

Abstract
Combined MEK‐BRAF inhibition is a well‐established treatment strategy in BRAF‐mutated cancer, most prominently in malignant melanoma with durable responses being achieved through this targeted therapy. However, a subset of patients face primary unresponsiveness despite presence of the activating mutation at position V600E, and others acquire resistance under treatment. Underlying resistance mechanisms are largely unknown, and diagnostic tests to predict tumor response to BRAF‐MEK inhibitor treatment are unavailable.
Multiple myeloma represents the second most common hematologic malignancy, and point mutations in BRAF are detectable in about 10% of patients. Targeted inhibition has been successfully applied, with mixed responses observed in a substantial subset of patients mirroring the widespread spatial heterogeneity in this genomically complex disease. Central nervous system (CNS) involvement is an extremely rare, extramedullary form of multiple myeloma that can be diagnosed in less than 1% of patients. It is considered an ultimate high‐risk feature, associated with unfavorable cytogenetics, and, even with intense treatment applied, survival is short, reaching less than 12 months in most cases. Here we not only describe the first patient with an extramedullary CNS relapse responding to targeted dabrafenib and trametinib treatment, we furthermore provide evidence that a point mutation within the capicua transcriptional repressor (CIC) gene mediated the acquired resistance in this patient.
Key Points
BRAF mutations constitute an attractive druggable target in multiple myeloma. This is the first genomic dissection of the central nervous system involvement in a multiple myeloma patient harboring a druggable BRAF V600E mutation. Deep genomic characterization of the extramedullary lesion prompted a personalized therapeutic approach.
Acquisition of CIC mutation confers a mechanism of BRAF‐MEK inhibitor drug resistance in multiple myeloma.
The in silico interrogation of the CoMMpass clinical study revealed 10 patients with somatic mutations of CIC and its downregulation at gene expression level in multiple myeloma.
CIC gene silencing decreases the sensitivity of multiple myeloma cells to BRAF‐MEK inhibition in vitro. The correlation between CIC downregulation and ETV4/5 nuclear factor expression in multiple myeloma BRAF‐mutant cells is shown for the first time.
CIC mutation, its downregulation, and the related downstream effect on MMP24 support disseminative potential providing new clues in the extramedullary biology definition.
Keywords: Multiple myeloma, Extramedullary disease, Capicua transcriptional repressor, Drug resistance, BRAF mutation
Short abstract
This article presents a case report of a patient with multiple myeloma (MM) and extramedullary localization at the central nervous system (CNS) at relapse. This is the first genomic characterization of CNS MM localization and provides evidence that a point mutation within the CIC gene is a possible mechanism of acquired resistance.
Patient History
An 81‐year‐old patient with κ light chain multiple myeloma (MM) was referred to our center after having a seizure and increasing M‐proteins. MM had been diagnosed 2 years before and the patient had undergone nine cycles of bortezomib‐based combination therapy (VMP) resulting in an initial good disease control. Magnetic resonance imaging of the brain and additional experimental whole body 11C‐methionine positron emission tomography (PET)‐computed tomography (CT) scan, which increased MM imaging sensitivity 1, demonstrated metabolic active disease supra‐ and infratentorial in the clivus, as well as in the right femur as the underlying cause of the clinical scenario (Fig. 1A). A high load of clonal plasma cells (PCs) was detected in his cerebrospinal fluid (CSF), whereas in the bone marrow (BM), only a few CD138pos MM cells could be detected. Thus, we diagnosed extramedullary central nervous system (CNS) relapse, and intrathecal triple therapy (methotrexate, cytarabine, and dexamethasone) along with age‐adjusted systemic chemotherapy (cytarabine and thiotepa) was initiated.
Figure 1.
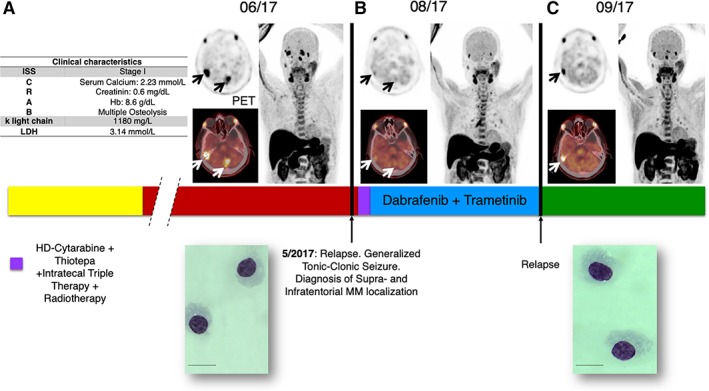
Patient disease history schematic view. (A): Clinical characteristics (left panel). 11C‐methionine positron emission tomography (PET)‐computed tomography central nervous system showing disease manifestation at first relapse (right panel): remission (B), and second brain recurrence (C).
Abbreviation: MM, multiple myeloma.
Molecular Tumor Board 1
Genotyping Results and Interpretation of the Molecular Results
Given the peculiar clinical course and the poor prognosis associated with a CNS localization, with limited effective therapeutic options available, we performed a deep molecular characterization of CSF and BM tumoral plasma cells.
DNA extracted from CD138pos purified cells obtained from CSF and BM paired samples was analyzed by next‐generation sequencing (NGS). We applied the M3P (v3.0) panel 2, 3, a disease‐specific in‐house customized, NGS‐targeted deep sequencing panel for MM (Ion torrent platform) that includes an 88‐gene selection of most commonly mutated genes such as TP53, DIS3, FAM46C, CYLD, MAF, XBP1, MYC, MAX 4, 5, actionable drug targets (i.e., NRAS, KRAS, BRAF) 6, and genes being associated with drug resistance 7, 8 (e.g., CRBN, IKZF1/3, NR3C1, PSMB5). The average sequencing depth increased 700×. The CSF cells harbored a clonal BRAF V600E mutation (allele frequency variance 52%) that was absent in the BM, highlighting spatial genomic heterogeneity 9; no other somatic point mutations were detected within the M3P genes and no circulating PCs were identified by peripheral blood flow‐cytometry analysis.
Functional and Clinical Significance
The BRAF V600E mutation in exon 15 of BRAF gene is present in between 4% and 10% of patients with MM at diagnosis 4, 5, 6, 10, rising to almost 20% at relapse 7, displaying a role in the extramedullary disease and exerting a negative impact overall survival (45 vs. 105 months, p = .04) 11, 12. Applying the M3P gene panel, we sequenced 608 MM patients at different disease stages. Concerning BRAF, we have identified 59 (9.7%) mutated patients with a total of 25 distinctive mutations. Among our patient cohort, 21 of 59 (35%) harbored the BRAF V600E mutation; within the remaining 38 patients with 24 BRAF non‐V600E mutations, we found 12 alterations conferring a kinase domain activation, comprising also a rare K601 mutation, and 12 leading to BRAF functional impairment (supplemental online Fig. 1) 13. BRAF exon 15 mutations confer sensitivity to target therapies such as vemurafenib, dabrafenib, and trametinib 14, 15. Heuck et al. reported BRAF‐MEK targeted therapy approach in 58 patients with MM with either BRAF, NRAS, and KRAS mutations or high‐risk gene expression profiling; out of 58 patients, 11 displayed an extramedullary localization. Ten patients received a combination therapy with trametinib, and two of them were additionally treated with dabrafenib or vemurafenib. Interestingly, within the patients with a measurable disease (40 patients), 16 patients achieved a reduction of at least 50% of the MM protein, and 9 achieved a complete remission evaluated by PET‐CT. Raab et al. described a case of a patient with MM with a disseminated disease harbor a BRAF V600E mutation. The patient was successfully treated with vemurafenib upfront and with a subsequent combination therapy with bortezomib at disease relapse owing to a clonal selection of NRAS mutants’ resistant subclones. 16, 17. These published real‐life based evidences and ongoing clinical trials (i.e., BIRMA trial) combining BRAF and MEK inhibitors, highlight the clinical relevance of circumventing the paradoxical RAS pathway activation upon BRAF inhibition already described in melanoma 18, 19. Lohr et al. tested in vitro the combination of trametinib and dabrafenib in several MM cell lines harboring distinct BRAF or RAS mutations; the U266 BRAF K601N proved most sensitive and displayed a similar paradoxical feedback loop of RAS‐activation 5.
Recently a combination of dabrafenib and trametinib effectively eliminated in BRAF V600E mutant melanoma brain metastases, demonstrating that the drug can cross the blood brain barrier 20, 21.
Patient Update
Although neutropenic, because of the cytarabine‐based chemotherapy, the patient developed a Gram‐positive septicemia. Taking into account the risk profile, the therapy‐related infectious episode, and the sequencing results, and according to German law and ethical approval (Einzelheilversuch), the patient started a combinational targeted therapy with continuous BRAF‐MEK inhibitor (dabrafenib 150 mg twice daily and trametinib 2 mg daily). Neurological examination revealed a significant clinical improvement on the basis of the absence of pathological signs and symptoms, which was confirmed by 11C‐methionine PET subtotal tumor shrinkage (Fig. 1B).
Regrettably, only 3 months after the treatment initiation, 11C‐methionine PET revealed local MM recurrence and disseminated bone while on continuous therapy (Fig. 1C). To confirm the disease relapse, we repeated the CSF assessment, revealing, as expected, a high mononucleated tumoral plasma cells load. The patient underwent palliation with hyperfractionated radiotherapy of the cerebrum (cumulative irradiation dose: 30 Gy); because of compromised performance status of the patient, no further systemic therapy could be applied, and best supportive care was adopted until patient exitus occurred at the end of October 2017.
Molecular Tumor Board 2
Genotyping Results and Interpretation of the Molecular Results
To investigate the underlying mechanisms of resistance development upon targeted MEK‐BRAF inhibitor therapy, we performed a whole exome DNA sequencing (Illumina platform) on the pretherapy sample and on CD138pos purified MM cells obtained from the CSF after confirmed disease relapse. Sequencing depth of 115× was applied. A total number of 97 nonsilent coding variants (missense, nonsense, indels, splice) with an allele frequency higher than 5% were identified, of which 74 were shared between the timepoints. Sequencing revealed 19 additional point mutations acquired at relapse. According to published guidelines, we performed an extensive literature revision, and we systematically selected four potential clinically relevant nonsynonymous point mutations (Table 1) 22. Dispatched RND transporter family member 2 (DISP2; p.P1271L) is a key regulator of the hedgehog signaling pathway 23, 24 and has been associated with the development of bortezomib resistance in MM 25. It further impacts fibroblast growth factor receptor 3 signaling to RAS pathway, thus potentially mediating the paradoxical activation of the downstream pathway in a BRAF‐independent manner 26. CREB binding protein (CREBBP; p.V429F) represents an epigenetic modulator able to control the TP53 apoptosis machinery activation 27 and the downstream regulation of the RAS‐RAF pathway 28. The pyrimidinergic receptor P2Y4 (P2RY4, p.R314Q) is an upstream regulator of PLCβ/PI3K pathway able to cross‐talk with the EGFR‐RAS pathway 29, a well‐known mechanism of resistance described in BRAF V600E mutated melanoma 30. We also identify a missense mutation in capicua transcriptional repressor (CIC; p.A984P) mapped on chromosome 19 with an allelic variance of 17% (Table 1). CIC represents a transcriptional repressor gene directly involved in the downstream regulation of the RAS‐RAF pathway able to drive the development of BRAF‐MEK inhibitor resistance 31. Based on this correlation, we hypothesized that the acquisition of CIC mutation may mechanistically underlie the BRAF‐MEK resistance in our patient.
Table 1.
Clinically relevant single nucleotide variations
| Chr | h19 position | SNV base change | Gene | SIFT | Polyphen2 | Mut. taster | Effect | Amino acid | dbSNP |
|---|---|---|---|---|---|---|---|---|---|
| 15 | 40662125 | c.3812 C>T | DISP2 | Damaging | Damaging | Damaging | Missense | p.P1271L | |
| 16 | 3842027 | c.1285 C>A | CREBBP | Damaging | Damaging | Damaging | Missense | p.V429F | |
| 19 | 42796301 | c.2950 G>C | CIC | Damaging | Damaging | Damaging | Missense | p.A984P | |
| X | 69478534 | c.941 C>T | P2RY4 | Tolerated | Damaging | Damaging | Missense | p.R314Q | rs775064288 |
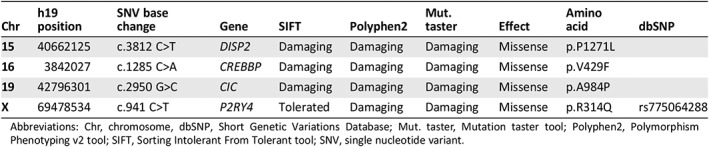
Abbreviations: Chr, chromosome, dbSNP, Short Genetic Variations Database; Mut. taster, Mutation taster tool; Polyphen2, Polymorphism Phenotyping v2 tool; SIFT, Sorting Intolerant From Tolerant tool; SNV, single nucleotide variant.
Functional and Clinical Significance of CIC in Cancer
Next, we aimed to functionally validate the molecular significance of CIC alteration in mediating resistance to BRAF and MEK inhibitors. Wang et al. demonstrated in lung, colon, pancreatic 31, 32, and melanoma 31, 33 human cancer models the pivotal role of low CIC expression in inducing resistance to vemurafenib and trametinib; however, evidences of its role of resistance induction in MM or other hematological malignancies are lacking. Le Blanc et al. reported that CIC missense mutations result in a gene expression downregulation 34. Interrogating the Multiple Myeloma Research Foundation CoMMpass study we found a similar correlation: the 10 patients that harbored a CIC mutation (Fig. 2A) had a significant gene downregulation compared with the unmutated ones (p = .03). Mutations in the proline‐rich (Fig. 2B) region are reported to impair the protein expression 35. Therefore, we established a CIC knockdown in vitro model, using a small interfering RNA as specific gene silencing technique. We employed the U266 MM cell line harboring an activating BRAF K601N mutation, usually sensitive to BRAF‐MEK inhibition 5, as a commercially available MM model harboring a BRAF activating mutation. Of note, upon CIC gene silencing, we observed drug resistance induction to BRAF‐MEK inhibition (Fig. 3A); in detail, we cultured the silenced and not‐silenced MM cells with trametinib and dabrafenib, either as single agents or in combination, and we observed resistance induction to the combination of the two drugs (row factor, 91.16%; p < .0001, two‐way ANOVA test). Next, we investigated whether this drug‐resistance phenotype also coincided with a more invasive behavior. Thus, we performed a motility and migration assay. CIC knockdown in U266 BRAF K601N cells significantly enhanced MM migration in a scratch wound healing assay (Fig. 3B). These findings prompted us to investigate potential mechanism able to explain the CIC‐impairment‐related biological effects. In particular, CIC is the direct master regulator of several transcription factors such as ETV4 and ETV5 two oncogenes able to modulate the RAS downstream pathway 31, 32, 33. Moreover, indirectly CIC induces an invasiveness related protein namely MMP24 32, 33. Consequently, we confirmed by Western blotting an upregulation of ETV4, ETV5, and MMP24 protein expression in the CIC‐knockdown U266, as mediators of drug resistance and MM invasiveness (Fig. 3C). The upregulation of these transcription factors can activate the MAPK signaling, in an independent p‐MEK manner 31, providing an escape mechanism from BRAF‐MEK inhibition (Fig. 3D) 36.
Figure 3.
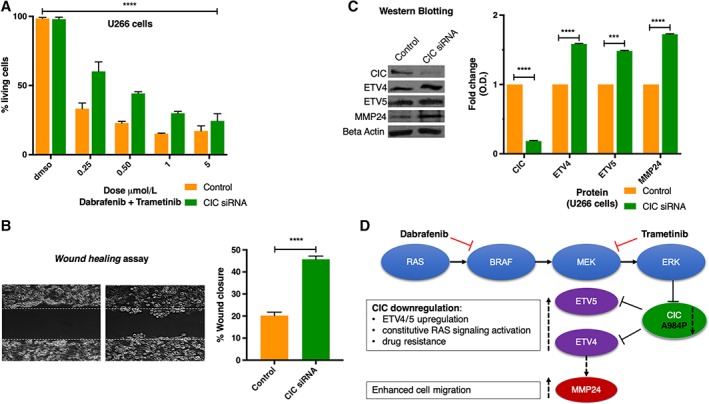
Functional validation of capicua transcriptional repressor (CIC) downregulation biological effect in multiple myeloma. (A): Cell viability assay measured with bioluminescence upon drug treatment with escalating doses of trametinib and dabrafenib in U266 human multiple myeloma cell line: scrambled versus CIC small interfering RNA (siRNA) transduced percentage of living cells are compared by ANOVA test. Experiments were conducted in three biological and technical replicates following manufacturer's instructions (CellTiter‐Glo Luminescent Cell Viability Assay; Promega, Madison, WI). (B): Scratch‐wound healing assay was performed as previously described 44, 45. Briefly, wound areas were analyzed with ImageJ Lab 1.51 software and quantified as percentage of total surface. (C): Western‐Blot analysis after CIC knockdown on U266 cells and corresponding densitometric quantification. (D): Dabrafenib and trametinib targets on RAS‐RAF pathway overview and CIC axis schematic interaction with the RAS‐RAF downstream signaling. ***p < .001; ****p < .0001.
Figure 2.
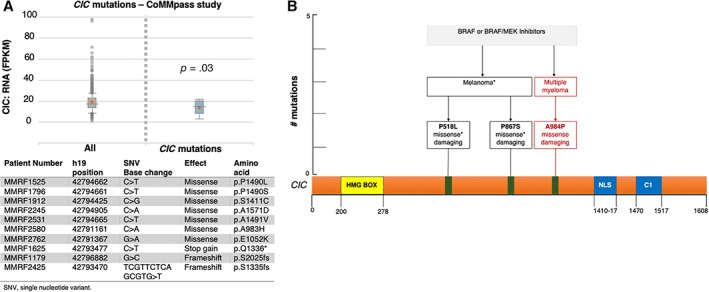
Capicua transcriptional repressor (CIC) is altered in multiple myeloma and is mutated after BRAF target treatment in cancer. (A): Report from in silico interrogation on CoMMpass study data set: comparison between RNA expression levels of CIC wild‐type and mutated patients (upper panel); list of single nucleotide variations among patients enrolled in the CoMMpass study (lower panel, t test performed); (B): CIC somatic mutations acquired in melanoma and central nervous system multiple myeloma after BRAF inhibition therapy.
Abbreviations: *, stop codon; FPKM, fragments per kilobase of exon model per million reads mapped.
Potential Strategies to Target the Pathway and Implications for Clinical Practice
CIC has recently been identified as a candidate gene related to MEK‐BRAF resistance development 31, 33. Our clinical observations, the subsequent in vitro MM model, and the public datasets interrogations support that the acquisition of CIC mutation and its subsequent downregulation confers MEK‐BRAF inhibitors resistance for the first time in MM.
As large BRAF‐RAS treated cohorts in MM are not available, we screened for published datasets to answer the question of whether mutation acquisition in CIC under BRAF‐RAS targeted therapy can be observed in a significant number of patients being resistant to BRAF inhibitors. Remarkably, Van Allen et al. recently published a comprehensive genomic characterization of 45 patients with metastatic melanoma resistant to BRAF inhibitors; 5 of them (11%) harbored a somatic CIC mutation (4 missense and 1 frame shift). Intriguingly, two of the single nucleotide variations out of these five were acquired at time of relapse (Fig. 2B). One out of these five patients harboring a pretherapy CIC mutation experienced a very early disease relapse under dabrafenib therapy 37. Given that almost 30% of patients with MM harbor mutations affecting the BRAF‐RAS pathway, this may represent a potential biomarker to predict therapy response. Based on prior published findings 31, 33, 34, public available datasets 37 and previous 31, 32, 33, 34 and original in vitro validations pinpoint CIC mutation as one of the mechanisms of drug resistance of BRAF‐MEK inhibition therapy. Extramedullary (EMD) dissemination in MM typically correlates with very poor prognosis, especially when the clinical onset manifests at disease relapse 12, 38, 39, 40, 41, 42. Furthermore, given the scanty evidences available about the disease biology behind the EMD 43, CIC onco‐suppressive functions might be also expressed as ancillary mechanism that sustain the EMD phenotype in MM 39.
Prospective clinical trials including the BRAF and MEK inhibition are ongoing in multiple myeloma in Europe (NCT02834364) and in the U.S. (NCT03091257) as well as in different solid cancers such as melanoma and colon cancer (NCT02974803, NCT03668431); these ongoing studies represent the ideal opportunity to determine and validate the role of CIC mutations as potential disease biomarker in a large clinical and controlled‐prospective setting.
Glossary of Genomic Terms and Nomenclature
- Allelic frequency
percentage of reads referred to the mutated allele
- Average sequencing depth
mean number of unique reads for each single nucleotide aligned to a reference sequence
- Spatial genomic heterogeneity
presence of distinctive genomic alterations in different anatomical sites
- BRAF
B‐Raf Proto‐Oncogene, Serine/Threonine Kinase
- MEK
Mitogen‐Activated Protein Kinase Kinase 1
- CIC
Capicua Transcriptional Repressor
- DISP2
Dispatched RND Transporter Family Member 2
- CREBBP
CREB Binding Protein
- P2RY4
Pyrimidinergic receptor P2Y4
- ETV4
ETS Variant 4
- ETV5
ETS Variant 5
- MMP24
Matrix Metallopeptidase 24
- TP53
Tumor Protein P53
- DIS3
DIS3 Homolog, Exosome Endoribonuclease And 3'‐5' Exoribonuclease
- FAM46C
Terminal Nucleotidyltransferase 5C
- CYLD
CYLD Lysine 63 Deubiquitinase
- MAF
MAF BZIP Transcription Factor
- XBP1
X‐Box Binding Protein 1
- MYC
MYC Proto‐Oncogene, BHLH Transcription Factor
- MAX
MYC Associated Factor X
- KRAS
Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog
- NRAS
Neuroblastoma Ras viral oncogene homolog
- CRBN
Cereblon
- IKZF1
IKAROS Family Zinc Finger 1
- IKZF3
IKAROS Family Zinc Finger 3
- NR3C1
Nuclear Receptor Subfamily 3 Group C Member 1
- PSMB5
Proteasome Subunit Beta 5
- PLCβ/PI3K
Phospholipase C beta/Phosphatidil‐Inositol‐3‐Kinase pathway
- EGFR
Epidermal grow factor receptor
Author Contributions
Conception/design: Matteo Claudio Da Vià, Antonio Giovanni Solimando, Andoni Garitano‐Trojaola, Hermann Einsele, K. Martin Kortüm
Provision of study material or patients: Matteo Claudio Da Vià, Antonio Giovanni Solimando, Susanne Strifler, Constantin Lapa, Andreas Beilhack, Leo Rasche, Hermann Einsele, K. Martin Kortüm
Collection and/or assembly of data: Matteo Claudio Da Vià, Antonio Giovanni Solimando, Andoni Garitano‐Trojaola, Santiago Barrio, Umair Munawar, Larissa Haertle, Nadine Rhodes, Eva Teufel, Cornelia Vogt, Constantin Lapa, K. Martin Kortüm
Data analysis and interpretation: Matteo Claudio Da Vià, Antonio Giovanni Solimando, Andoni Garitano‐Trojaola, Santiago Barrio, Umair Munawar, Leo Rasche, K. Martin Kortüm
Manuscript writing: Matteo Claudio Da Vià, Antonio Giovanni Solimando, Leo Rasche, Hermann Einsele, K. Martin Kortüm
Final approval of manuscript: Matteo Claudio Da Vià, Antonio Giovanni Solimando, Andoni Garitano‐Trojaola, Santiago Barrio, Umair Munawar, Susanne Strifler, Larissa Haertle, Nadine Rhodes, Eva Teufel, Cornelia Vogt, Constantin Lapa, Andreas Beilhack, Leo Rasche, Hermann Einsele, K. Martin Kortüm
Disclosures
Hermann Einsele: Bristol‐Myers Squibb, Novartis, Takeda (C/A), Celgene, Janssen (RF, SAB), Celgene, Janssen, Bristol‐Myers Squibb, Novartis, Takeda (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Figure S1 Heatmap of BRAF mutation in M3P sequenced cohort [2, 7, 13].
Acknowledgments
This work was supported by the following: Borsa di Perfezionamento financed by Italian Society for Experimental Hematology to M.D.V.; Progetto Professionalità financed by Banca del Monte di Lombardia Foundation to M.D.V.; GLOBALDOC Project ‐ CUP H96J17000160002 approved with A.D. n. 9 of January 18, 2017, from Puglia Region, financed under the Action Plan for Cohesion approved with Commission decision C (2016) 1417 of 3.03.2016 to A.G.S.; the Apulian Regional project: medicina di precisione to A.G.S.; ZKF Würzburg (Z‐3R/2) to L.R. and M.D.V.; and a BTHA grant to K.M.K. The German SKELMET/μBone consortium supported by the German Research Council (DFG FOR 1586, SPP 2084) through an Investigator grant to A.B. Bayerische Forschungsstiftung consortium FortiTher (WP2TP3), the Deutsche Forschungsgemeinschaft mBone consortium (2084/1, 401253051 to A.B.).
Disclosures of potential conflicts of interest may be found at the end of this article.
Footnotes
For Further Reading: Winnie S. Liang, Jo‐Anne Vergilio, Bodour Salhia et al. Comprehensive Genomic Profiling of Hodgkin Lymphoma Reveals Recurrently Mutated Genes and Increased Mutation Burden. The Oncologist 2018;24:219–228; first published August 14, 2018.
Implications for Practice: This study provides the first evidence that comprehensive genomic profiling can be performed to map the genomic landscape of Hodgkin lymphoma and that a subpopulation of patients has mutations in TP53, B2M, XPO1, and other genes. It was found that 15% of patients have high mutation burden, which, in cancers such as melanoma, may indicate sensitivity to immune checkpoint inhibitors, and may thus be explored for Hodgkin lymphoma. Lastly, this work demonstrates that changes in the mutant allele frequency of XPO1 in serially collected plasma cell‐free DNA samples correspond with treatment outcomes measured with conventional radiographic imaging.
References
- 1. Lapa C, Garcia‐Velloso MJ, Lückerath K et al. (11)C‐Methionine‐PET in multiple myeloma: A combined study from two different institutions. Theranostics 2017;7:2956–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kortum KM, Langer C, Monge J et al. Longitudinal analysis of 25 sequential sample‐pairs using a custom multiple myeloma mutation sequencing panel (M(3)P). Ann Hematol 2015;94:1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barrio S, DáVia M, Bruins L et al. Protocol for M3P: A comprehensive and clinical oriented targeted sequencing panel for routine molecular analysis in multiple myeloma. Methods Mol Biol 2018;1792:117–128. [DOI] [PubMed] [Google Scholar]
- 4. Bolli N, Avet‐Loiseau H, Wedge DC et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014;5:2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lohr JG, Stojanov P, Carter SL et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014;25:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walker BA, Boyle EM, Wardell CP et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol 2015;33:3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kortum KM, Mai EK, Hanafiah NH et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016;128:1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barrio S, Stühmer T, Da‐Viá M et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2018;33:447–456. [DOI] [PubMed] [Google Scholar]
- 9. Rasche L, Chavan SS, Stephens OW et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi‐region sequencing. Nat Commun 2017;8:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruiz‐Heredia Y, Sánchez‐Vega B, Onecha E et al. Mutational screening of newly diagnosed multiple myeloma patients by deep targeted sequencing. Haematologica 2018;103:e544–e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andrulis M, Lehners N, Capper D et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov 2013;3:862–869. [DOI] [PubMed] [Google Scholar]
- 12. Varettoni M, Corso A, Pica G et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: A longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–330. [DOI] [PubMed] [Google Scholar]
- 13. Kortuem KM, Braggio E, Bruins L et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J 2016;6:e397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim KB, Kefford R, Pavlick AC et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF‐mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol 2013;31:482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dahlman KB, Xia J, Hutchinson K et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov 2012;2:791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heuck CJ, Jethava Y, Khan R et al. Inhibiting MEK in MAPK pathway‐activated myeloma. Leukemia 2016;30:976–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raab MS, Lehners N, Xu J et al. Spatially divergent clonal evolution in multiple myeloma: Overcoming resistance to BRAF inhibition. Blood 2016;127:2155–2157. [DOI] [PubMed] [Google Scholar]
- 18. Flaherty KT, Robert C, Hersey P et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med 2012;367:107–114. [DOI] [PubMed] [Google Scholar]
- 19. Poulikakos PI, Zhang C, Bollag G et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature 2010;464:427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davies MA, Saiag P, Robert C et al. Dabrafenib plus trametinib in patients with BRAF(V600)‐mutant melanoma brain metastases (COMBI‐MB): A multicentre, multicohort, open‐label, phase 2 trial. Lancet Oncol 2017;18:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Puszkiel A, Noé G, Bellesoeur A et al. Clinical pharmacokinetics and pharmacodynamics of dabrafenib. Clin Pharmacokinet 2019;58:451–467. [DOI] [PubMed] [Google Scholar]
- 22. Services NUSDoHH . PubMed Tutorial. 2018. Available at https://www.nlm.nih.gov/bsd/disted/pubmedtutorial/cover.html.
- 23. Shou Y, Robinson DM, Amakye DD et al. A five‐gene hedgehog signature developed as a patient preselection tool for hedgehog inhibitor therapy in medulloblastoma. Clin Cancer Res 2015;21:585–593. [DOI] [PubMed] [Google Scholar]
- 24. Martello M, Remondini D, Borsi E et al. Opposite activation of the Hedgehog pathway in CD138+ plasma cells and CD138‐CD19+ B cells identifies two subgroups of patients with multiple myeloma and different prognosis. Leukemia 2016;30:1869–1876. [DOI] [PubMed] [Google Scholar]
- 25. Alonso S, Hernandez D, Chang YT et al. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. J Clin Invest 2016;126:4460–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katoh M, Nakagama H. FGF receptors: Cancer biology and therapeutics. Med Res Rev 2014;34:280–300. [DOI] [PubMed] [Google Scholar]
- 27. Lee CW, Ferreon JC, Ferreon AC et al. Graded enhancement of p53 binding to CREB‐binding protein (CBP) by multisite phosphorylation. Proc Natl Acad Sci USA 2010;107:19290–19295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dixon ZA, Nicholson L, Zeppetzauer M et al. CREBBP knockdown enhances RAS/RAF/MEK/ERK signaling in Ras pathway mutated acute lymphoblastic leukemia but does not modulate chemotherapeutic response. Haematologica 2017;102:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erb L, Weisman GA. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal 2012;1:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun C, Wang L, Huang S et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014;508:118–122. [DOI] [PubMed] [Google Scholar]
- 31. Wang B, Krall EB, Aguirre AJ et al. ATXN1L, CIC, and ETS transcription factors modulate sensitivity to MAPK pathway inhibition. Cell Rep 2017;18:1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okimoto RA, Breitenbuecher F, Olivas VR et al. Inactivation of Capicua drives cancer metastasis. Nat Genet 2017;49:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dissanayake K, Toth R, Blakey J et al. ERK/p90(RSK)/14‐3‐3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicúa. Biochem J 2011;433:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. LeBlanc VG, Firme M, Song J et al. Comparative transcriptome analysis of isogenic cell line models and primary cancers links capicua (CIC) loss to activation of the MAPK signalling cascade. J Pathol 2017;242:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu HC, Tan Q, Rousseaux MW et al. Disruption of the ATXN1‐CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat Genet 2017;49:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu J, Pfarr N, Endris V et al. Molecular signaling in multiple myeloma: Association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017;6:e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Allen EM, Wagle N, Sucker A et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bladé J, Fernández de Larrea C, Rosiñol L et al. Soft‐tissue plasmacytomas in multiple myeloma: Incidence, mechanisms of extramedullary spread, and treatment approach. J Clin Oncol 2011;29:3805–3812. [DOI] [PubMed] [Google Scholar]
- 39. Ghobrial IM. Myeloma as a model for the process of metastasis: Implications for therapy. Blood 2012;120:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rasche L, Bernard C, Topp MS et al. Features of extramedullary myeloma relapse: High proliferation, minimal marrow involvement, adverse cytogenetics: A retrospective single‐center study of 24 cases. Ann Hematol 2012;91:1031–1037. [DOI] [PubMed] [Google Scholar]
- 41. Varettoni M, Marchioni E, Bonfichi M et al. Successful treatment with Rituximab and Bendamustine in a patient with newly diagnosed Waldenstrom's Macroglobulinemia complicated by Bing‐Neel syndrome. Am J Hematol 2015;90:E152–153. [DOI] [PubMed] [Google Scholar]
- 42. Jurczyszyn A, Grzasko N, Gozzetti A et al. Central nervous system involvement by multiple myeloma: A multi‐institutional retrospective study of 172 patients in daily clinical practice. Am J Hematol 2016;91:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Egan JB, Kortuem KM, Kurdoglu A et al. Extramedullary myeloma whole genome sequencing reveals novel mutations in Cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br J Haematol 2013;161:748–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Solimando AG, Brandl A, Mattenheimer K et al. JAM‐A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia 2018;32:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rao L, De Veirman K, Giannico D et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: A preclinical study. Oncotarget 2018;9:13366–13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Figure S1 Heatmap of BRAF mutation in M3P sequenced cohort [2, 7, 13].