongly with the cross-linked 12H-RGO/rutile (110) composite (Figure 3c), which has multiple interfacial hydrogen bonds with small (≤20°) deviations from the ideal angle. This lack of interfacial hydrogen bond formation can explain the low binding energy in the GO/TiO2 composite. We note that we did not attempt to achieve optimal GO/rutile (110) interfacial bonding by tuning the structure of this interface or the position of the GO above the TiO2 slab. We expect that stronger bonding may be achieved in alternative interface structures; however, our results show that a high concentration of oxygen functional groups is not a necessary or sufficient requirement for strong interfacial binding in such composites.
Figure 3.

(a–c) Hydrogen bonding interactions (blue dashed lines) in the (a,b) GO/rutile (110) and (c) 12H-RGO/rutile (110) (CL) composites, visualized with a maximum distance cutoff of 3 Å and maximum O–H···O bond angle deviations of (a,c) 20° and (b) 50° from the ideal value of 180°. (d) Isosurface of the electron density difference upon formation of the cross-linked 12H-RGO/rutile (110) composite shown in panel (c), calculated using the HSE06 functional (rendered at 0.001 eÅ−3). Charge depletion is shown in yellow, and charge accumulation is shown in blue.
To investigate the nature of the interaction in these composite systems further, the electron density difference was mapped for the 12H-RGO/rutile (110) (CL) system (Figure 3d). This difference was defined as the difference in electron density of the composite and the isolated 12H-RGO and rutile (110) parts in their composite geometries. Large rearrangements of electron density can be seen. Overall, there is charge depletion (shown in yellow) in RGO and charge accumulation (shown in blue) in TiO2 and at the interface, indicating overall charge transfer from RGO to TiO2. A significant aspect is the interfacial hydrogen bonding seen as alternating charge depletion and accumulation areas. Some rearrangement of charge can be seen in the rutile (110) slab: accumulation of density in nonbonding orbitals and slight depletion in the bonding orbitals of the surface and subsurface oxygen atoms. Interestingly, while there is a widespread depletion of charge in the π orbitals of the sp2 carbon atoms in RGO, there is also a slight increase in charge density in the σ orbitals of the same atoms, indicating a shift in electron density from the π system to local σ bonding upon forming the interface. Furthermore, there are clear differences between the oxygen functional groups on the upper face of the RGO sheet, which do not take part in interfacial bonding: there is noticeable accumulation of charge density on the epoxide groups but almost none on the hydroxyl group. This shows that the epoxide groups have a much greater electron-withdrawing effect on the surrounding carbon sp2 atoms. These charge density variations are notably different from those observed in the graphene/rutile (110) system studied in our earlier work32 where the dominant effect was the transfer of charge from graphene to rutile (110) oxygen atoms. In the case of the RGO composite, the predominant effect is the donation of electron density from RGO to the interfacial bonds.
Our findings on the nature of interfacial binding are consistent with earlier experimental studies of RGO/TiO2 composites, which suggested that oxygen defects play an important role in the interfacial binding in these materials.21,36 The results presented in this section show that the formation of cross-links and interfacial hydrogen bonds is the important means by which strong interfacial binding is achieved. High local concentrations of functional groups (such as in the case of GO) do not necessarily promote interfacial binding, as it will be less likely that GO will approach TiO2 closely and form strong hydrogen bonds and covalent bonds with TiO2. Instead, lower local concentrations of functional groups (as in RGO) lead to strong interfacial binding. In particular, the formation of Ti–O–C and Ti–O–H···O–C cross-links (as seen in Figure 2c,d) significantly improves the interfacial binding compared to hydrogen bonding alone. It can be expected that the formation of these cross-links would require overcoming an energy barrier. High-temperature (over 150 °C) processes such as hydrothermal reduction or high-temperature annealing are likely to favor the formation of cross-links during the reduction process. On the other hand, evidence of Ti–O–C and Ti–C bonding is seen even in samples of RGO/TiO2 that were chemically reduced using hydrazine prior to combination with TiO2 and kept at relatively low temperatures (40 to 80 °C).46 Studies of the kinetics of formation of such cross-links, similar to modeling studies of transformations of RGO structures,44 would be needed to determine the mechanisms of cross-link formation and the favorable experimental conditions; however, this is beyond the scope of this work.
Electronic Properties of the Graphene Oxide/Rutile (110) Interface
To obtain insight into the enhanced photocatalytic efficiency of TiO2 composites with GO and RGO, the electronic structure of the composites shown in Figure 2 was calculated using the HSE06 functional. Electronic properties of the GO/rutile (110) composite are presented in Figure 4 and Figure S2. The density of states (DoS) spectra (large-scale image in Figure 4a and small-scale overview image in Figure S2a) and the band structure plot (Figure 4b) indicate that GO in this composite has an electronic structure similar to that of an isolated molecule, with discrete occupied GO levels in the TiO2 band gap, and the HOMO of GO (labeled “VBM” in Figure 4a) in this instance is situated just below the rutile (110) conduction band minimum (CBM). These positions of the GO HOMO and rutile CBM originate from the electronic structure or the isolated GO and rutile components shown in Figure S2b,c. This unusual alignment of the composite’s electronic energy levels results in an almost zero band gap (Table S1); however, it is clear from the band structure and from the composition of the bands that this system is far from metallic and is better described as semiconducting. The HOMO is almost entirely composed of π orbitals of sp2 carbon atoms, while the corresponding π* orbital is roughly 2.1 eV higher in energy and is positioned deep within the TiO2 CB (as seen in Figure S2a). No mid-gap states with mixed TiO2 and GO character are found in this system, indicating that there is very little interaction across the interface. This composite does not have any cross-links with the surface of rutile (110). Without such strong interactions with the surface, it can be expected that mixed TiO2/GO electronic states would not form, and therefore, charge transfer excitation is unlikely to take place. It is still possible that visible-light excitation of GO could then lead to electron injection to the rutile (110) conduction band as a second step; however, the weak interactions between GO and rutile (110) would likely lead to slow rates of charge transfer. Therefore, it is unlikely that this type of interfacial binding arrangement will show enhanced absorption or strong charge separation compared to pure TiO2; thus, this system is unlikely to show enhanced photocatalytic properties.
Figure 4.

(a) DoS spectrum of the highest occupied and lowest unoccupied levels of the GO/rutile (110) composite, showing discrete GO levels in the TiO2 band gap. The color scheme is shown in the legend here and in all following DoS plots, contributions of C atoms are shown as shades of blue, O atoms as shades of red and yellow, and Ti atoms as shades of green. (b) Band structure of the GO/rutile (110) composite. Blue bands are formally occupied; red bands are formally unoccupied. The zero energy is the energy of the electron in vacuum. The highest occupied level (labeled “VBM” in the DoS spectrum) in this system is just below the TiO2 CBM.
Electronic Properties of Reduced Graphene Oxide/Rutile (110) Interfaces
Effect of Interfacial Binding on the Electronic Properties
Next, we consider the electronic properties of composites based on 12-RGO, as its composition is representative of experimentally achievable oxygen concentrations. Composites with different interfacial binding were compared: the hydrogen-bonded 12H-RGO/rutile (110) (NCL) structure (Figure 2b), the cross-linked hydrogen-bonded 12H-RGO/rutile (110) (CL) structure (Figure 2c), and the covalently bonded 12C-RGO/rutile (110) structure (Figure 2d).
The DoS and band structure of the least strongly bound 12H-RGO/rutile (110) (NCL) composite are shown in Figure 5 and Figure S3. The electronic structure is similar to the pure graphene/TiO2 composite:32 the graphene Dirac point is present in the band structure (between the Y and Γ k-points) and is positioned in the TiO2 band gap, indicating that RGO is semimetallic in this system. Localized states with large contributions of the RGO oxygen functional groups can be seen lower in the VB (below −8 eV) and higher in the CB (above −5 eV, see Figure S3). TiO2 CB states begin to appear as low-dispersion bands at −5.5 eV, and TiO2 VB states begin to appear at −9 eV. The way that the RGO bands intersect the rutile (110) bands without obvious signs of interaction indicates that there is no strong electronic coupling between the two, similar to the case of graphene/rutile (110) described in our earlier work32 and GO/rutile (110) described above. The DoS similarly shows no evidence of mixing of electronic states between the two materials. Therefore, charge transfer excitation is not expected to be prominent in this weakly bonded composite, and it is not expected that this type of local chemical environment would be the source of the enhanced visible-light photocatalytic properties seen in experimental RGO/TiO2 composite systems.
Figure 5.

(a) DoS spectrum of the highest occupied and lowest unoccupied levels of the 12H-RGO/rutile (110) composite with no cross-link. The color scheme is shown in the legend. (b) Band structure of the same composite. Blue bands are formally occupied; red bands are formally unoccupied. The zero energy is the energy of the electron in vacuum.
For the cross-linked 12H-RGO/rutile (110) system, the electronic structure is very different (Figure 6 and Figure S4). The system is not semimetallic any more but has an indirect band gap of 0.23 eV (see Table S1). A new unoccupied band can be seen between −5.9 and −5.6 eV in the gap between the valence band maximum (VBM) of RGO and the rutile (110) CB. This particular band now forms the lowest unoccupied band of the system. This band has an unusual dependence on electron momentum: its energy is highest at the Γ point and lowest much farther away in the Brillouin zone near k-point S. This band has similar shape and orbital characteristics to the first VB: both bands are largely based on RGO sp2 carbons (as seen in the DoS spectrum Figure 6a). The upper part of the CB (near Γ) is highly dispersed (graphene-like) and shows roughly equal contributions from sp2 carbons and epoxide oxygen atoms, while away from the Γ point it becomes much less dispersed (molecule-like) and acquires greater contributions from sp3 carbon and oxygen functional groups, including very small contributions from the Ti and O atoms involved in the cross-link.
Figure 6.

(a) DoS spectrum of the highest occupied and lowest unoccupied levels of the 12H-RGO/rutile (110) (CL) composite. The color scheme is shown in the legend. (b) Band structure of the same composite. Blue bands are formally occupied; red bands are formally unoccupied. “K740” refers to the k point (in the 12 × 12 × 1 grid) where the CBM is observed. The zero energy is the energy of the electron in vacuum.
The nature of this lowest unoccupied band was investigated further by visualizing the crystalline orbitals (COs) of this composite at k-points Γ (band maximum) and S (close to the band minimum) (Figure 7). It can be seen that, overall, the compositions of the CB at Γ and S (Figure 7a,c, respectively) are quite similar, both containing a combination of carbon, RGO oxygen, and some titanium atomic orbitals (AOs). However, there are subtle differences: there is a greater contribution of carbon sp2 AOs at Γ than at S; the carbon sp2 AOs contributing to the CO at k-point S are much more localized and mostly belong to carbon atoms nearest to the epoxide oxygen atoms. This explains the DoS and band structure results for this CB in Figure 6: although sp2 carbons contribute to this band at all energies, the band is delocalized over many sp2 carbon atoms near the Γ point but is localized only on a few sp2 carbon atoms at the S point.
Figure 7.

(a,c) Visualizations of the crystalline orbitals (COs) of the 12H-RGO/rutile (110) (CL) composite, for the first conduction band at k-points Γ (panel (a)) and S (panel (c)). (b) Reference structure of the 12H-RGO/rutile (110) (CL) composite showing the atom species (dark gray: C, light gray: Ti, red: O, white: H), to compare with panels (a) and (c).
The VB shows the same distinction between the mainly carbon-based, highly dispersed, spatially delocalized states (top of the first VB) and largely oxygen-based localized states (lower-lying bands in the VB), as seen in Figure 6a and Figure S4. Overall, the band structure and DoS spectrum of the 12H-RGO/rutile (CL) composite show that the RGO electronic states in this system are more delocalized than those in GO/rutile (110), as a consequence of the partial restoration of the sp2 network, but it has more localized electronic states than the weakly bound (non-cross-linked) composite shown in Figure 5 and the composite containing defect-free graphene.32
These changes in the band structure, in particular, the gap opening and the new lowest unoccupied band with the inverted shape, show that the strong interfacial binding has a significant effect on the electronic structure of the RGO/TiO2 composite. The indirect nature of the band gap has important implications for the photoexcited electron and hole properties: this difference in momentum between the CBM and VBM is expected to greatly slow the recombination of photogenerated charge carriers and lead to long-lived excited states.
Finally, the electronic structure of the chemisorbed 12C-RGO/rutile (110) system was analyzed by the same means (Figure S5). The most obvious feature is that, similar to the 12H-RGO/rutile (CL) composite, this structure has an indirect band gap and qualitatively the same RGO-based lowest-unoccupied band, with a similar shape and composition: carbon sp2, carbon sp3, epoxide, and hydroxyl. There are slight differences, especially in the occupied bands: the widths of the first unoccupied band and the occupied bands decrease in the 12C-RGO composite (the band gap consequently increases to 0.56 eV), indicating that there is more disruption to the carbon sp2 system and greater localization than seen in the 12H-RGO (CL) composite. DoS spectra show much stronger contributions from sp3 carbons and weaker contributions from sp2 carbons in the 12C-RGO composite; this can be explained, first, simply by the larger number of sp3 carbons present in this system and, second, by the electron-withdrawing epoxide groups drawing electron density from sp2 carbons and producing more localized states. Notably, contributions of the Ti atom involved in the interfacial bonding can be seen in the predominantly RGO-based VB and first CB, indicating strong through-bond coupling between the TiO2 and RGO components. The overlap between these states could facilitate charge transfer photoexcitation in this composite.
The key conclusion reached by comparing the electronic structure of the 12C-RGO composite and the cross-linked and non-cross-linked 12H-RGO composites is that the presence of strong interfacial bonds provides the necessary chemical environment to form the new RGO-based CB of the system and open an indirect band gap. In contrast, in the absence of a cross-link, a graphene-like electronic structure is observed. This suggests that strong interfacial binding at the interface transforms the RGO electronic structure from semimetallic to semiconducting and produces this new CB state that may act as an electron trap state.
Effect of the Oxygen Content in RGO on the Electronic Properties
To determine whether the formation of the cross-link or the presence of oxygen defects is primarily responsible for the formation of the new CB, the 12H-RGO/rutile (CL) composite was modified by sequentially removing oxygen functional groups. First, two hydroxyl groups (of the original total of 4) were removed (structure shown in Figure 2e), leaving two epoxide groups and the cross-linking hydroxyl groups to form the 18H-RGO/rutile (110) composite. The electronic structure is shown in Figure S6, and it strongly resembles that of 12H-RGO/rutile (CL) shown in Figure 6 and Figure S4: the RGO lowest unoccupied band seen in the 12H-RGO (CL) composite is still present, and its width does not change significantly. The band maximum is still centered at Γ, while the minimum has shifted to be exactly at the S point. The indirect band gap is very slightly reduced from 0.23 to 0.21 eV (Table S1) as a result of the expansion of the carbon sp2 network. The composition of this band is also similar to the 12H-RGO (CL) composite: there is a majority contribution from sp2 carbons around the maximum, while at the lowest energy the band is predominantly composed of epoxide oxygens. Thus, reduction in the oxygen content by removing weakly interfacially bonded RGO hydroxyl groups has only a minor influence on the electronic structure of the composite: the RGO-based CB is still formed and is expected to act as an electron trap.
Removing the final two epoxide groups from the 18H-RGO structure, thus yielding the 36H-RGO/rutile (110) composite (Figure 2f), leads to a greater change in the electronic structure (Figure 8 and Figure S7). The VB is now more graphene-like (cf. the band structure of the pure graphene/TiO2 composite32 shown in Figure 8c), and the energy gap between the CBM and VBM has closed sufficiently to make the system an indirect zero-gap semiconductor. The energy gap between the carbon π and π* bands (between the VBM and the bottom of the CB + 1) has decreased significantly to around 0.48 eV, and carbon sp2 and oxygen functional group states are more distinct from each other in this DoS than in the previous structures. This shows that the carbon sp2 network has expanded following the removal of the epoxide groups. The much stronger effect of removing the epoxide groups compared to removing hydroxyl groups can be attributed to the greater electron-withdrawing ability of the epoxide groups (as illustrated in the electron density difference plot in Figure 3d). However, despite these differences in the band structure, the key feature of inverted RGO-based CB and the indirect gap is preserved in this system as in the other cross-linked composites, in clear contrast to the semimetallic pure graphene/TiO2 system.32
Figure 8.

(a) DoS spectrum of the highest occupied and lowest unoccupied levels of the 36H-RGO/rutile (110) composite. The color scheme is shown in the legend. (b) Band structure of the same composite. (c) Band structure of the graphene/rutile (110) composite initially studied elsewhere.32 Blue bands are formally occupied; red bands are formally unoccupied. The zero energy is the energy of the electron in vacuum.
Comparison of these systems with varied oxygen content confirms that the presence of cross-linking oxygen defects, even at very low concentrations (up to a C/O ratio of 36:1 in this example), has a very strong effect on the electronic structures of RGO/TiO2 composites and in all cases leads to the formation of an indirect band gap and a possible trap state (while at the same time all structures have subtle differences in their electronic structure caused by the differences in their chemical structures). As such, it is essential that any computational modeling of RGO-based composites must take into account both the presence of functional groups and cross-links formed at the interfaces.
We can also compare our systems to the RGO/anatase (101) composite with a single epoxide cross-linking group studied by Ferrighi et al.49 That work found enhanced density of carbon states at the bottom of the CB; these carbon states extended to lower energies than the Ti CB states and formed the CBM of the composite. Based on the DoS spectra in that work, it is possible that a similar RGO-based lowest unoccupied band was formed as in our strongly bound systems, but it cannot be verified because the band structure was not produced.49 Thus, band structure analysis is essential for understanding the nature of electronic states in such composites, in particular for revealing the indirect band gap and the inverted RGO-based lowest unoccupied band—a potential photoelectron trap state.
Electronic Structure of Isolated RGO
In order to separate the effects of the RGO oxygen groups from those caused by the interfacial binding in the RGO composites, the electronic structures of the isolated RGO sheets taken from the cross-linked 12H-RGO and 36H-RGO/rutile (110) composites were analyzed. Both RGO systems were optimized with the same method as for the composite systems. The hydroxyl group that is transferred to the rutile (110) surface was not included in these RGO structures, as it was considered to be part of the rutile (110) component rather than the RGO component, thus creating spin-polarized RGO systems. Band structures for these RGO structures (Figure 9, plotted for both spin states, where the α spin state contains the unpaired electron) show that these structures are semiconducting, as opposed to semimetallic in the case of pristine graphene, and have a singly occupied (α spin) or first unoccupied (β spin) band, which closely resembles the lowest unoccupied band of their respective composite systems (Figure 6b and Figure 8b). The compositions of these α-spin highest occupied band and the β-spin lowest unoccupied band are also similar to the lowest unoccupied bands of the RGO/rutile (110) composites: mainly sp2 carbon around the Γ point and sp3 and sp2 carbon, hydroxyl, and epoxide oxygen between Y–S–X.
Figure 9.

α- and β-spin band structures of the isolated 12H-RGO ((a,b), respectively) and 36H-RGO ((c,d), respectively). The zero energy is the energy of the electron in vacuum.
The β-spin lowest unoccupied band, in particular, can be seen as the precursor to the lowest unoccupied band of the composite. This RGO-based band is unoccupied in the composites due to the transfer of electron density from RGO to rutile (110). The width of this band in isolated RGO is much smaller than that in the composite, and the band gap is correspondingly larger (Table S1). These changes in the band width and band gap can be explained by considering the band composition. The width of this band and the indirect energy gap are largely dependent on the energy of the RGO oxygen and carbon states, which make up the minimum of the lowest unoccupied band; these are the atoms involved in the interaction with titanium atoms of rutile (110) in the composite. It can be concluded that stabilizing electronic interaction between the RGO oxygen functional groups of the cross-link and surface titanium atoms lowers the energy of the unoccupied RGO oxygen states relative to the carbon sp2 states. This increases the lowest unoccupied band width in the composite compared to isolated RGO and accentuates the inverted curve of the lowest unoccupied band. Thus, the origins of the inverted shape of this band can be already seen in the RGO β-spin band structure, but this shape becomes more pronounced when cross-links to TiO2 are formed in the composites.
Discussion: Relationship between the Composites’ Electronic Structures and Photocatalytic Properties
This work has identified, broadly speaking, two types of RGO/rutile (110) composites that differ in the type of interfacial binding and the electronic structure: (i) the weakly bound (purely hydrogen-bonded or physisorbed) composites, where the electronic structure of the RGO component is similar to that of graphene and there is little electronic coupling between the RGO and the TiO2 component, and (ii) the strongly bound composites with either hydrogen-bonded or covalent cross-links, which possess an indirect band gap and an RGO-based lowest unoccupied state, which is energetically separated from the TiO2 CB. The latter structures, which display strong RGO-TiO2 electronic coupling, are interesting to discuss in relation to photocatalytic properties of such composites.
First, in each cross-linked system studied in this work, the RGO oxygen-based bands contain small contributions from the surface titanium atom forming the rutile (110) terminus of the cross-link—labeled as Ti (Ti–OH) or Ti (Ti–O–C) in the DoS plots. This is clear evidence of electronic coupling between the orbitals of the oxygen functional groups in RGO and the Ti atoms involved in the cross-link. Several experimental studies proposed that the enhanced visible-light photocatalytic properties of such composite systems are due to the formation of Ti–O–C cross-links, which provide direct means of promoting electrons from RGO to the TiO2 conduction band with visible-light photons.14,46 The results in this work support this hypothesis: our results show that the formation of a cross-link changes the electronic behavior of the RGO component and couples the electronic structures of RGO and rutile (110). We can see from the DoS plots that RGO bands are suitably positioned to enable UV- and visible-light excitation. While in this work we did not calculate the oscillator strengths of transitions, it would be reasonable to assume, based on the band energies and compositions, that these oxygen functional group bands may be involved in charge transfer excitation from RGO to TiO2. This is consistent with experimental observations that show that oxygen-containing RGO/TiO2 composites show enhanced visible-light excitation22,29,68 and that these composites have measurable quantities of interfacial cross-links.22,46
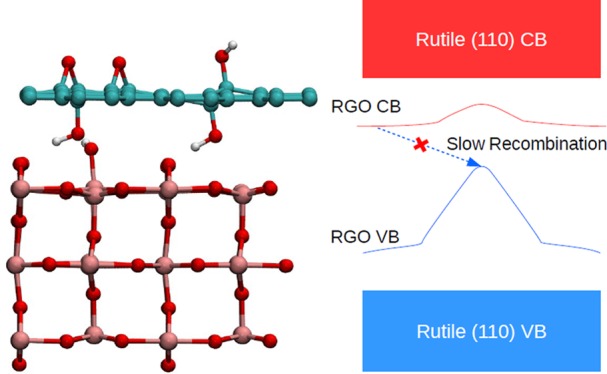
The most unusual and important property of the RGO/TiO2 composite systems discovered in this study is the shape of the RGO-based lowest unoccupied band (CB) in the strongly bound composites. The maximum of this CB is at the Γ point, and its minimum is at around the S point. As the energy profile of the CB is qualitatively similar to that of the VB, the band gap of the system is indirect with a large difference in electron momentum between the top of the VB and the bottom of the CB. The RGO CB is also below the rutile (110) CB and is energetically separated from the TiO2 CB by 0.2–0.4 eV. Therefore, it can be expected that this type of unoccupied band would act to trap photoexcited electrons in such a way that would hinder electron–hole recombination. Figure 10 schematically shows our proposed model for electronic processes in strongly coupled RGO/TiO2 composite systems. Upon UV light excitation, electrons are promoted from the TiO2 VB to the TiO2 CB. However, photogenerated electrons are able to decay from the TiO2 CB to the lower lying RGO CB; this is facilitated by the RGO-TiO2 electronic coupling (in particular, the contribution of the surface Ti to the RGO CB). Similarly, photogenerated holes decay from the TiO2 VB to the higher-lying RGO VB. Because of the indirect band gap between the RGO VB and CB, recombination between the holes and electrons in these bands is slow; therefore, photogenerated charges remain trapped. Upon long-wavelength visible-light excitation, electrons are promoted from the RGO VB to the RGO CB; similarly, recombination is hindered. Long-wavelength visible-light excitation may also promote electrons from the RGO VB to the TiO2 CB, and this again would lead to photoelectron relaxation to the RGO CB and hindered recombination, similar to the scenario of UV-light excitation.
Figure 10.
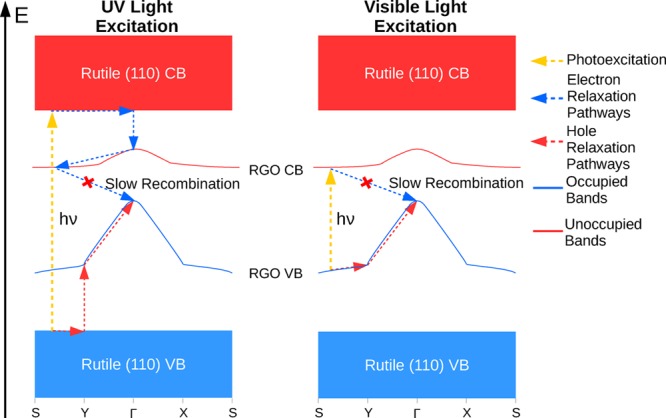
Proposed schematic of photoexcitation and charge trapping processes in the RGO/TiO2 composite system. UV-light excitation produces photogenerated electrons in the TiO2 CB and holes in the TiO2 VB, which decay to the RGO CB and RGO VB, respectively. Visible-light excitation produces photogenerated electrons and holes in the RGO CB and RGO VB. In both scenarios, the recombination process would be between electrons in the RGO CB and holes in the RGO VB. It can be seen that the large difference in electron momentum between the two charge carriers is what slows recombination and results in long excited-state lifetimes for such composites.
While there is no experimental verification of the shape of the band structure or the energy distribution of the CB of RGO/TiO2 composites, there is some support from experimental studies of charge recombination and photocurrent responses. Studies have shown that the lifetime of the excited state is increased, charge recombination is reduced, and the UV photocurrent increases when GO or RGO is added to TiO2,24−26 that the photocurrent increases as oxygen functional groups are removed during the reduction from GO to RGO,26 and that RGO/TiO2 composites also yield visible-light photocurrent29,68 (as opposed to no visible-light photocurrent in pure TiO2 samples), although it is much lower than the photocurrent observed under UV illumination. Photocurrent has been attributed to both photogenerated electrons and holes; the increase in UV photocurrent points to reduced charge carrier recombination, which is ascribed to the trapping of photogenerated electrons.25 This is consistent with the results of this work: the RGO-based CB observed in this work is a likely electron trap state, and its shape hinders recombination of the electrons with holes in the VB. Our results can also explain the weak visible-light photocurrent: the band positions allow visible-light excitation from the RGO VB to the RGO and TiO2 CB; however, excitation to the TiO2 CB is expected to be weak because of the relatively weak coupling between the TiO2 and RGO subsystems (evidenced by the small contributions of Ti atoms to the RGO VB), while RGO VB to CB excitation does not yield photocurrent.29 Therefore, overall, the visible-light photocurrent can be expected to be low.
Finally, it should be noted that the weakly bound 12H-RGO/rutile (110) (NCL) composite shows little electronic coupling between RGO and TiO2 and its graphene-like band structure suggests fast recombination between RGO-based electrons and holes. Such weakly bound composites are likely to form easily, as they do not involve large energy barriers that would be required to create interfacial cross-links. However, our calculations show that even a very small density of interfacial cross-links (as seen in the composite based on RGO with a C/O ratio of 36:1, well below the typical oxygen content in RGO) is sufficient to create the energetically separated RGO-based CB. Therefore, even if a small fraction of oxygen groups of RGO form cross-links with TiO2, this would be sufficient to produce this band, which acts as a photoelectron trap.
Conclusions
In this work, the nature of interfacial binding between graphene oxide or reduced graphene oxide and rutile (110) and the relationship between the electronic properties, photocatalytic properties, and the local oxygen defect concentration of GO/ and RGO/TiO2 composites have been investigated using DFT simulations. From the analysis of binding energies, it is clear that the formation of cross-links, such as Ti–O–C and Ti–O–H···O–C bonds, between RGO and TiO2 is a key factor in achieving strong binding in the composite. Hydrogen bonding has also been identified as an important aspect of the interfacial binding in these composites. It is found that higher concentrations of oxygen functional groups do not always promote the formation of interfacial hydrogen bonds and that, in the extreme case of very high concentrations of oxygen functional groups (such as our GO/rutile (110) system), these groups predominantly participate in noninterfacial hydrogen bonding within GO itself instead. The trends in binding energies also show that some oxygen functional groups of RGO, which do not participate in interfacial binding, such as epoxide oxygen, slightly weaken the interfacial interaction. By analyzing the electron density difference in the 12H-RGO/rutile (110) composite, it is concluded that the influence of nonbinding epoxide groups on binding energies is caused by these groups drawing electron density away from the cross-linking hydroxyl groups and their associated sp3 carbon atoms. It can be expected, based on the results of this work, that interfacial binding would be strongest in a system containing more hydroxyl groups than epoxide groups. The kinetics of formation of the interfacial cross-links is still unknown however, and further work in this area will be needed to understand how these cross-links are formed during synthesis of such composites.
The electronic structure results presented here help to explain numerous experimentally observed unusual behaviors of the RGO/TiO2 composite, such as increased UV- and visible-light photocatalytic performance, the long recorded lifetime of the excited state, and changes in measured UV- and visible-light photocurrent. Crucially, it has been demonstrated that covalent bonding between RGO and TiO2, both Ti–O–C and Ti–O–H···O–C motifs, is associated with the formation of a new unoccupied band that is predominantly localized on RGO below the conduction band of TiO2. This inverted RGO-based lowest unoccupied band is a key feature consistently observed in our strongly bound RGO/TiO2 composites. The energy profile of this band is such that it would promote the trapping of photoexcited electrons and thus hinder charge carrier recombination and extend the lifetime of the excited state. The energy of this band allows for visible-light photoexcitation of electrons to this band directly from occupied RGO bands. It is also possible that electrons in occupied RGO bands with strong oxygen character may be photoexcited to the TiO2 CB, as there is some orbital overlap between RGO oxygen and the TiO2 terminus of the interfacial cross-link, and then may decay to the lowest unoccupied band. The presence of higher unoccupied RGO bands also allows photosensitization (photoexcitation of RGO followed by transfer of photoelectrons from RGO to TiO2).
It is clear from the results in this work that oxygen functional groups have a major effect on the electronic properties of RGO and RGO-based composites. Therefore, a variety of oxygen functional groups should be included in any modeling studies of such composites, while a pure graphene-based composite is insufficient to describe the full range of possible interactions present in this composite system. Overall, our results suggest that oxygen functional groups in RGO, far from being a detrimental component, are responsible for the unusual electronic properties and for the enhancement of photocatalytic properties of this type of composite material, in particular their long electron and hole lifetimes.
This work considered only the interfaces of GO and RGO with the rutile phase of TiO2. The anatase phase is more widely used in photocatalysis experiments, and therefore, modeling of RGO/anatase composites is an important direction for further studies. The most stable (101) surface of anatase, similar to the rutile (110) surface considered here, exposes undercoordinated atoms (5-coordinated Ti and 2-coordinated O atoms)69 and therefore is likely to form cross-links with oxygen functional groups of RGO, similar to cross-links formed at our rutile-based interfaces (one example is the epoxide-linked structure reported elsewhere49). Therefore, we can expect similar RGO-based lowest unoccupied states to form in RGO/anatase composites. One difference between the two TiO2 phases is that the CBM of anatase is ∼0.2 eV lower than that in rutile;70 therefore, the gap between the RGO-based lowest unoccupied states and the TiO2-based CB is expected to be smaller, or possibly disappear, in anatase-based composites. In the latter situation, the lowest unoccupied RGO-based states would form the CBM of the composite, but they may still act as electron traps. Further calculations are needed to clarify the exact nature of the electronic structure of RGO/anatase composites.
We note that the band gaps of our calculated RGO/rutile (110) composites are very low, at ≤0.5 eV. This means that the band gap energy would fall within the infrared region of the spectrum and would thus be hard to determine using current experimental or spectroscopic techniques. The electronic structure data presented here could be complemented by calculating excitation spectra for each system, for example, using time-dependent DFT (TDDFT) methods; however, at present, such calculations have not been practically achievable because of the large size of the system.
It should also be emphasized that the RGO and GO structures proposed in this study are a sample of the wide range of possible structures that these amorphous materials could form. Further work therefore is needed to investigate the effect of other important structural features that exist in this type of composite (e.g., carbonyl groups44 and carbon vacancies in RGO71) and how domains with different arrangements and concentrations of functional groups interact with each other, for example, the interplay between cross-linked and non-cross-linked regions of the composites. Modeling a variety of RGO domains will provide more in-depth explanations of the properties of these systems, such as their photocatalytic and photocurrent performances.
Acknowledgments
P.G. would like to thank the University of Sheffield and EPSRC (UK) for his Ph.D. studentship (grant EP/K503149/1). This work used the HPC facilities centrally provided by the University of Sheffield (ShARC cluster) and the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk); the latter was accessed via membership of the UK’s HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.9b09235.
Images of the 6H- and 6C-RGO/rutile (110) composites; small-scale DoS spectra of all composites; DoS spectra of the highest occupied and lowest unoccupied levels and band structures for the 12C- and 18H-RGO/rutile (110) composites; calculated band gap energies of the systems used in this study; technical notes regarding the CP2K calculations performed in this work (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fujishima A.; Honda K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- Chang X.; Wang T.; Gong J. CO2 photo-reduction: Insights into CO2 activation and Reaction on Surfaces of Photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. 10.1039/C6EE00383D. [DOI] [Google Scholar]
- Etacheri V.; Di Valentin C.; Schneider J.; Bahnemann D.; Pillai S. C. Visible-Light Activation of TiO2 Photocatalysts: Advances in Theory and Experiments. J. Photochem. Photobiol., C 2015, 25, 1–29. 10.1016/j.jphotochemrev.2015.08.003. [DOI] [Google Scholar]
- Maeda K. Z-Scheme Water Splitting Using Two Different Semiconductor Photocatalysts. ACS Catal. 2013, 3, 1486–1503. 10.1021/cs4002089. [DOI] [Google Scholar]
- Chen X.; Shen S.; Guo L.; Mao S. S. Semiconductor-based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. 10.1021/cr1001645. [DOI] [PubMed] [Google Scholar]
- Kudo A.; Miseki Y. Heterogeneous Photocatalyst Materials for water Splitting. Chem. Soc. Rev. 2009, 38, 253–278. 10.1039/B800489G. [DOI] [PubMed] [Google Scholar]
- Gaya U. I.; Abdullah A. H. Heterogeneous Photocatalytic Degradation of Organic Contaminants over Titanium Dioxide: A Review of Fundamentals, Progress and Problems. J. Photochem. Photobiol., C 2008, 9, 1–12. 10.1016/j.jphotochemrev.2007.12.003. [DOI] [Google Scholar]
- Schneider J.; Matsuoka M.; Takeuchi M.; Zhang J.; Horiuchi Y.; Anpo M.; Bahnemann D. W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. 10.1021/cr5001892. [DOI] [PubMed] [Google Scholar]
- Dahl M.; Liu Y.; Yin Y. Composite Titanium Dioxide Nanomaterials. Chem. Rev. 2014, 114, 9853–9889. 10.1021/cr400634p. [DOI] [PubMed] [Google Scholar]
- Kumar S. G.; Devi L. G. Review on Modified TiO2 Photocatalysis under UV/Visible Light: Selected Results and Related Mechanisms on Interfacial Charge Carrier Transfer Dynamics. J. Phys. Chem. A 2011, 115, 13211–13241. 10.1021/jp204364a. [DOI] [PubMed] [Google Scholar]
- Park H.; Kim H.-I.; Moon G.-H.; Choi W. Photoinduced Charge Transfer Processes in Solar Photocatalysis Based on Modified TiO2. Energy Environ. Sci. 2016, 9, 411–433. 10.1039/C5EE02575C. [DOI] [Google Scholar]
- Cowan A. J.; Durrant J. R. Long-lived Charge Separated States in Nanostructured Semiconductor Photoelectrodes for the Production of Solar Fuels. Chem. Soc. Rev. 2013, 42, 2281–2293. 10.1039/C2CS35305A. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Yang C.-F. Recent Advances in the TiO2/CdS Nanocomposite Used for Photocatalytic Hydrogen Production and Quantum-dot-sensitized Solar Cells. Renewable Sustainable Energy Rev. 2016, 54, 1048–1059. 10.1016/j.rser.2015.10.100. [DOI] [Google Scholar]
- Woan K.; Pyrgiotakis G.; Sigmund W. Photocatalytic Carbon-Nanotube-TiO2 Composites. Adv. Mat. 2009, 21, 2233–2239. 10.1002/adma.200802738. [DOI] [Google Scholar]
- Leary R.; Westwood A. Carbonaceous Nanomaterials for the Enhancement of TiO2 Photocatalysis. Carbon 2011, 49, 741–772. 10.1016/j.carbon.2010.10.010. [DOI] [Google Scholar]
- Xiang Q.; Yu J.; Jaroniec M. Graphene-based Semiconductor Photocatalysts. Chem. Soc. Rev. 2012, 41, 782–796. 10.1039/C1CS15172J. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Yang M.-Q.; Liu S.; Sun Y.; Xu Y.-J. Waltzing with the Versatile Platform of Graphene to Synthesize Composite Photocatalysts. Chem. Rev. 2015, 115, 10307–10377. 10.1021/acs.chemrev.5b00267. [DOI] [PubMed] [Google Scholar]
- Williams G.; Seger B.; Kamat P. V. TiO2-Graphene Nanocomposites. UV-Assisted Photocatalytic Reduction of Graphene Oxide. ACS Nano 2008, 2, 1487–1491. 10.1021/nn800251f. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Lv X.; Li Y.; Wang Y.; Li J. P25-Graphene Composite as a High Performance Photocatalyst. ACS Nano 2009, 4, 380–386. 10.1021/nn901221k. [DOI] [PubMed] [Google Scholar]
- Pastrana-Martínez L. M.; Morales-Torres S.; Likodimos V.; Figueiredo J. L.; Faria J. L.; Falaras P.; Silva A. M. T. Advanced Nanostructured Photocatalysts based on Reduced Graphene Oxide-TiO2 Composites for Degradation of Diphenhydramine Pharmaceutical and Methyl Orange Dye. Appl. Catal. B 2012, 123-124, 241–256. 10.1016/j.apcatb.2012.04.045. [DOI] [Google Scholar]
- Pastrana-Martínez L. M.; Morales-Torres S.; Likodimos V.; Falaras P.; Figueiredo J. L.; Faria J. L.; Silva A. M. T. Role of Oxygen Functionalities on the Synthesis of Photocatalytically Active Graphene-TiO2 Composites. Appl. Catal. B 2014, 158-159, 329–340. 10.1016/j.apcatb.2014.04.024. [DOI] [Google Scholar]
- Sun M.; Li W.; Sun S.; He J.; Zhang Q.; Shi Y. One-step in Situ Synthesis of Graphene–TiO2 Nanorod Hybrid Composites with Enhanced Photocatalytic Activity. Mater. Res. Bull. 2015, 61, 280–286. 10.1016/j.materresbull.2014.10.040. [DOI] [Google Scholar]
- Cruz-Ortiz B. R.; Hamilton J. W.; Pablos C.; Díaz-Jiménez L.; Cortés-Hernández D. A.; Sharma P. K.; Castro-Alférez M.; Fernández-Ibañez P.; Dunlop P. S.; Byrne J. A. Mechanism of Photocatalytic Disinfection Using Titania-Graphene Composites under UV and Visible Irradiation. Chem. Eng. J. 2017, 316, 179–186. 10.1016/j.cej.2017.01.094. [DOI] [Google Scholar]
- Bell N. J.; Ng Y. H.; Du A.; Coster H.; Smith S. C.; Amal R. Understanding the Enhancement in Photoelectrochemical Properties of Photocatalytically Prepared TiO2-Reduced Graphene Oxide Composite. J. Phys. Chem. C 2011, 115, 6004–6009. 10.1021/jp1113575. [DOI] [Google Scholar]
- Morais A.; Longo C.; Araujo J. R.; Barroso M.; Durrant J. R.; Nogueira A. F.; Wang D.; Manivannan A.; Bristow A. D.; Wu N. Q. Nanocrystalline Anatase TiO2/Reduced Graphene Oxide Composite Films as Photoanodes for Photoelectrochemical Water Splitting Studies: the Role of Reduced Graphene Oxide. Phys. Chem. Chem. Phys. 2016, 18, 2608–2616. 10.1039/C5CP06707C. [DOI] [PubMed] [Google Scholar]
- Manga K. K.; Zhou Y.; Yan Y.; Loh K. P. Multilayer Hybrid Films Consisting of Alternating Graphene and Titania Nanosheets with Ultrafast Electron Transfer and Photoconversion Properties. Adv. Funct. Mater. 2009, 19, 3638–3643. 10.1002/adfm.200900891. [DOI] [Google Scholar]
- Williams K. J.; Nelson C. A.; Yan X.; Li L.-S.; Zhu X. Hot Electron Injection from Graphene Quantum Dots to TiO2. ACS Nano 2013, 7, 1388–1394. 10.1021/nn305080c. [DOI] [PubMed] [Google Scholar]
- Long R.; English N. J.; Prezhdo O. V. Photo-Induced Charge Separation across the Graphene-TiO2 Interface Is Faster than Energy Losses: A Time-Domain ab Initio Analysis. J. Am. Chem. Soc. 2012, 134, 14238–14248. 10.1021/ja3063953. [DOI] [PubMed] [Google Scholar]
- Du A.; Ng Y. H.; Bell N. J.; Zhu Z.; Amal R.; Smith S. C. Hybrid Graphene/Titania Nanocomposite: Interface Charge Transfer, Hole Doping, and Sensitization for Visible Light Response. J. Phys. Chem. Lett. 2011, 2, 894–899. 10.1021/jz2002698. [DOI] [PubMed] [Google Scholar]
- Li X.; Gao H.; Liu G. A LDA+U Study of the Hybrid Graphene/Anatase TiO2 Nanocomposites: Interfacial Properties and Visible Light Response. Comput. Theor. Chem. 2013, 1025, 30–34. 10.1016/j.comptc.2013.10.006. [DOI] [Google Scholar]
- Masuda Y.; Giorgi G.; Yamashita K. DFT Study of Anatase-Derived TiO2 nanosheets/graphene Hybrid Materials. Phys. Status Solidi B 2014, 251, 1471–1479. 10.1002/pssb.201451089. [DOI] [Google Scholar]
- Gillespie P. N. O.; Martsinovich N. Electronic Structure and Charge Transfer in the TiO2 Rutile (110)/Graphene Composite Using Hybrid DFT Calculations. J. Phys. Chem. C 2017, 121, 4158–4171. 10.1021/acs.jpcc.6b12506. [DOI] [Google Scholar]
- Dreyer D. R.; Park S.; Bielawski C. W.; Ruoff R. S. The Chemistry of Graphene Oxide. Chem. Soc. Rev. 2010, 39, 228–240. 10.1039/B917103G. [DOI] [PubMed] [Google Scholar]
- Hunt A.; Dikin D. A.; Kurmaev E. Z.; Boyko T. D.; Bazylewski P.; Chang G. S.; Moewes A. Epoxide Speciation and Functional Group Distribution in Graphene Oxide Paper-Like Materials. Adv. Funct. Mater. 2012, 22, 3950–3957. 10.1002/adfm.201200529. [DOI] [Google Scholar]
- Wu Z.-S.; Ren W.; Gao L.; Liu B.; Jiang C.; Cheng H.-M. Synthesis of High-quality Graphene with a Pre-determined Number of Layers. Carbon 2009, 47, 493–499. 10.1016/j.carbon.2008.10.031. [DOI] [Google Scholar]
- Stankovich S.; Dikin D. A.; Piner R. D.; Kohlhaas K. A.; Kleinhammes A.; Jia Y.; Wu Y.; Nguyen S. T.; Ruoff R. S. Synthesis of Graphene-based Nanosheets via Chemical Reduction of Exfoliated Graphite Oxide. Carbon 2007, 45, 1558–1565. 10.1016/j.carbon.2007.02.034. [DOI] [Google Scholar]
- Shin H.-J.; Kim K. K.; Benayad A.; Yoon S.-M.; Park H. K.; Jung I.-S.; Jin M. H.; Jeong H.-K.; Kim J. M.; Choi J.-Y.; Lee Y. H. Efficient Reduction of Graphite Oxide by Sodium Borohydride and Its Effect on Electrical Conductance. Adv. Funct. Mater. 2009, 19, 1987–1992. 10.1002/adfm.200900167. [DOI] [Google Scholar]
- Tian S.; Sun J.; Yang S.; He P.; Ding S.; Ding G.; Xie X. Facile Thermal Annealing of Graphite Oxide in Air for Graphene with a Higher C/O Ratio. RSC Adv. 2015, 5, 69854–69860. 10.1039/C5RA09388K. [DOI] [Google Scholar]
- Shin D. S.; Kim H. G.; Ahn H. S.; Jeong H. Y.; Kim Y.-J.; Odkhuu D.; Tsogbadrakh N.; Lee H.-B.-R.; Kim B. H. Distribution of Oxygen Functional Groups of Graphene Oxide Obtained from Low-temperature Atomic Layer Deposition of Titanium Oxide. RSC Adv. 2017, 7, 13979–13984. 10.1039/C7RA00114B. [DOI] [Google Scholar]
- Boukhvalov D. W.; Katsnelson M. I. Modeling of Graphite Oxide. J. Am. Chem. Soc. 2008, 130, 10697–10701. 10.1021/ja8021686. [DOI] [PubMed] [Google Scholar]
- Yan J.-A.; Xian L.; Chou M. Y. Structural and Electronic Properties of Oxidized Graphene. Phys. Rev. Lett. 2009, 103, 086802 10.1103/PhysRevLett.103.086802. [DOI] [PubMed] [Google Scholar]
- Bagri A.; Mattevi C.; Acik M.; Chabal Y. J.; Chhowalla M.; Shenoy V. B. Structural Evolution During the Reduction of Chemically Derived Graphene Oxide. Nat. Chem. 2010, 2, 581–587. 10.1038/nchem.686. [DOI] [PubMed] [Google Scholar]
- Kim S.; Zhou S.; Hu Y.; Acik M.; Chabal Y. J.; Berger C.; de Heer W.; Bongiorno A.; Riedo E. Room-temperature Metastability of Multilayer Graphene Oxide Films. Nat. Mater. 2012, 11, 544–549. 10.1038/nmat3316. [DOI] [PubMed] [Google Scholar]
- Kumar P. V.; Bernardi M.; Grossman J. C. The Impact of Functionalization on the Stability, Work Function, and Photoluminescence of Reduced Graphene Oxide. ACS Nano 2013, 7, 1638–1645. 10.1021/nn305507p. [DOI] [PubMed] [Google Scholar]
- Sun P.; Wang Y.; Liu H.; Wang K.; Wu D.; Xu Z.; Zhu H. Structure Evolution of Graphene Oxide during Thermally Driven Phase Transformation: Is the Oxygen Content Really Preserved?. PLoS One 2014, 9, e111908 10.1371/journal.pone.0111908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umrao S.; Abraham S.; Theil F.; Pandey S.; Ciobota V.; Shukla P. K.; Rupp C. J.; Chakraborty S.; Ahuja R.; Popp J.; Dietzek B.; Srivastava A. A Possible Mechanism for the Emergence of an Additional Band Gap due to a Ti-O-C Bond in the TiO2-Graphene Hybrid System for Enhanced Photodegradation of Methylene Blue under Visible Light. RSC Adv. 2014, 4, 59890–59901. 10.1039/C4RA10572A. [DOI] [Google Scholar]
- Huang Q.; Tian S.; Zeng D.; Wang X.; Song W.; Li Y.; Xiao W.; Xie C. Enhanced Photocatalytic Activity of Chemically Bonded TiO2/Graphene Composites Based on the Effective Interfacial Charge Transfer through the C–Ti Bond. ACS Catal. 2013, 3, 1477–1485. 10.1021/cs400080w. [DOI] [Google Scholar]
- Rakkesh R. A.; Durgalakshmi D.; Balakumar S. Effcient Sunlight-driven Photocatalytic Activity of Chemically Bonded GNS–TiO2 and GNS–ZnO Heterostructures. J. Mater. Chem. C 2014, 2, 6827–6834. 10.1039/C4TC01195C. [DOI] [Google Scholar]
- Ferrighi L.; Fazio G.; Di Valentin C. Charge Carriers Separation at the Graphene/(101) Anatase TiO2 Interface. Adv. Mater. Interfaces 2016, 3, 1500624. 10.1002/admi.201500624. [DOI] [Google Scholar]
- Long R.; Casanova D.; Fang W.-H.; Prezhdo O. V. Donor–Acceptor Interaction Determines the Mechanism of Photoinduced Electron Injection from Graphene Quantum Dots into TiO2: π-Stacking Supersedes Covalent Bonding. J. Am. Chem. Soc. 2017, 139, 2619–2629. 10.1021/jacs.6b09598. [DOI] [PubMed] [Google Scholar]
- Bukowski B.; Deskins N. A. The Interactions Between TiO2 and Graphene with Surface Inhomogeneity Determined using Density Functional Theory. Phys. Chem. Chem. Phys. 2015, 17, 29734–29746. 10.1039/C5CP04073F. [DOI] [PubMed] [Google Scholar]
- Aryasetiawan F.; Gunnarsson O. The GW Method. Rep. Prog. Phys. 1998, 61, 237–312. 10.1088/0034-4885/61/3/002. [DOI] [Google Scholar]
- Finazzi E.; Di Valentin C.; Pacchioni G.; Selloni A. Excess Electron States in Reduced Bulk Anatase TiO2: Comparison of Standard GGA, GGA+U, and Hybrid DFT Calculations. J. Chem. Phys. 2008, 129, 154113. 10.1063/1.2996362. [DOI] [PubMed] [Google Scholar]
- Landmann M.; Rauls E.; Schmidt W. G. The Electronic Structure and Optical Response of Rutile, Anatase and Brookite TiO2. J. Phys.: Condens. Matter 2012, 24, 195503. 10.1088/0953-8984/24/19/195503. [DOI] [PubMed] [Google Scholar]
- Vandevondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. 10.1016/j.cpc.2004.12.014. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Heyd J.; Scuseria G. E.; Ernzerhof M. Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 2006, 124, 219906. 10.1063/1.2204597. [DOI] [Google Scholar]
- Grimme S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Hutter J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. 10.1063/1.2770708. [DOI] [PubMed] [Google Scholar]
- Goedecker S.; Teter M.; Hutter J. Separable Dual-space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. 10.1103/PhysRevB.54.1703. [DOI] [PubMed] [Google Scholar]
- Guidon M.; Hutter J.; Vandevondele J. Auxiliary Density Matrix Methods for Hartree-Fock Exchange Calculations. J. Chem. Theory Comput. 2010, 6, 2348–2364. 10.1021/ct1002225. [DOI] [PubMed] [Google Scholar]
- Van Duijneveldt F. B.; van Duijneveldt-van de Rijdt J. G.; van Lenthe J. H. State of the Art in Counterpoise Theory. Chem. Rev. 1994, 94, 1873–1885. 10.1021/cr00031a007. [DOI] [Google Scholar]
- Dovesi R.; Orlando R.; Erba A.; Zicovich-Wilson C. M.; Civalleri B.; Casassa S.; Maschio L.; Ferrabone M.; De La Pierre M.; D’Arco P.; Noël Y.; Causà M.; Rérat M.; Kirtman B. CRYSTAL14: A Program for the Ab Initio Investigation of Crystalline Solids. Int. J. Quantum Chem. 2014, 114, 1287–1317. 10.1002/qua.24658. [DOI] [Google Scholar]
- Dovesi R.; Erba A.; Orlando R.; Zicovich-Wilson C. M.; Civalleri B.; Maschio L.; Rérat M.; Casassa S.; Baima J.; Salustro S.; Kirtman B. Quantum-mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1360 10.1002/wcms.1360. [DOI] [Google Scholar]
- Peintinger M. F.; Oliveira D. V.; Bredow T. Consistent Gaussian Basis Sets of Triple-Zeta Valence with Polarization Quality for Solid-State Calculations. J. Comput. Chem. 2013, 34, 451–459. 10.1002/jcc.23153. [DOI] [PubMed] [Google Scholar]
- Luo Y. R.Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Chen C.; Cai W.; Long M.; Zhou B.; Wu Y.; Wu D.; Feng Y. Synthesis of Visible-Light Responsive Graphene Oxide/TiO2 Composites with p/n Heterojunction. ACS Nano 2010, 4, 6425–6432. 10.1021/nn102130m. [DOI] [PubMed] [Google Scholar]
- Martsinovich N.; Jones D. R.; Troisi A. Electronic Structure of TiO2 Surfaces and Effect of Molecular Adsorbates using Different DFT Implementations. J. Phys. Chem. C 2010, 114, 22659–22670. 10.1021/jp109756g. [DOI] [Google Scholar]
- Scanlon D. O.; Dunnill C. W.; Buckeridge J.; Shevlin S. A.; Logsdail A. J.; Woodley S. M.; Catlow C. R. A.; Powell M. J.; Palgrave R. G.; Parkin I. P.; Watson G. W.; Keal T. W.; Sherwood P.; Walsh A.; Sokol A. A. Band Alignment of Rutile and Anatase TiO2. Nat. Mater. 2013, 12, 798–801. 10.1038/nmat3697. [DOI] [PubMed] [Google Scholar]
- Datteo M.; Liu H.; Di Valentin C. Water on Graphene-Coated TiO2: Role of Atomic Vacancies. ACS Appl. Mater. Interfaces 2018, 10, 5793–5804. 10.1021/acsami.7b18087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.