

Summary
Following T‐cell antigen receptor (TCR) engagement, rearrangement of the actin cytoskeleton supports intracellular signal transduction and T‐cell activation. The non‐catalytic region of the tyrosine kinase (Nck) molecule is an adapter protein implicated in TCR‐induced actin polymerization. Further, Nck is recruited to the CD3ε subunit of the TCR upon TCR triggering. Here we examine the role of actin polymerization in the recruitment of Nck to the TCR. To this end, Nck binding to CD3ε was quantified in Jurkat cells using the proximity ligation assay. We show that inhibition of actin polymerization using cytochalasin D delayed the recruitment of Nck1 to the TCR upon TCR triggering. Interestingly, CD3ε phosphorylation was also delayed. These findings suggest that actin polymerization promotes the recruitment of Nck to the TCR, enhancing downstream signaling, such as phosphorylation of CD3ε.
Keywords: actin polymerization, CD3ε, cytochalasin D, Nck, T‐cell activation
Actin polymerization promotes the recruitment of Nck to the TCR, enhancing downstream signaling, such as phosphorylation of CD3ε.
Abbreviations
- CD3ε
cluster of differentiation 3ε
- CREB
cyclic AMP‐response element binding protein
- CytD
cytochalasin D
- Erk
extracellular signal‐regulated kinase
- F‐actin
filamentous actin
- LAT
linker of activated T cells
- Lck
lymphocyte‐specific protein tyrosine kinase
- MHC
major histocompatibility complex
- Nck
non‐catalytic region of tyrosine kinase
- PLA
proximity ligation assay
- PRS
proline‐rich sequence
- SH domain
Src homology domain
- SLP‐76
SH domain containing leukocyte protein of 76 000 MW
- TCR
T‐cell receptor
- WAsP
Wiskott‐–Aldrich syndrome protein
- ZAP70
ζ chain‐associated protein kinase of 70 000 MW
Introduction
T cells use their T‐cell receptor (TCR) to interact with antigenic peptides presented by major histocompatibility complex (MHC) molecules on the antigen‐presenting cells, in order to carry out their effector functions. This TCR–peptide–MHC binding results in intracellular signaling that involves a dynamic rearrangement of the actin cytoskeleton. The latter event is important for T‐cell cytokine production, proliferation, survival, metabolism and differentiation.1
Filamentous actin (F‐actin) is enriched at the interface of the conjugation between the T‐cell and antigen‐presenting cells, the so‐called immunological synapse. Prevention of actin reorganization by using actin polymerization inhibitors such as cytochalasin D (CytD) can result in impaired synapse formation, intracellular signaling and so T‐cell activation.2, 3
Understanding of the molecular mechanisms that regulate actin rearrangement has expanded in the past years. Various signaling molecules have been described that are involved in intracellular signaling from the TCR to actin rearrangement. Engagement of the TCR by antigen‐ or anti‐TCR/CD3 antibodies stabilizes the TCR in an active CD3 conformation that allows the lymphocyte‐specific protein tyrosine kinase (Lck) to access the tyrosines in the cytoplasmic tails of the signaling subunits of the TCR, namely CD3ζ, CD3ε, CD3δ and CD3γ.4, 5 This results in the phosphorylation of CD36 and of multiple effector molecules, including the ζ chain‐associated protein kinase of 70 000 MW (ZAP70), the linker of activated T cells (LAT) and the Src homology (SH)‐2 domain containing leukocyte protein of 76 000 MW (SLP‐76).7, 8, 9 An adaptor protein called the non‐catalytic region of tyrosine kinase (Nck) and its associated molecule the Wiskott–Aldrich syndrome protein (WASp) are recruited to phosphorylated SLP‐76 in the vicinity of the activated TCR.10, 11 Here, WASp is activated by the Rho family GTPase Cdc42 and subsequently leads to the initiation of actin filament formation.10 Previous studies have confirmed the involvement of Nck recruitment associated with LAT, SLP‐76 and WASp in the process of actin polymerization induced by TCR engagement.12, 13
Nck is ubiquitously expressed and integrates signals from transmembrane receptors to downstream effectors to regulate actin cytoskeletal rearrangement in most, if not all, cell types.14 In human and mice, two Nck isoforms exist (Nck1/Nckα and Nck2/Nckβ).15, 16 Both isoforms consist of three SH3 domains (SH3·1, SH3·2 and SH3·3) and one SH2 domain, and so potentially interact with proline‐rich sequence (PRS)‐bearing proteins and tyrosine phosphorylated proteins, respectively.17, 18, 19 In T cells, Nck uses its SH3·2 and SH3·3 domains to bind to WASp,18 which subsequently controls actin polymerization.20 Nck also functions as a linker for the recruitment and activation of other proteins in multiple intracellular signaling pathways.17 In addition, upon TCR triggering, Nck is recruited directly to the CD3ε subunit of the TCR.21, 22 In fact, in TCRs in the active conformation the CD3ε’s PRS is exposed and binds to the SH3·1 domain of Nck.4, 23 However, this is not sufficient to stably recruit Nck to the TCR. In addition, the Nck SH2 domain needs to bind to the phosphorylated first tyrosine of the immunoreceptor‐tyrosine based activation motifs of CD3ε.22 Interestingly, Nck bound to the TCR might bind simultaneously to WASp and thereby recruit WASp to the TCR,13 regulating actin polymerization. Indeed, inhibiting the Nck–TCR interaction reduced actin polymerization,24 showing that the Nck pool at the TCR significantly contributes to actin rearrangements. However, the opposite scenario, i.e. whether actin polymerization affects Nck recruitment to the TCR, has not been investigated so far.
Recently, we have demonstrated that Nck1, rather than Nck2, plays a major role in TCR signaling in human T cells.25 Hence, in this work we focus on Nck1, examining the involvement of actin polymerization in Nck recruitment to the TCR. We show that inhibition of actin polymerization is associated with a delayed recruitment of Nck1 to the TCR upon TCR triggering. This might be explained by delayed phosphorylation of CD3ε.
Materials and methods
Antibodies and reagents
In this present study, the following antibodies were used: rabbit anti‐Nck1, rabbit anti‐ZAP70, rabbit anti‐phospho‐ZAP70 (Y319) and rabbit anti‐GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA), the anti‐idiotypic TCR antibody (C305) was from Millipore (Merk KGaA, Darmstadt, Germany), mouse anti‐CD3 (OKT‐3) antibody was from eBioscience (San Diego, CA), and goat anti‐CD3ε M20 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit anti‐CD3ζ antiserum 448 and the rabbit anti‐phospho‐CD3ε antiserum (anti‐phospho‐εY1) have been described previously.26, 27 The inhibitor of actin polymerization CytD was purchased from Sigma‐Aldrich (St. Louis, MO) and the rhodamine‐coupled phalloidin for F‐actin staining was purchased from Invitrogen (Waltham, MA).
Generation of Nck1‐deficient Jurkat T cells
The Jurkat variant N1KO (deficient in Nck1) was generated by transiently expressing Cas9, a guide sgRNA against the Nck1 region. The Nck1 sgRNA oligonucleotide sequence used was 5′‐GTCGTCAATAACCTAAATAC‐3′. These guide sgRNAs were cloned into the GeneArt® CRISPR Nuclease (OPF) vector (Invitrogen, #A21174) that was then electroporated into Jurkat T cells using Amaxa Nucleofector II using the manufacturer's protocol X‐005. The stable Nck1‐deficient single cells were obtained from these lines by limiting dilution. Successful positive knockout individual clones were selected by fluorescence analysis and screening with immunoblot probed for anti‐Nck1 antibody. The selected clones had 100% down‐regulation of endogenous Nck1 and TCR expression was evaluated by flow cytometry. Clones with similar expression levels of TCR were selected for further studies. Here, we chose one clone each that was named N1KO.
T‐cell culture and stimulation
The human Jurkat T‐cell line (E6‐1 clone) obtained from the American Type Culture Collection (Rockville, MD) and the N1KO Jurkat cells produced in our laboratory were used for studying TCR signaling and T‐cell activation. Both cell types were cultured in RPMI‐1640 medium (Gibco Thermo Fisher Scientific, Waltham, MA) supplemented with 10% heat‐inactivated fetal bovine serum (Gibco), 2 mm l‐glutamine and 100 U/ml penicillin G and 100 µg/ml streptomycin (HyClone, Fisher Scientific, Loughborough, UK) in a humidified incubator with 5% CO2 at 37°. The cells were harvested and starved in RPMI‐1640 medium without fetal bovine serum for 1 hr at 37°. Subsequently they were either stimulated with the C305 antibody diluted 1 : 50 for the indicated time‐points or left untreated as a control. This antibody dilution corresponded to ∼0·12 mg/ml of C305.
Inhibitor treatment and fluorescence microscopic visualization
To determine the optimal inhibitory concentration to inhibit actin polymerization, Jurkat cells were treated with CytD for 30 min at concentrations of 5 and 10 µm at 37° before cell stimulation. The investigation of F‐actin formation inhibition was performed as previously known for application in fixed cells.28 Briefly, chambered coverglasses (LabTek; Thermo Scientific) pretreated with 0·01% weight/volume poly‐l‐lysine solution (Merk KGaA) were coated with anti‐CD3 antibody (1 μg/ml) and incubated at 4° overnight. After washing with phosphate‐buffered saline (PBS), 2 × 105 cells in 200 µl medium containing 10% fetal calf serum and 10 mm HEPES were seeded into the chambers and incubated for 5 min. Cells were then fixed with 4% paraformaldehyde and permeabilized with 0·1% Triton X‐100. After washing, the samples were blocked for 1 hr with 10% fetal calf serum and 0·02% sodium azide in PBS. Subsequently, the chambers were incubated with rhodamine‐phalloidin staining (diluted in PBS at 1 : 200) and they were observed for red fluorescence signals under a fluorescence microscope (Nikon Eclipse Ti, Nikon®, Melville, NY) using NIS‐Elements D software with 2560 × 1920 record pixels. Three to five high‐power fields in each slide were always included for visualization to avoid slide‐to‐slide variation.
Multiplex TCR signaling assay
Ten million Jurkat T cells were pretreated with 5 µm CytD for 30 min at 37° before being stimulated with C305 antibody (1 : 50) at 37° for the indicated time‐points (30 seconds, 1, 5, 10 and 30 min) or left untreated as a control. After stimulation, cell pellets were lysed in 1 ml of MILLIPLEX® MAP Lysis Buffer (Merck KGaA) containing freshly prepared protease inhibitors. The working concentration of protein for the assay was 7·5 µg of total protein/well (25 µl/well at 300 µg/ml). Subsequently, the levels of phosphorylation of each protein were determined using a T‐Cell Receptor Magnetic Bead Kit 96‐well Plate Assay, Milliplex Map Kit (Merck KGaA), following the manufacturer's instructions and the mean fluorescence intensity was measured with the MAGPIX, Luminex® system (Austin, TX).
In situ proximity ligation assay
Jurkat T cells were grown on diagnostic microscopic slides (ThermoScientific). Cells were pretreated with 5 µm CytD for 30 min at 37°. Then, cells were stimulated with the anti‐TCR antibody C305 (1 : 50) at 37° for 5 or 10 min, or left untreated as a control. Subsequently, cells were fixed with paraformaldehyde, permeabilized with 0·5% saponin, and blocked with blocking solution. Then, cells were co‐incubated with goat anti‐CD3ε M20 antibody (Santa Cruz Biotechnology) and rabbit anti‐Nck1 antibody (Cell Signaling Technology). A proximity ligation assay (PLA) between the CD3ε and Nck1 molecules was performed using the Duolink kit (Olink Bioscience, Uppsala, Sweden) according to the manufacturer's instructions. The PLA signals appeared as red fluorescence dots. Cell nuclei were stained with DAPI. A fluorescence microscope (Nikon Eclipse Ti) was used for imaging and analysis. The number of the PLA signal dots was scored with the Blob‐Finder program (Uppsala University).
Immunoprecipitation and Western blotting
Jurkat T cells that were either pretreated or not with CytD for 30 min were stimulated with anti‐TCR antibody (C305, 1 : 50) at 37° for 3, 10 and 30 min or left unstimulated. Cells were then lysed in 100 μl lysis buffer (20 mm Tris–HCl pH 8·0, 137 mm NaCl, 2 mm EDTA, 10% glycerol, protease inhibitor mixture (Sigma‐Aldrich), 1 mm phenylmethylsulfonyl fluoride, 5 mm iodoacetamide, 0·5 mm sodium orthovanadate, 1 mm NaF, and 0·3% Brij96V) for 30 min on ice. TCRs from the total cellular lysates were then immunoprecipitated with 1 mg anti‐CD3ε antibody (OKT3) ‐coupled protein‐G Sepharose beads (GE Healthcare Life Sciences, Uppsala, Sweden). The sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting were performed with the antibodies indicated, and visualization was carried out using a CCD camera (ImageQuant LAS4000; GE Healthcare Life Sciences, Pittsburgh, PA). Band intensity was assessed by The ImageJ software (Rasband, W.S., U. S. National Institutes of Health, Bethesda, MA).
Statistical analysis
Data were analyzed using SPSS software and presented as means ± SEM. Differences between two experimental groups were analyzed with Student's t‐test. Significant differences were considered when P‐values were < 0·05.
Results
Analysis of F‐actin distribution in Jurkat T cells treated with CytD
To study the function of actin polymerization in the recruitment of Nck1 to the TCR, CytD was used to disturb actin polymerization. To confirm that the CytD used was having the expected inhibitory effect on actin polymerization, Jurkat T cells were treated with 5 and 10 µm CytD for 30 min before TCR stimulation or not. Subsequently, the TCR was stimulated with the anti‐idiotypic monoclonal antibody C305, which binds to the variable regions of the endogenous TCR expressed by Jurkat cells.29 Then the amount of polymerized actin (F‐actin) was probed with rhodamine‐conjugated phalloidin and was assessed by fluorescence microscopy (Fig. 1a,b). As anticipated, TCR stimulation led to actin polymerization showing an apical ring of F‐actin. CytD treatment prevented actin polymerization compared with the staining pattern observed in C305‐stimulated cells without CytD. The result suggested that 5 µm of CytD was sufficient to inhibit actin polymerization under our experimental system and this concentration was used in all following experiments.
Figure 1.
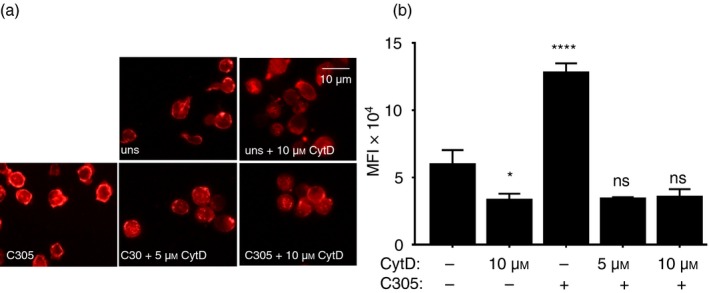
Inhibition of actin polymerization by cytochalasin D (CytD). Jurkat T cells were treated without or with 5 or 10 µm of CytD for 30 min at 37°. Subsequently, the cells were stimulated for 5 min at 37° with the anti‐T‐cell receptor (TCR) antibody C305 (1 : 50) or left untreated (uns) (a). Cells were stained with rhodamine‐coupled phalloidin to visualize F‐actin formation using fluorescence microscopy. Original magnification is 600×. The fluorescence intensities of cells were quantified by imagej and the mean fluorescence intensity is displayed. Significant differences were determined by unpaired, two‐tailed Student's t‐test. Results were the average of two independent experiments (ns, non‐significant, *P < 0·05, ****P < 0·0001) (b).
Requirement of actin polymerization for an efficient TCR‐induced Nck1‐CD3ε interaction
A possible involvement of actin polymerization in the recruitment of Nck to CD3ε has not been studied so far. To determine this, we used the in situ PLA, which is a technique that allows visualization of the close proximity between endogenous proteins in fixed cells by red fluorescent dot detection.30 Recently, we established the PLA to quantify the proximity of Nck with the cytoplasmic tail of CD3ε in T cells using anti‐Nck1 and anti‐CD3ε antibodies22 (Fig. 2a). Here, we left Jurkat T cells untreated as a control or treated them with CytD to prevent actin polymerization. Subsequently, the cells were stimulated with the anti‐TCR antibody C305 for 5 and 10 min at 37° or left unstimulated. PLA was performed with anti‐Nck1 and anti‐CD3ε antibodies and analyzed by fluorescence microscopy. The red fluorescent dots indicative of close proximity between Nck and TCR were counted (Fig. 2b). Without CytD treatment and as reported before,22 we detected an increase in the number of red dots when the TCR was stimulated for 5 min compared with unstimulated cells, showing that endogenous Nck was recruited to CD3ε upon TCR stimulation. At 10 min of stimulation, Nck recruitment to the TCR ceased, demonstrating that this is a very transient event following TCR triggering (Fig. 2c).
Figure 2.
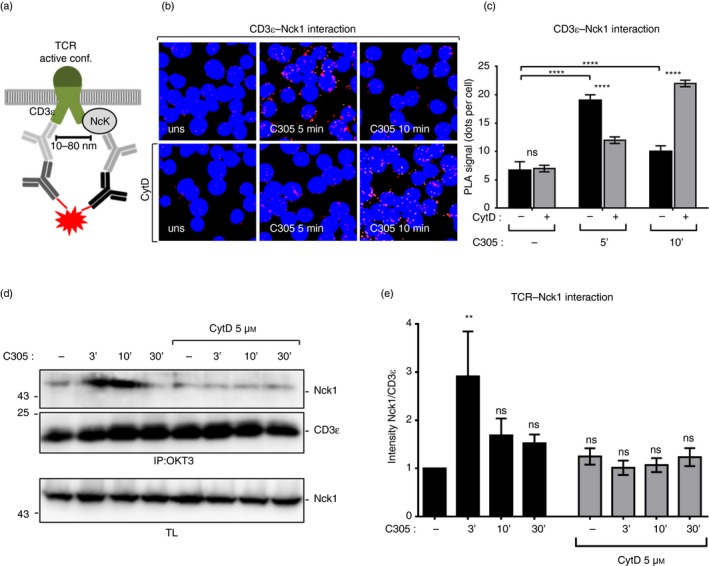
Involvement of actin polymerization in tyrosine phosphorylation of CD3ε and the recruitment of non‐catalytic region of tyrosine kinase 1 (Nck1) to the T‐cell receptor (TCR). Schematic of the in situ proximity ligation assay (PLA) using anti‐CD3ε and anti‐Nck1 antibodies (a). The proximity between Nck1 and the TCR was detected by in situ PLA in intact fixed Jurkat cells. Jurkat T cells were either left untreated or treated with 5 µm cytochalasin D (CytD) for 30 min at 37°. Subsequently, cells were stimulated with the anti‐TCR antibody C305 (1 : 50) for 5 and 10 min or left unstimulated. After fixation and permeabilization, PLA was performed using goat anti‐CD3ε (M20ε) and rabbit anti‐Nck1 primary antibodies, followed by secondary antibodies. Nuclei were stained with DAPI. Cells were imaged using a Nikon Eclipse Ti‐U fluorescence microscope. Original magnification is 600× (b). The corresponding quantification of the red PLA dots from (b) and the mean ± SEM is displayed; statistical analysis was done by two‐tailed Student's t‐test. For each condition an average of 500 cells were analyzed. Three independent experiments were performed (c). Jurkat cells were pretreated with 5 µm CytD for 30 min at 37° and stimulated as in (b). (d) Cell lysates were subjected to immunoprecipitation with the anti‐CD3 antibody (OKT3). After sodium dodecyl sulfate–polyacrylamide gel electrophoresis, the Western blot was developed with anti‐Nck1 and anti‐CD3ε antibody. The corresponding lysates were developed with anti‐Nck1 antibody. Data are representative of four independent experiments. The intensity of the Nck1 and CD3ε bands in the immunoprecipitation was quantified using imagej software and is presented as a ratio of Nck1 to CD3ε normalized to the unstimulated/untreated cells. The data represent the mean ± SEM (ns, non‐significant, **P < 0·01, ****P < 0·0001) (e).
In the presence of CytD, the anti‐TCR‐induced proximity between Nck and CD3ε was delayed, so that Nck recruitment was hardly detectable after 5 min, but prominent after 10 min of stimulation (Fig. 2b,c). Hence, actin cytoskeletal rearrangement is necessary for a fast recruitment of Nck1 to the TCR, including a fast shut‐off of the signal. To test whether the induced Nck–TCR proximity was caused by Nck binding to the TCR, Jurkat cells were stimulated under the same conditions as in Fig. 2(b) and subjected to immunoprecipitation with anti‐CD3 antibody. Consistent with the data from the PLA, Nck binding to the TCR was increased upon TCR triggering (Fig. 2d). Importantly, lower amounts of Nck were co‐immunoprecipitated with the TCR from anti‐TCR C305 antibody‐stimulated and CytD‐pretreated cells compared with C305‐stimulated cells alone (Fig. 2d,e). Collectively, these data indicate that actin polymerization is required for an efficient recruitment of Nck to the TCR.
Antibody‐mediated CD3ε phosphorylation was delayed under CytD treatment
Nck recruitment to the TCR requires both an interaction of Nck with the exposed PRS of CD3ε and with a phosphorylated tyrosine in CD3ε.22 Since the exposure of the CD3ε PRS occurs concomitantly with ligand‐binding to the TCR,4, 23 we hypothesized that CD3ε phosphorylation might decrease under CytD treatment. To explore this possibility, we tested for CD3ε phosphorylation using C305 as a TCR stimulus and immunoprecipitation of the TCR (Fig. 3a). In the absence of CytD, CD3ε phosphorylation was maximal at 3 and 10 min of stimulation and decreased at 30 min. In contrast, in the presence of CytD, there were lower amounts of tyrosine phosphorylation at CD3ε at all stimulation time‐points (Fig. 3a). Hence, CytD prevented tyrosine phosphorylation of CD3ε after TCR antibody‐mediated engagement.
Figure 3.
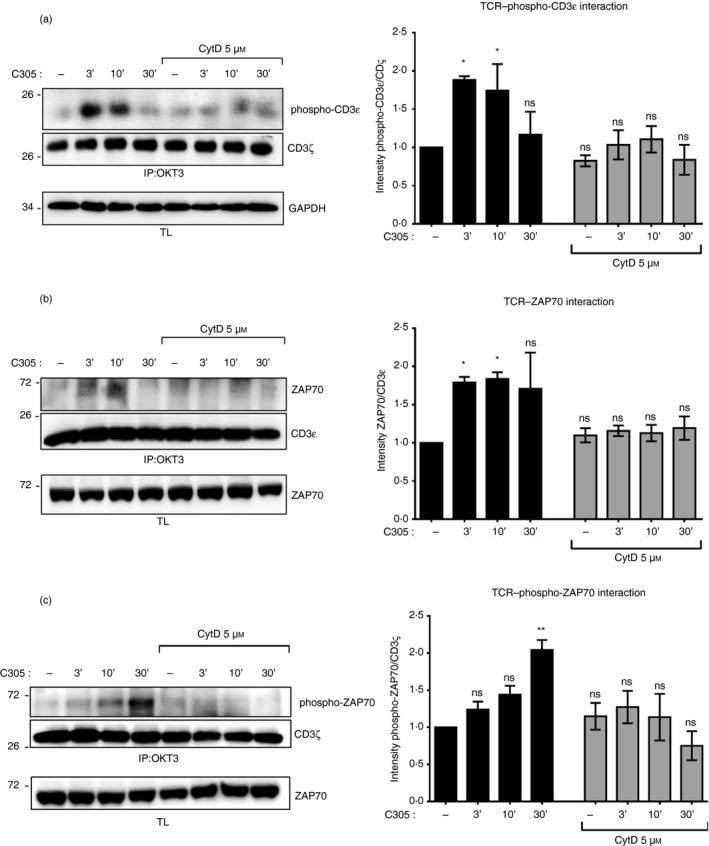
Inhibition of the actin polymerization impairs tyrosine phosphorylation at CD3ε and recruitment of ζ chain‐associated protein kinase of 70 000 MW (ZAP70) to the T‐cell receptor (TCR). Jurkat cells were left untreated or pretreated with 5 µm cytochalasin D (CytD) for 30 min at 37° before stimulation with the anti‐TCR antibody C305 (1 : 50) at 37° for the indicated time‐points or left unstimulated (uns) as in Fig. 2(d). After stimulation, cell lysates were subjected to immunoprecipitation with the anti‐CD3 antibody OKT3. After sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) the Western blot was developed with anti‐phospho‐CD3ε and anti‐CD3ζ antibodies. The lysates were developed with anti‐GAPDH antibody (a). Jurkat cells were stimulated as in (a). Cell lysates were subjected to immunoprecipitation with the anti‐CD3 antibody OKT3. After SDS–PAGE the Western blot was detected with anti‐ZAP70 and anti‐CD3ɛ antibodies (b), and anti‐phospho‐ZAP70 and anti‐CD3ζ antibodies (c). The corresponding lysates were developed with an anti‐ZAP70 antibody. The intensity of the phospho‐CD3ε and CD3ζ bands (a), the ZAP70 and CD3ɛ bands (b), and the phospho‐ZAP70 and CD3ζ bands (c) in the immunoprecipitation was quantified using imagej software and is presented as a ratio of phospho‐CD3ε to CD3ζ (a), of ZAP70 to CD3ɛ (b), and of phospho‐ZAP70 to CD3ζ (c) normalized to the unstimulated/untreated cells. Data are representative of four experiments, and the statistical analysis was performed as in Fig. 2(e) (ns, non‐significant, *P < 0·1, **P < 0·01).
To substantiate this finding, CD3ε tyrosine phosphorylation in total cell lysates was measured using the Luminex system and Western blotting. To this end, Jurkat T cells were stimulated with the antibody C305 for the indicated time‐points in the absence or presence of CytD and the levels of tyrosine phosphorylated CD3ε were quantified (Fig. 4a,b, respectively). Consistent with the immunoprecipitation data, the phosphorylation of CD3ε was a transient event, peaking at around 5 min. In sharp contrast, CytD treatment resulted in a delay of phosphorylation of CD3ε with the peak occurring at 10 min (Fig. 4c). Together this suggested that inhibition of actin polymerization was associated with a delayed and reduced tyrosine phosphorylation at CD3ε upon TCR triggering.
Figure 4.
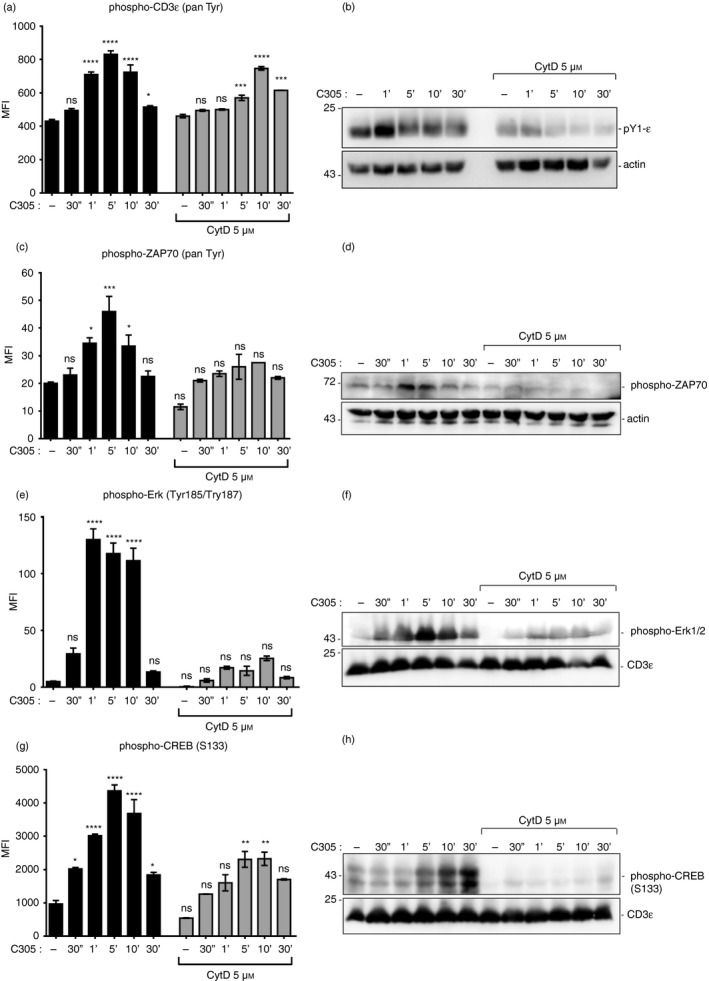
Down‐regulation of the proximal and downstream signaling proteins upon disturbance of polymerized actin. Phosphorylation of CD3ɛ (pan‐phospho‐Tyr CD3ε) (a), ζ chain‐associated protein kinase of 70 000 MW (ZAP70; pan‐phospho‐Tyr ZAP70) (c), Erk (phospho‐Tyr185/Tyr187) (e) and CREB (phospho‐Ser133) (g) was measured with the Luminex system. Jurkat T cells that were either left untreated or pretreated with 5 µm cytochalasin D (CytD) for 30 min at 37° before stimulation with anti‐T‐cell receptor (TCR) antibody C305 (1 : 50) at 37° for the indicated time‐points or left unstimulated. Cells were then lysed and the mean fluorescence intensity (MFI) was measured using the MAGPIX, Luminex® system. The graph represents the MFI of duplicate wells from two independent experiments. Jurkat cells were stimulated and lysed as in the Luminex experiments (a,c,e,g). Total cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot. Membranes were detected with anti‐phospho‐CD3ɛ (b), anti‐phospho‐ZAP70 (d), anti‐phospho Erk1/2 (f) and anti‐phospho‐CREB (h) antibodies. The corresponding lysates were detected with an anti‐actin or anti‐CD3ε antibody. Data are representative of three independent experiments. Significant differences were determined by ordinary one‐way analysis of variance (ns, non‐significant, *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001).
Actin polymerization is required for the stimulation‐induced interaction between ZAP70 and the TCR
To test whether the interaction between TCR and ZAP70 was altered by inhibiting actin polymerization, we performed a time–course experiment of ZAP70 co‐immunoprecipitation with the TCR using the anti‐TCR C305 antibody as a stimulus in the presence or absence of CytD (Fig. 3b). Binding of ZAP70 to TCR was decreased upon TCR triggering in the presence of CytD compared with C305‐stimulated cells alone (Fig. 3b). This was correlated with a decrease of phosphorylated ZAP70 which was co‐immunoprecipitated with TCR in the presence of CytD (Fig. 3c). These data indicate that actin polymerization is required for an efficient recruitment of ZAP70 to the antibody‐stimulated TCR.
Defective actin filament formation correlates with impaired TCR‐induced phosphorylation of intracellular signaling proteins
Following the proximal TCR phosphorylation data of Fig. 3, we hypothesized that upon TCR triggering, actin polymerization would also be involved in more downstream signaling. To investigate this, Jurkat T cells were stimulated with the anti‐TCR antibody C305 at 37° for the indicated time‐points in the absence or presence of CytD. Then, the phosphorylation of ZAP70, Extracellular‐signal regulated kinase (Erk) and the transcription factor cyclic AMP‐response element binding protein (CREB) in total cell lysates was measured using the Luminex system (Fig. 4c,e,g, respectively) and Western blotting (Fig. 4d,f,h, respectively). As predicted, the impaired actin polymerization was associated with decreased and delayed phospho‐ZAP70, decreased phospho‐Erk, and decreased phospho‐CREB. Therefore, actin polymerization promoted the phosphorylation of ZAP70, Erk and CREB upon TCR triggering.
Phosphorylation of CD3ε depends on the presence of Nck
Next, we tested whether CD3ε phosphorylation was caused by the recruitment of Nck to the TCR. To this end, we stimulated Nck1‐CRISPR/Cas9 knockout (N1KO) and wild‐type Jurkat cells with the anti‐TCR antibody C305 or left cells unstimulated. All cells expressed similar amounts of TCR on their cell surface, as shown by flow cytometry (Fig. 5a). We show that an efficient phosphorylation of CD3ε required the presence of Nck1, as this phosphorylation was reduced in N1KO cells after TCR stimulation compared with the control Jurkat cells containing Nck1. These data indicate that Nck recruitment to the TCR is required for an efficient phosphorylation of CD3ε (Fig. 5b). Therefore, this finding places the recruitment of Nck to the TCR upstream of CD3ε phosphorylation.
Figure 5.
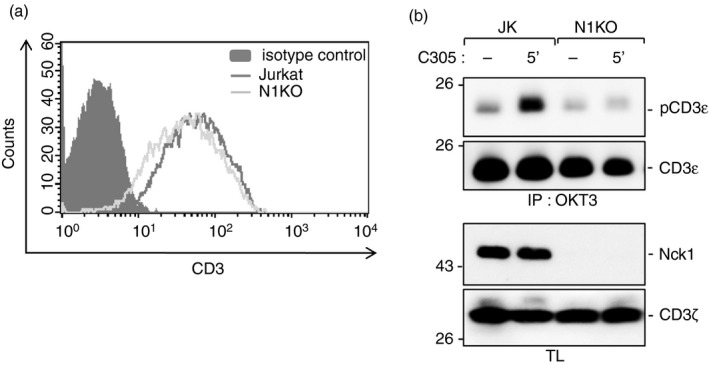
Non‐catalytic region of tyrosine kinase 1 (Nck1) is required for the phosphorylation of CD3ε. Jurkat wild‐type and Nck1‐CRISPR/Cas9 knockout (N1KO) Jurkat cells were stained with allophycocyanin‐conjugated anti‐CD3ε antibody UCHT1. The expression levels of surface CD3 molecule of the cell clones were evaluated by flow cytometry. The clones with similar expression of T‐cell receptor (TCR) were selected for further studies (a). The cells were either stimulated with C305 or left unstimulated for 5 min at 37°. Cell lysates were collected and TCRs were immunoprecipitated with the anti‐CD3 antibody OKT3. After sodium dodecyl sulfate–polyacrylamide gel electrophoresis, the Western blot was developed with anti‐phospho‐CD3εY1 (pCD3ε) and anti‐CD3ε antibodies. The corresponding lysates were developed with anti‐Nck1 and anti‐ζ antibodies (b).
Discussion
In this study, we describe the role of actin polymerization in proximal TCR signaling involving the recruitment of the adaptor protein Nck to the TCR upon TCR triggering. Our data show that disruption of actin polymerization resulted in a delayed recruitment of Nck1 to the TCR. This was correlated with a delay in tyrosine phosphorylation of the CD3ε. In the present finding, Nck was recruited as early as 5 min after TCR ligation. A previous work has demonstrated that a mechanotransduction caused by TCR engagement at the immune synapse leads to actin polymerization.31 Hence, together with our findings, Nck is required for CD3ε phosphorylation by Lck, in which F‐actin acts to link Nck and Lck to initiate this phosphorylation. Nck and F‐actin, therefore, could function to promote each other at proximal TCR signaling.
Inhibition of actin polymerization might delay CD3ε phosphorylation by the following mechanisms. First, on TCR triggering, Nck interacts with a proline motif of the unique domain of the lymphocyte‐specific protein tyrosine kinase Lck,32 the interaction being enhanced by a specific T‐cell adaptor protein TSAd.33 Therefore, it could be that disruption of actin polymerization impairs recruitment of Nck‐TSAd‐Lck to the TCR, leading to delayed CD3 phosphorylation. Second, proximal TCR signaling involves very early and simultaneous binding between Nck, CD3ε, and Lck. This signaling complex formation might require actin polymerization in stabilizing the recruitment of either of these molecules. Hence, inhibition of actin polymerization then causes a delayed formation of the signaling complex and as a consequence the delayed CD3ε phosphorylation. Third, actin polymerization was shown to be important for the directed movement of TCRs upon TCR triggering. A study used CytD to inhibit actin retrograde flow. It was demonstrated that actin retrograde flow is required for centripetal TCR microcluster transport and drives the receptor cluster dynamics at the immunological synapse in Jurkat T cells.34 Importantly, TCR clustering enhances phosphorylation of CD3.35, 36 Hence, without actin polymerization TCR clustering might be delayed, resulting in less CD3 phosphorylation.
T‐cell antigen receptor microcluster formation by transgenic TCR T cells on synthetic lipid bilayers with MHC–peptide agonist and intercellular adhesion molecule‐1 (ICAM‐1) depends upon F‐actin.37 Another report using latrunculin‐A to inhibit F‐actin formation showed that TCR microclusters stopped translocating immediately after latrunculin‐A treatment. This inhibitory effect was primarily on disorganization of the peripheral supramolecular activation complex (pSMAC), but very little on the central supramolecular activation complex (cSMAC), showing that the stability of the cSMAC might not depend on F‐actin. In addition, the newly formed TCR microclusters could be inhibited by F‐actin inhibitor, whereas those already generated were resistant to F‐actin inhibitor.38 These findings are in accordance with another report suggesting that TCR microcluster formation is resistant to inhibition by Src family kinase inhibitor PP2, but can be inhibited by latrunculin‐A.37 In our present study, phosphorylation of CD3ɛ was reduced when F‐actin was inhibited, hence, it is likely that F‐actin supports Src kinase‐independent formation of TCR microclusters in response to MHC–peptide agonist for TCR signal amplification.
Actin drives the process of cell polarization and maintains the cell–cell contact; its polymerization also likely provides a scaffold for clustering, translocation and spatial segregation of proteins, these are key steps to amplify and maintain T‐cell signaling.12, 39, 40, 41, 42 These data were consistent with our study showing that actin polymerization is required for an efficient recruitment of ZAP70 and Nck1 to the antibody‐stimulated TCR. Therefore, the actin polymerization is essential for the translocation of signaling proteins, especially for the recruitment of proximal signaling molecules to TCR to sustain initial T‐cell signaling. The present finding shows that the presence of Nck is required for an efficient phosphorylation of CD3ε. However, F‐actin has been demonstrated to be essential for micro‐adhesion rings, LAT and SLP76 clusters, but not for TCR microclusters and ZAP70 clusters.43 F‐actin might support phospho‐Phospholipase C gamma (PLC‐γ) as actin foci similar to the F‐actin localized at the core of the synapse‐like structure that supports SLP76 clusters and the micro‐adhesion ring.3, 43 This is in agreement with the present study demonstrated that the inhibition of actin filament formation resulted in the impaired TCR‐induced phosphorylation of intracellular signaling proteins including ZAP70, Erk and CREB. Previous studies have also shown the involvement of F‐actin in the function of calcium signaling molecules and calcium influx.38, 44, 45 It has also been suggested that actin polymerization acts very early at immune synapses when TCR is ligated and it also serves as a scaffold to which signaling molecules would bind and so be protected from degradation by proteasomal pathways.31, 45, 46
In summary, our present study reveals that the TCR engagement leads to actin polymerization, this might be caused by fast and very‐low‐level downstream signaling. The polymerized actin stabilizes the exposure of the PRS of CD3ε and allows the early binding of the SH3·1–Nck to the PRS. This transient and weak interaction promotes CD3ε phosphorylation, possibly because Nck can interact with Lck. Then, Nck can bind to TCR stably using its SH3 and SH2 domains, resulting in a subsequent induction of the downstream signaling cascade (Fig. 6). Taken together, our data highlight the important mechanism of actin polymerization in the recruitment of the endogenous Nck1 molecule to phosphorylated CD3ε contributing to T‐cell activation.
Figure 6.
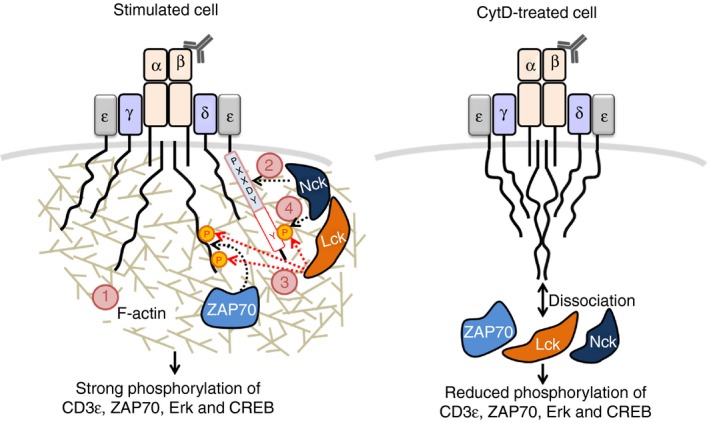
Our model of the role of actin polymerization in promoting non‐catalytic region of tyrosine kinase (Nck) ‐binding to the ligand‐/antibody‐engaged T‐cell receptor (TCR). Engagement of the TCR first leads to actin remodeling (1); this might be caused by fast and very‐low‐level downstream signaling. The polymerized actin promotes binding of the SH3·1(Nck) domain to the proline‐rich sequence (light blue box) in the CD3ε (2). This transient and weak interaction promotes the phosphorylation of CD3, possibly due to the interaction of Nck with Lck (3). If the second immunoreceptor‐tyrosine based activation motif (ITAM) tyrosine of CD3ε is phosphorylated, Nck can bind using its SH3·1 and SH2 domains in a cooperative manner (4). This binding mode is strong and together with ζ chain‐associated protein kinase of 70 000 MW (ZAP70) binding to doubly phosphorylated ITAMs leads to T‐cell activation (left panel). If actin polymerization is blocked, the following downstream events do not occur in an efficient manner (right panel).
Author contributions
PW and NMW performed experiments and JN, PP and SP designed experiments. SM and WWS gave suggestions on phosphorylation study. PW, SM, WWS and SP wrote or edited the manuscript. SP conceived the project.
Disclosures
The authors declare no conflicting interests.
Acknowledgements
This work was supported by Naresuan University (R2562C012) to SP; the Royal Golden Jubilee PhD scholarship of the Thailand Science Research and Innovation (TSRI) to PW; the Deutsche Forschungsgemeinschaft (DFG) (EXC294 Center for Biological Signaling Studies, BIOSS) to SM and WWS; grant CRC1381 (project A9) by the DFG to WWS; and the TSRI (BRG6180010) to SP.
References
- 1. Porciello N, Kunkl M, Viola A, Tuosto L. Phosphatidylinositol 4‐phosphate 5‐kinases in the regulation of T cell activation. Front Immunol 2016; 7:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor‐induced spreading: a role for the signal transduction adaptor LAT. Immunity 2001; 14:315–29. [DOI] [PubMed] [Google Scholar]
- 3. Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta 2014; 1838:546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swamy M, Beck‐Garcia K, Beck‐Garcia E, Hartl FA, Morath A, Yousefi OS et al A cholesterol‐based allostery model of T cell receptor phosphorylation. Immunity 2016; 44:1091–101. [DOI] [PubMed] [Google Scholar]
- 5. Schamel WW, Alarcon B, Hőfer T, Minguet S. The allostery model of TCR regulation. J Immunol 2017; 198:47–52. [DOI] [PubMed] [Google Scholar]
- 6. Lin J, Weiss A. T cell receptor signalling. J Cell Sci 2001; 114:243–4. [DOI] [PubMed] [Google Scholar]
- 7. Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci 2018; 43:108–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guy CS, Vignali DA. Organization of proximal signal initiation at the TCR:CD3 complex. Immunol Rev 2009; 232:7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wunderlich L, Faragó A, Downward J, Buday L. Association of Nck with tyrosine‐phosphorylated SLP‐76 in activated T lymphocytes. Eur J Immunol 1999; 29:1068–75. [DOI] [PubMed] [Google Scholar]
- 10. Ramesh N, Anton IM, Martinez‐Quiles N, Geha RS. Waltzing with WASP. Trends Cell Biol 1999; 9:15–9. [DOI] [PubMed] [Google Scholar]
- 11. Griffiths EK, Penninger JM. Communication between the TCR and integrins: role of the molecular adaptor ADAP/Fyb/Slap. Curr Opin Immunol 2002; 14:317–22. [DOI] [PubMed] [Google Scholar]
- 12. Barda‐Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol 2005; 6:80–9. [DOI] [PubMed] [Google Scholar]
- 13. Paensuwan P, Ngoenkam J, Khamsri B, Preechanukul K, Sanguansermsri D, Pongcharoen S. Evidence for inducible recruitment of Wiskott–Aldrich syndrome protein to T cell receptor–CD3 complex in Jurkat T cells. Asian Pac J Allergy Immunol 2015; 33:189–95. [DOI] [PubMed] [Google Scholar]
- 14. Pauker MH, Reicher B, Fried S, Perl O, Barda‐Saad M. Functional cooperation between the proteins Nck and ADAP is fundamental for actin reorganization. Mol Cell Biol 2011; 31:2653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lehmann JM, Reithmüller G, Johnson JP. Nck, a melanoma cDNA encoding a cytoplasmic protein consisting of the src homology units SH2 and SH3. Nucleic Acids Res 1990; 18:1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park D. Cloning, sequencing, and overexpression of SH2/SH3 adaptor protein Nck from mouse thymus. Mol Cells 1997; 7:231–6. [PubMed] [Google Scholar]
- 17. Buday L, Wunderlich L, Tamás P. The Nck family of adaptor proteins: regulators of actin cytoskeleton. Cell Signal 2002; 14:723–31. [DOI] [PubMed] [Google Scholar]
- 18. Lettau M, Pieper J, Janssen O. Nck adaptor proteins: functional versatility in T cells. Cell Commun Signal 2009; 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pesti S, Balázs A, Udupa R, Szabó B, Fekete A, Bőgel G et al Complex formation of EphB1/Nck/Caskin1 leads to tyrosine phosphorylation and structural changes of the Caskin1 SH3 domain. Cell Commun Signal 2012; 10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaki SP, Rivera GM. Integration of signaling and cytoskeletal remodeling by Nck in directional cell migration. Bioarchitecture 2013; 3:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alarcón B, Gil D, Delgado P, Schamel WW. Initiation of TCR signaling: regulation within CD3 dimers. Immunol Rev 2003; 191:38–46. [DOI] [PubMed] [Google Scholar]
- 22. Paensuwan P, Hartl FA, Yousefi OS, Ngoenkam J, Wipa P, Beck‐Garcia E et al Nck binds to the T cell antigen receptor using its SH3.1 and SH2 domains in a cooperative manner, promoting TCR functioning. J Immunol 2016; 196:448–58. [DOI] [PubMed] [Google Scholar]
- 23. Gil D, Schamel WW, Montoya M, Sánchez‐Madrid F, Alarcón B. Recruitment of Nck by CD3 epsilon reveals a ligand‐induced conformational change essential for T cell receptor signaling and synapse formation. Cell 2002; 109:901–12. [DOI] [PubMed] [Google Scholar]
- 24. Borroto A, Reyes‐Garau D, Jiménez MA, Carrasco E, Moreno B, Martínez‐Pasamar S et al First‐in‐class inhibitor of the T cell receptor for the treatment of autoimmune diseases. Sci Transl Med 2016; 8:370ra184. [DOI] [PubMed] [Google Scholar]
- 25. Ngoenkam J, Paensuwan P, Preechanukul K, Khamsri B, Yiemwattana I, Beck‐García E et al Non‐overlapping functions of Nck1 and Nck2 adaptor proteins in T cell activation. Cell Commun Signal 2014; 12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. San José E, Sahuquillo AG, Bragado R, Alarcón B. Assembly of the TCR/CD3 complex: CD3ε/d and CD3ε/g dimers associate indistinctly with both TCR a and TCR b chains. Evidence for a double TCR heterodimer model. Eur J Immunol 1998; 28:12–21. [DOI] [PubMed] [Google Scholar]
- 27. Dopfer EP, Schöpf B, Louis‐Dit‐Sully C, Dengler E, Höhne K, Klescová A et al Analysis of novel phospho‐ITAM specific antibodies in a S2 reconstitution system for TCR‐CD3 signalling. Immunol Lett 2010; 130:43–50. [DOI] [PubMed] [Google Scholar]
- 28. Melak M, Plessner M, Grosse R. Actin visualization at a glance. J Cell Sci 2017; 130:525–30. [DOI] [PubMed] [Google Scholar]
- 29. Weiss A, Stobo JD. Requirement for the coexpression of T3 and the T cell antigen receptor on a malignant human T cell line. J Exp Med 1984; 160:1284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Söderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J et al Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3:995–1000. [DOI] [PubMed] [Google Scholar]
- 31. Comrie WA, Burkhardt JK. Action and traction: cytoskeletal control of receptor triggering at the immunological synapse. Front Immunol 2016; 7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vazquez ML. Biological consequences of the phosphorylation of serine 59 on the tyrosine kinase Lck. ProQuest Dissertations and Theses, IN, USA: Purdue University, 2007. [Google Scholar]
- 33. Hem CD, Sundvold‐Gjerstad V, Granum S, Koll L, Abrahamsen G, Buday L et al T cell specific adaptor protein (TSAd) promotes interaction of Nck with Lck and SLP‐76 in T cells. Cell Commun Signal 2015; 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yi J, Wu XS, Crites T, Hammer JA 3rd. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell 2012; 23:834–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Minguet S, Swamy M, Alarcón B, Luescher IF, Schamel WW. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity 2007; 26:43–54. [DOI] [PubMed] [Google Scholar]
- 36. Taylor MJ, Husain K, Gartner ZJ, Mayor S, Vale RD. A DNA‐based T cell receptor reveals a role for receptor clustering in ligand discrimination. Cell 2017; 169:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campi G, Varma R, Dustin ML. Actin and agonist MHC–peptide complex–dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med 2005; 202:1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor‐proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 2006; 25:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Billadeau DD, Nolz JC, Gomez TS. Regulation of T‐cell activation by the cytoskeleton. Nat Rev Immunol. 2007; 7:131–43. [DOI] [PubMed] [Google Scholar]
- 40. Dustin ML. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr Opin Cell Biol 2007; 19:529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gomez TS, Billadeau DD. T cell activation and the cytoskeleton: you can't have one without the other. Adv Immunol 2008; 97:1–64. [DOI] [PubMed] [Google Scholar]
- 42. Beemiller P, Krummel MF. Mediation of T‐cell activation by actin meshworks. Cold Spring Harb Perspect Biol 2010; 2:a002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hashimoto‐Tane A, Sakuma M, Ike H, Yokosuka T, Kimura Y, Ohara O et al Micro‐adhesion rings surrounding TCR microclusters are essential for T cell activation. J Exp Med 2016; 213:1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huppa JB, Gleimer M, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat Immunol 2003; 4:749–55. [DOI] [PubMed] [Google Scholar]
- 45. Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T‐cell receptors by a few peptide–MHC complexes. Nature 1995; 375:148–51. [DOI] [PubMed] [Google Scholar]
- 46. Penninger JM, Crabtree GR. The actin cytoskeleton and lymphocyte activation. Cell 1999; 96:9–12. [DOI] [PubMed] [Google Scholar]