

Abstract
Interferon (IFN) regulatory factors (IRFs) are important determinants of the innate response to infection. We evaluated the role(s) of combined and individual IRF deficiencies in the outcome of infection of C57BL/6 mice with Sindbis virus, an alphavirus that infects neurons and causes encephalomyelitis. The brain and spinal cord levels of Irf7, but not Irf3 mRNAs, were increased after infection. IRF3/5/7−/− and IRF3/7−/− mice died within 3–4 days with uncontrolled virus replication, similar to IFNα receptor-deficient mice, while all wild-type (WT) mice recovered. IRF3−/− and IRF7−/− mice had brain levels of IFNα that were lower, but brain and spinal cord levels of IFNβ and IFN-stimulated gene mRNAs that were similar to or higher than WT mice without detectable serum IFN or increases in Ifna or Ifnb mRNAs in the lymph nodes, indicating that the differences in outcome were not due to deficiencies in the central nervous system (CNS) type I IFN response. IRF3−/− mice developed persistent neurological deficits and had more spinal cord inflammation and higher CNS levels of Il1b and Ifnγ mRNAs than WT mice, but all mice survived. IRF7−/− mice died 5–8 days after infection with rapidly progressive paralysis and differed from both WT and IRF3−/− mice in the induction of higher CNS levels of IFNβ, tumour necrosis factor (TNF) α and Cxcl13 mRNA, delayed virus clearance and more extensive cell death. Therefore, fatal disease in IRF7−/− mice is likely due to immune-mediated neurotoxicity associated with failure to regulate the production of inflammatory cytokines such as TNFα in the CNS.
Keywords: Sindbis virus, mice, tumor necrosis factor, CXCL13, CNS inflammation, cell death
Introduction
Arthropod-borne viruses, most importantly alphaviruses and flaviviruses, cause widespread epidemics of fever, encephalitis and arthritis, and pose increasing threats to human populations through expansion into new geographical areas [1]. Encephalomyelitis due to arbovirus infection of the nervous system is a particularly important global cause of morbidity and mortality because neuronal damage can lead to congenital malformations, progressive disease with long-term disability and acute fatal disease [2]. Neurotropic alphaviruses [chikungunya (CHIKV) and Venezuelan, western and eastern equine encephalitis viruses] and flaviviruses (West Nile, St Louis encephalitis and Zika viruses) are now endemic in the Americas. There are no treatments for these infections, and vaccines are not available for most of them.
Our studies in mice of Sindbis virus (SINV), the prototypic alphavirus, have shown that the outcome of neuronal infection is determined by both virus and host factors [3, 4]. Early activation of innate responses in infected neurons and local production of type I IFN are major host determinants of outcome. Mice that are unable to respond to IFNα/β due to the lack of the α chain of the IFN receptor (IFNAR) or the IFNAR-activated STAT1 transcription factor needed to induce IFN-stimulated antiviral genes fail to control alphavirus replication and develop rapidly fatal disease [5–9]. The innate sensing pathways for RNA viruses include the endosomal toll-like receptors (TLRs) 3, 7/8 and 9 and cytoplasmic RNA helicases (RIG-I, MDA5) that activate latent transcription factors NF-κB and IFN regulatory factor (IRF) 3. NF-κB is important for inducing the transcription of several cytokines and chemokines in response to virus infection, while IRF3 is required in addition to NF-κB for the induction of IFNβ.
IRFs belong to a family of transcription factors that are characterized by a conserved N-terminal DNA-binding domain and a more variable C terminal regulatory region [10]. IRF3 is constitutively present at varying levels in the cytoplasm of most cells, including neurons [11]. Serine phosphorylation of the auto-inhibitory region by innate sensing pathway-induced kinases leads to dimerization and nuclear translocation [12]. In the nucleus, IRF3 dimers complex with co-activator CBP/p300 and cooperate with NF-κB and ATF2/c-Jun for the transcriptional activation of IFNβ and other innate response genes [13, 14]. The amplification of this response with the production of IFNαs and IFNλs, in addition to IFNβ, is dependent on the activation of IRF7 [15–17]. Except in plasmacytoid dendritic cells [18], IRF7 is present at very low levels and is one of the many genes induced by IRF3. IRF7 associates with the toll-like receptor (TLR) adaptor proteins MyD88 or TRIF and the ubiquitin ligase TRAF6 and is activated by TLR signalling through TRAF6-mediated lysine ubiquitylation followed by IKKε-mediated phosphorylation [19]. Activated IRF7 can homodimerize or heterodimerize with phosphorylated IRF3 and is responsible for the slow phase of IFN production and the induction of IFN-stimulated genes (ISGs) [15, 16, 20–23].
Mice that are triply deficient in IRFs 3, 5 and 7 are highly susceptible to multiple virus infections with failure to restrict replication in peripheral organs or the CNS, similar to mice deficient in type I IFN signalling (IFNAR−/−) [24, 25]. Dual IRF3/7 deficiency leads to 100 % mortality after infection with West Nile virus (WNV), CHIKV and Ross River virus [26–28]. However, the effects of single IRF deficiencies on outcome are distinctive, organ-specific and less clearly related to the induction of type I IFN. For instance, single IRF3 deficiency increases susceptibility to infection of the CNS, but not peripheral infection, with herpes simplex virus (HSV)[29, 30], and increased susceptibility to WNV infection is IFN-dependent in the CNS and IFN-independent in the periphery [31]. Single IRF7 deficiency increases susceptibility to HSV [29] and to WNV in both the CNS and the periphery in a manner that is correlated with the induction of IFNα [32]. Both IRF3−/− and IRF7−/− mice survive peripheral infection with CHIKV [27].
To determine the roles of IRFs in the well-characterized nonfatal mouse model of SINV encephalomyelitis caused by the infection of neurons in the brain and spinal cord, we have analysed the effects of single and combined deficiencies of IRF3 and IRF7 on outcomes for mice infected intracerebrally with the TE strain of SINV. Infection resulted in the death of IRF3/5/7−/− and IRF3/7−/− mice by day 4, similar to the outcome in IFNAR−/− mice, while all WT mice survived. IRF3−/− mice survived but developed persistent evidence of neurological disease, while IRF7−/− mice showed initial control of infection but then rapid progression of paralysis and death by day 7–8 without virus clearance.
Results
Effect of SINV infection on levels of IRF3 and 7 mRNAs
To determine how SINV infection and IRF deficiency affected the induction of IRFs, we assessed the levels of Irf3 and Irf7 mRNAs in brains (Fig. 1a) and spinal cords (Fig. 1b) after infection. Irf3 mRNA was not induced by infection in either WT or IRF7−/− mice, while Irf7 mRNA was quickly and similarly upregulated in both the brains and spinal cords of WTB6 and IRF3−/− mice with a peak at 5 days after infection.
Fig. 1.

Effect of SINV infection on the expression of Irf3 and Irf7 mRNAs in the nervous system. WT, IRF3−/− and IRF7−/− mice were infected intracranially with 103 p.f.u. SINV TE. Levels of Irf3 and Irf7 mRNAs were measured in the brain (a) and spinal cord (b) by qRT-PCR. The primers used for the detection of Irf3 mRNA (IDT) can detect residual transcripts in IRF3−/− mice. Data normalized to gapdh are presented as the mean fold change relative to uninfected WT mice±SEM of three mice/time point/group.
Effects of combined and single IRF deficiencies on clinical outcome from SINV encephalomyelitis
To determine the effects of individual and combined IRF deficiencies on the outcome of SINV encephalomyelitis, we inoculated 4–6 week-old IRF3−/−, IRF7−/−, IRF3/7−/− and IRF3/5/7−/− mice with 103p.f.u. of the TE strain of SINV for comparison with similarly infected WTB6 mice and mice deficient in type I IFN signalling due to lack of the α chain of the IFN receptor (IFNAR−/−) (Fig. 2). WT mice developed mild clinical signs, including abnormal hind limb movement (Fig. 2a), and lost approximately 5 % of their initial body weight (Fig. 2b), but recovered by 14 days after infection with 100 % survival (Fig. 2c). In contrast, mice deficient in type I IFN signalling (IFNAR−/−) or mice deficient in the induction of type I IFN (IRF3/5/7−/−) had a rapid onset of disease (Fig. 2a) and died within 4 days (Fig. 2c; median survival=3 days). Mice dually deficient in IRF3 and IRF7 (IRF3/7−/−) survived somewhat longer (median survival=4 days), but mortality was also 100 % (Fig. 2c). IRF7−/− mice developed neurological signs at the same time as WT mice, but paralysis (Fig. 2a) and weight loss (Fig. 2b) were accelerated, and they died a median of 8 days after infection (Fig. 2c). IRF3−/− mice developed abnormal gaits that had not resolved 3 weeks after infection (Fig. 2a) and lost 15–20 % of their initial body weight. However, all mice survived (Fig. 2c) and their body weight eventually surpassed that of WT mice (Fig. 2b). Thus, IRFs 3, 5 and 7 separately and together determined the outcome from SINV encephalomyelitis. Triple deficiency of IRF3, 5 and 7 and double deficiency of IRF3 and 7 had effects that were equivalent to those from lack of IFNAR, while IRF3 was necessary for complete recovery and IRF7 was required for survival.
Fig. 2.

IRF7, but not IRF3, is required for survival following SINV infection. WT (n=19), IFNAR−/− (n=24), IRF3/5/7−/− (n=15), IRF3/7−/− (n=32), IRF3−/− (n=19) and IRF7−/− (n=19) 4–6-week-old male and female mice were infected intracranially with 103p.f.u. SINV TE. Mice were monitored daily for signs of clinical disease (a, c) and weight loss (b). (a) The clinical scale was: 0, no signs of disease; 1, abnormal hind limb and tail posture, ruffled fur and/or hunched back; 2, unilateral hind limb paralysis; 3, bilateral hind limb paralysis or full body paralysis; 4, dead. The data are presented as the mean±SEM. (b) Weights are presented as the mean±SEM percentage change in initial weight for each time point/strain. (c) Survival was assessed using a Kaplan–Meier curve and the log-rank test. The mean day of death was 3 for IFNAR−/− and IRF3/5/7−/− mice, 4 for IRF3/7−/− mice and 8 for IRF-7−/− mice. The data were pooled from three independent experiments.
Effect of combined and single IRF deficiencies on CNS virus replication and clearance
To determine how IRF deficiency affected the control of CNS virus replication and clearance, we measured the quantities of infectious virus in the brains and spinal cords (Fig. 3a) of infected mice by plaque formation in BHK cells. In WT mice, SINV p.f.u. peaked at 3 days in the brains and spinal cords and was cleared to below the level of detection by 9 days. In contrast, IRF 3/5/7−/− and IRF3/7−/− mice failed to control virus replication in either the brain or spinal cord, with 10–100-fold higher titres by 48 h. In IRF3−/− mice, the brain virus titres were higher than those for WT mice 24 h after infection, but infectious virus was cleared similarly. In the spinal cord, both the peak replication and clearance of infectious virus were similar to those for the WT. In IRF7−/− mice, the brain titres remained high and had not decreased by 7 days, when the mice began to die. The levels of infectious virus in the spinal cords of IRF7−/− mice were similar to that in WT mice at 3 days, but the initiation of clearance was delayed.
Fig. 3.

SINV replication and antibody responses in brain and spinal cord. WT, IRF3/5/7−/−, IRF3/7−/−, IRF3−/− and IRF7−/− mice were infected intracranially with 103p.f.u. TE. The quantities of infectious virus in brain and spinal cord (a) homogenates were determined by plaque assay on BHK cells. The data were pooled from two independent experiments and are presented as the mean±SEM of 6–9 mice for each time point per strain. SINV E2 genome copies in brain and spinal cord (b) were determined by RT-qPCR. RNA levels are expressed as the mean SINV copy number (log10)±SEM of 6–9 mice for each time point per strain. ****P<0.0001, WT vs IRF7−/−; ^^^^P<0.0001, IRF3−/− vs IRF7−/−, Tukey’s multiple comparison test. SINV-specific IgM (c) and IgG (d) in serum, brain and spinal cord were measured by EIA and are expressed as the mean optical density (OD) +/−SEM for three mice/time point. *P<0.05; **P<0.01; ***P<0.001 IRF3−/− vs WT.
Viral RNA in the brain peaked by 3 days after infection with subsequent decreases for WT and IRF3−/− mice, but not IRF7−/− mice (Fig. 3b). Viral RNA levels were similar in the spinal cords of WTB6, IRF3−/− and IRF7−/− mice through 5 days, with subsequent evidence of clearance in WT and IRF3−/− mice, but not in IRF7−/− mice, at 7 days (Fig. 3b). These data show that although combined deficiency leads to uncontrolled virus replication (Fig. 3a), individual deficiencies of IRF3 and IRF7 have a limited effect on early control of SINV replication in the CNS. However, IRF7 but not IRF3 is required for efficient clearance of infectious virus and viral RNA from both the brain and spinal cord, as well as survival.
Effect of IRF3 or IRF7 deficiency on the production of SINV-specific antibody
Because virus-specific antibody produced within the CNS is important for virus control and clearance [33, 34], we examined the levels of SINV-specific IgM (Fig. 3c) and IgG (Fig. 3d) antibody in serum, brain and spinal cord after SINV infection of WT, IRF3−/− and IRF7−/− mice. SINV-specific IgM was detected in serum by day 3 with a peak at day 5 and was detected in the CNS between day 5 and 7 for all groups. IgG was detected by day 5 in serum and day 7 in the CNS in all groups. IRF3−/− mice developed higher levels of SINV-specific IgG in the brain than WT mice by 10–14 days after infection. The levels of antibody early after infection were not different for IRF7−/− mice than for WT and IRF3−/− mice.
Effect of IRF3 or IRF7 deficiency on levels of IFN
IRF3 and IRF7 work in concert to transcriptionally direct type I IFN expression. IRF3 is important for the early induction of IFNβ, while IRF7 is important for the subsequent amplification of the IFN response with the induction of multiple IFNαs [15, 35]. To determine the effect of deficiency in IRF3 or IRF7 on IFN induction after SINV infection, sera and brain homogenates were analysed by enzyme immunoassay for IFNα, IFNβ and IFNγ proteins (Fig. 4a), and brain (Fig. 4b), spinal cord (Fig. 4c) and cervical lymph node (Fig. 4d) homogenates were assessed by qRT-PCR for Ifna1, Ifnb and Ifng mRNAs.
Fig. 4.

Effect of IRF3 and IRF7 deficiencies on the expression of IFN-α, IFN-β and IFN-γ after SINV infection. WT, IRF3−/− and IRF7−/− mice were infected intracranially with 103p.f.u. SINV TE. (a) The protein levels of IFN-α, IFN-β and IFN-γ in the brain were measured by EIA. The expression of Ifna1, Ifnb and Ifng mRNAs was measured by qRT-PCR in the brain (b), spinal cord (c) and draining cervical lymph nodes (d). The data are presented as the mean±SEM of 3–6 mice per strain per time point. Significant differences at each time point determined by Tukey’s (0–7 days p.i.) or Bonferroni’s (9 and 14 days p.i.) post-test are shown (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, WT compared to IRF7−/−; # P<0.05, ## P<0.01, ### P<0.001, #### P<0.0001, WT compared to IRF3−/−; ^P<0.05, ^^P<0.01, ^^^P<0.001, ^^^^P<0.0001, IRF3−/− compared to IRF7−/−).
In the CNS, the levels of Ifnb mRNA and IFNβ protein in the brains of WT and IRF3−/− mice were similar, but were higher in IRF7−/− mice 3–5 days p.i. (Fig. 4a,b). In the spinal cord, the level of Ifnb mRNA was higher in IRF7−/− mice than WT mice 3 days p.i. (Fig. 4c). In the periphery, serum levels of IFNβ were below the level of detection (data not shown) and lymph node Ifnb mRNA levels (Fig. 4d) did not change during infection.
In the CNS, Ifna1 mRNA expression and IFNα protein levels peaked at 3 days p.i. in the brains and spinal cords of WT mice and then steadily decreased to baseline levels at 7 days p.i. (Fig. 4a–c) when infectious virus had been cleared (Fig. 3a). Levels were generally lower in IRF3−/− and IRF7−/− mice and the induction of both Ifna1 mRNA and IFNα protein was slower in IRF7−/− mice, with an increase first detected at day 3 in the brain (Fig. 4a, b) and day 5 in the spinal cord (Fig. 4c). In the periphery, serum levels of IFNα were below the level of detection (data not shown) and the lymph node Ifna1 mRNA levels did not change (Fig. 4d).
Therefore, type I IFN was induced in the CNS, but not the periphery of all mice after intracerebral infection with SINV. IFN generally peaked at 3 days p.i., with the highest levels of IFNα in WT mice and the highest levels of IFNβ in IRF7−/− mice. Overall, compared to WT mice, IRF3 deficiency had little effect on the synthesis of IFNβ and modest effects on the synthesis of IFNα in the CNS, while IRF7 deficiency led to slower production of IFNα and higher production of IFNβ. Therefore, type I IFN was induced in the CNS despite the absence of IRF3 or IRF7, although the types of IFN produced differed.
We also investigated the expression of IFNγ, as type II IFN production and the T cell response to infection can be influenced by type I IFN [36]. In the brain, IFNγ protein and Ifng mRNA levels peaked at 5 days p.i. and were more highly expressed in IRF3−/− mice than WT mice (Fig. 4a, b). IRF3−/− mice also had the highest levels of Ifng mRNA in the spinal cord (Fig. 4c).
Effect of IRF3 or IRF7 deficiency on the induction of IFN-stimulated antiviral genes
IFN signalling directs the expression of hundreds of genes involved in the antiviral response [37], of which a subset is known to have a role in controlling alphavirus infection [38–42]. To determine whether IRF deficiency with altered proportions of IFNα and IFNβ in the CNS affected the induction of representative antiviral IFN-stimulated genes (ISGs) after infection, we assessed the brain levels of ISG mRNAs by RT-qPCR (Fig. 5). By 3 days p.i., Isg15, Isg56 and Isg54 were induced in the brains of all infected mice, with the highest levels in IRF7−/− mice (Fig. 5a). Similarly, ISGs induced by both type I and type II IFNs, Zc3hav1 and Rsad2, were induced in the brains of all infected mice, with the highest levels being observed in the brains of IRF7−/− mice (Fig. 5b).
Fig. 5.

Effect of IRF3 and IRF7 deficiencies on the expression of ISGs in the nervous system after SINV infection. WT, IRF3−/− and IRF7−/− mice were infected intracranially with 103p.f.u. SINV TE. The expression of (a) IFNα/β-responsive and (b) IFNα/β and IFNγ-responsive antiviral protein gene mRNAs in the brain was measured by qRT-PCR, normalized to Gapdh and compared to uninfected WT controls. The data were pooled from two independent experiments and are presented as the mean±SEM of six mice for each time point per strain. **P<0.01, ***P<0.001, ****P<0.0001 WT vs IRF7−/−; # P<0.05, ### P<0.001, #### P<0.0001 WT vs IRF3−/−; ^P<0.05, ^^P<0.01, ^^^P<0.001, ^^^^P<0.0001 IRF3−/− vs IRF7−/−, Tukey’s multiple comparison test.
Effect of IRF3 or IRF7 deficiency on induction of innate chemokines and cytokines
In addition to inducing IFN and ISGs, viral infection induces the early expression of chemokines and cytokines through similar innate signalling pathways that may involve IRF3 or 7 [43]. Chemokines attract inflammatory cells expressing the appropriate receptors into the CNS in response to infection and can also induce signalling that has detrimental effects in the nervous system [44]. In the brain (Fig. 6a) and spinal cord (Fig. 6b), Ccl2, Ccl5, Cxcl10 and Cxcl13 mRNAs were higher in IRF7−/− mice compared to WT mice. While levels of Ccl5 and Cxcl10 mRNAs in the brain and spinal cord were decreasing by 7 days p.i., Ccl2 mRNA expression was still increasing in the brains and remained high in the spinal cords of IRF7−/− mice. Cxcl13 mRNA levels were orders of magnitude higher in IRF7−/− mice than in WT or IRF3−/− mice from 3 to 7 days p.i. in both the brain and the spinal cord.
Fig. 6.

Effect of IRF3 and IRF7 deficiencies on the expression of chemokine and cytokine mRNAs and TNFα protein in the nervous system after SINV infection. WT, IRF3−/− and IRF7−/− mice were infected intracranially with 103p.f.u. SINV TE. The levels of expression of Ccl2, Ccl5, Cxcl10 and Cxcl13 chemokine mRNAs (a, b) and Il1b, Tnf, Il6 and Il10 cytokine mRNAs (c, d) were measured by qRT-PCR in the brain (a, c) and spinal cord (b, d), normalized to Gapdh and compared to uninfected WT controls. (e) Levels of TNFα protein in brain and spinal cord. The data were pooled from two independent experiments and are presented as the mean±SEM of six mice for each time point per strain. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 WT vs IRF7−/−; # P<0.05, ## P<0.01, ### P<0.001, #### P<0.0001 WT vs IRF3−/−; ^P<0.05, ^^P<0.01, ^^^P<0.001, ^^^^P<0.0001 IRF3−/− vs IRF7−/−; Tukey’s multiple comparison test (0–7 days p.i.), Bonferroni’s multiple comparison test (9 and 14 days p.i.).
To determine the effects of IRF deficiency on cytokine induction, we analysed the expression of the mRNAs for the innate cytokines IL-1β, TNFα, IL-6 and IL-10 in the brain (Fig. 6c) and spinal cord (Fig. 6d). In the brain, the expression of Il1b mRNA was similar, with the highest expression in IRF3−/− mice, while the expression of Tnf, Il6 and Il10 mRNAs tended to be higher in IRF3−/− and IRF7−/− mice compared to WT mice. All cytokine mRNAs except Tnf showed evidence of appropriate regulation, with peak expression at 5 days p.i. followed by a rapid decrease by 7 and 9 days p.i. In IRF7−/− mice, however, Tnf mRNA continued to increase at 7 days p.i. in both the brain and spinal cord, which was concurrent with the development of paralysis in these mice. Compared to WT and IRF3−/− mice, the levels of TNFα protein were significantly higher in the brains of IRF7−/− mice at 5 days p.i. and 4 to 5-fold higher in the spinal cord at all times examined (Fig. 6e).
Therefore, IRF3- and IRF7-deficient mice developed a robust expression of cytokines and chemokines in the CNS in response to SINV infection, but IRF7−/− mice failed to limit expression of TNFα, Ccl2 and Cxcl13.
CNS inflammation and cell death in the CNS of SINV-infected mice
To determine whether the inflammatory response to infection was influenced by IRF3 or IRF7 deficiency in the light of differences in chemokine expression, brain (Fig. 7a) and spinal cord (Fig. 7b) sections were stained with haematoxylin and eosin (H and E) and examined for inflammatory infiltrates. Inflammatory cells were primarily noted around vessels as perivascular cuffs and within the tissue parenchyma of infected mice and were minimal in mock-infected controls. Inflammation was quantified on coded slides using previously established scoring systems [45, 46]. In the brain, the scores for mock-infected mice were typically less than 1, while the brains of infected mice had scores greater than 2, with IRF3−/− brains having the highest scores at both 5 and 7 days (Fig. 7c). The inflammatory scores for spinal cords from mock-infected mice were all 0 regardless of mouse strain. At 5 days after infection, the scores were significantly higher in spinal cords from IRF3−/− and IRF7−/− mice compared to WT mice, and at 7 days the scores were highest in IRF3−/− mice (Fig. 7d).
Fig. 7.
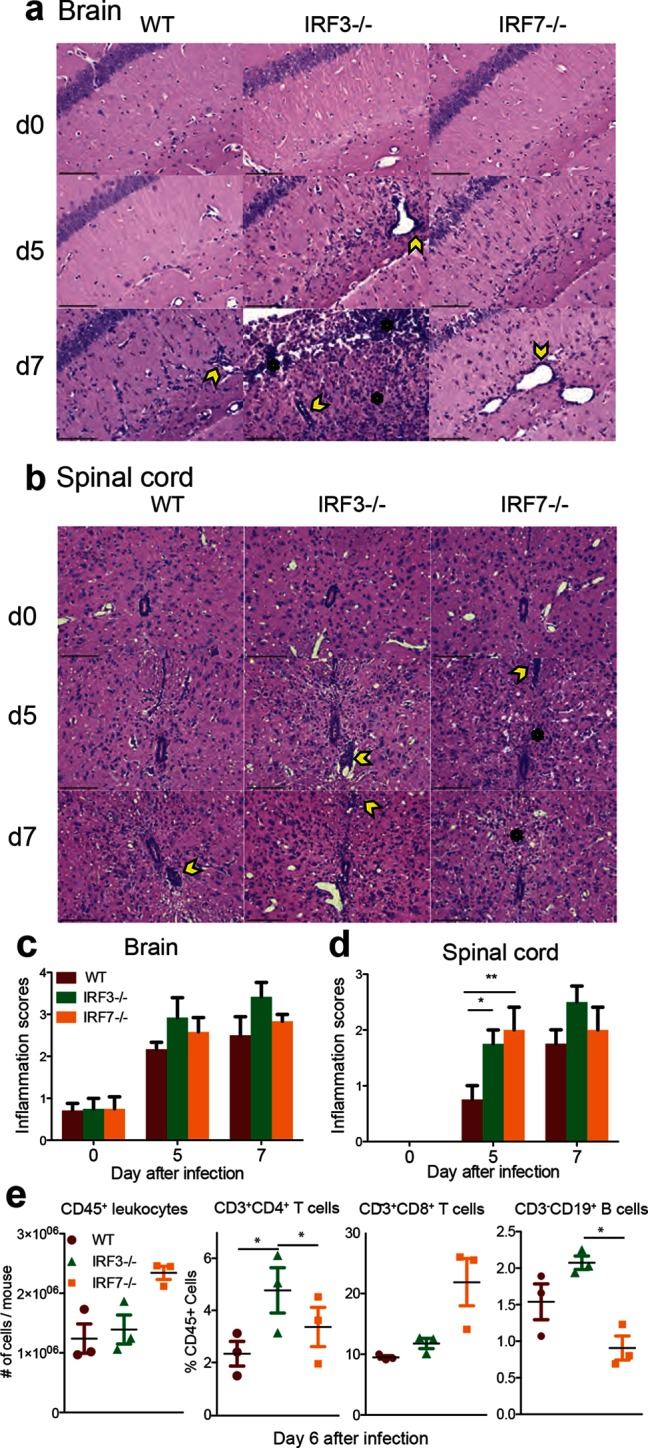
Effect of IRF3 or IRF7 deficiency on brain and spinal cord pathology and inflammation after SINV infection. WT, IRF3−/− and IRF7−/− mice were infected intracranially with 103p.f.u. SINV TE. Representative H and E-stained sections of brain (a) and spinal cord (b) from uninfected (d0) and SINV-infected WT, IRF3−/− and IRF7−/− mice at 5 and 7 days after infection. Two hundred× magnification; the scale bar represents 100 µm. The yellow arrowheads denote the perivascular cuffs of inflammatory cells and the black asterisks denote inflammatory cells infiltrating the parenchyma. For quantification, slides were coded and scored for inflammation using a 3 (4) point scale for brain (c) and a 2 (3) point scale for spinal cord (d). The data represent the results of two independent experiments, totalling four mice per strain per time point. *P<0.05, **P<0.01, Tukey’s multiple comparison test. Identification by flow cytometry of cells infiltrating the brain 6 days after infection (e). Numbers of CD45+ cells/brain and the percentages of CD45+ cells that were CD3+CD4+ T cells, CD3+CD8+ T cells or CD3-CD19+ B cells. The data are presented as the mean+/-SEM from three independent experiments with the cells pooled from four to six mice. *P<0.05.
To better characterize the inflammatory response, the leukocytes infiltrating the CNS 6 days after infection were analysed by flow cytometry (Fig. 7e). The numbers of CD45+ cells were not significantly different, but in IRF3−/− mice a higher proportion of these cells were CD4+ T cells compared to WT and IRF7−/− mice and CD19+ B cells compared to IRF7−/− mice. Although not significant, IRF7−/− mice tended to have more infiltrating CD45+ cells with a higher proportion of CD8+T cells than WT or IRF3−/− mice.
To determine whether prolonged neurological deficits in IRF3−/− mice and mortality in IRF7−/− mice were associated with cell death in the CNS, the brains (Fig. 8a) and spinal cords (Fig. 8b) of infected mice were examined by terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining. TUNEL-positive cells were rarely present in the brains and spinal cords of mock-infected mice, but were readily identified in tissues from SINV-infected mice. The numbers of TUNEL-positive cells (brown staining) were quantified in SINV-infected mouse brains (Fig. 8c) and spinal cords (Fig. 8d). IRF7−/− mice had significantly more TUNEL-positive cells in both the brain and spinal cord than WT or IRF3−/− mice. Many, but not all, of the TUNEL-positive cells were morphologically consistent with neurons, suggesting the death of neurons as well as bystander cells in the brains and spinal cords of IRF7−/− mice. Cell death was not correlated with the amount of inflammation, as IRF3−/− mice typically showed the most inflammation, while IRF7−/− mice had the largest number of TUNEL-positive cells.
Fig. 8.
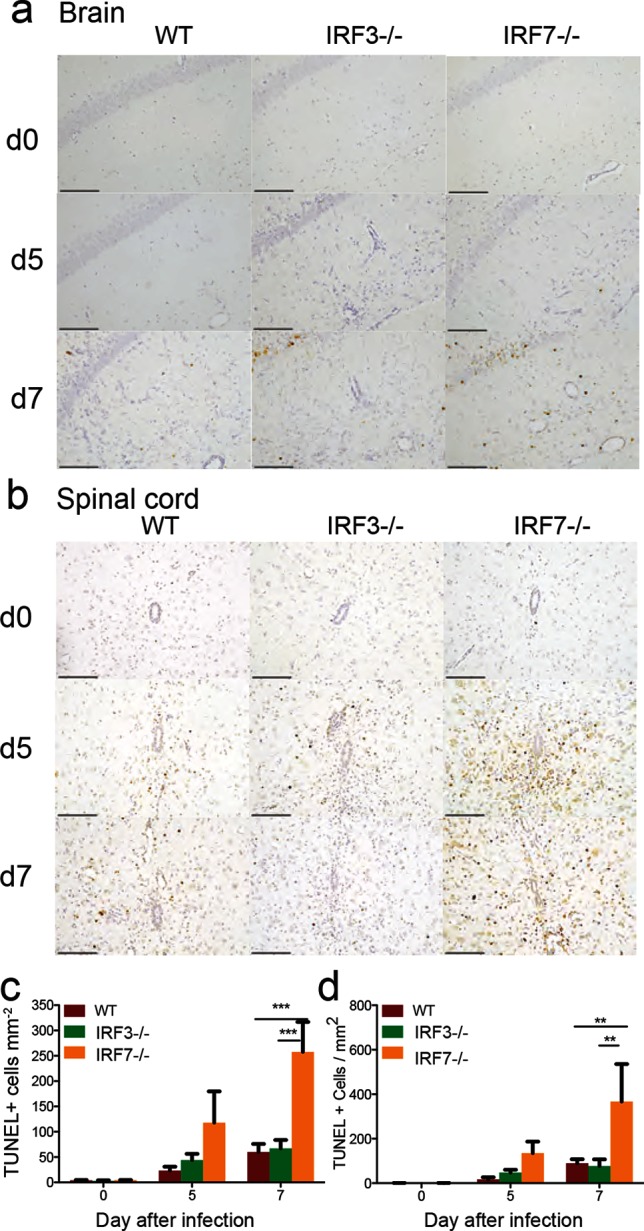
Effect of IRF3 or IRF7 deficiency on SINV-induced cell death in brain and spinal cord. WT, IRF3−/− and IRF7−/− mice were infected intracranially with 103p.f.u. SINV TE. Representative TUNEL-stained sections of brain (a) and spinal cord (b) from uninfected (d0) and SINV-infected WT, IRF3−/− and IRF7−/− mice at 5 and 7 days p.i. Two hundred× magnification; the scale bar represents 100 µm. TUNEL-positive cells have dark brown nuclear staining. The numbers of TUNEL-positive cells were quantified on coded slides from brains (c) and spinal cords (d). The data represent the results of two independent experiments, totalling four mice per strain per time point. **P<0.01, ***P<0.001, Tukey’s multiple comparison test.
Discussion
IRFs are transcription factors that regulate the induction of type I IFN and ISGs in response to viral infection, as well as other cellular responses [47, 48]. In these studies of alphavirus encephalomyelitis, we have shown that the outcome of infection in mice with single and combined deficiencies of IRFs 3, 5 and 7 is indicative of distinct roles in the pathogenesis of CNS infection. Lack of IRFs 3, 5, and 7 or IRFs 3 and 7 together resulted in an outcome that was comparable to the loss of IFN signalling in IFNAR−/− mice, with failure to control virus replication and rapid death. However, the effects of single deficiencies of IRF3 or IRF7 revealed the unique contributions of these two IRFs to the pathogenesis of viral encephalomyelitis. IRF3−/− mice survived with persistent neurological disease, while IRF7−/− mice developed rapidly fatal paralysis. Both IRF3−/− and IRF7−/− mice had lower levels of IFNα in the CNS than WT mice, but the levels of IFNβ, ISG mRNAs and antiviral antibody were similar or higher, indicating that the differences in outcome were not due to deficiencies in the type I IFN or antibody responses in the CNS. IRF3-deficient mice had somewhat more inflammation, with higher proportions of CD4+ T cells and B cells than WT mice, but similar amounts of cell death in the CNS. IRF7-deficient mice differed from both WT and IRF3−/− mice in showing higher levels of IFNβ, TNFα and Cxcl13, failing to downregulate the expression of brain Ccl2 and brain and spinal cord Tnf and Cxcl13, and developing more extensive cell death. Thus, IRF3 is important for the resolution of neurological disease, while IRF7 is important for the control of inflammatory cytokine production and neuronal survival.
IRF3 and IRF7 are expressed in the normal mouse CNS at low levels [11, 49, 50]. IRF3 is constitutively present in the cytoplasm of most cells, while IRF7 must be induced [51–53]. In response to SINV infection, CNS levels of Irf3 mRNA did not change, and levels of Irf7 were similarly increased in both IRF3−/− and WT mice, consistent with the previously reported virus-induced IRF7 expression by microglia, neurons and infiltrating inflammatory cells that is dependent on STAT2 [49, 50].
Both IRF3 and IRF7 are activated by phosphorylation, and their functions with respect to the induction of IFN and ISGs are overlapping [12, 54–58], so that cells deficient in both IRF3 and IRF7 do not produce IFN [16]. Mice with combined deficiencies of IRFs 3, 5 and 7 or IRFs 3 and 7 had a phenotype similar to that seen in IFNAR−/− mice [9]. SINV infection was rapidly fatal, with failure to control virus replication, indicating the importance of early induction of IFN for survival. Similar outcomes have been observed for IRF3/5/7−/− mice after infection with WNV, Zika virus and murine norovirus [24, 59], but mice infected with dengue virus survive [60]. Previous studies with IRF3/7−/− mice have had more variable outcomes, with uniformly fatal encephalitis after CNS infection with CHIKV [27, 61], WNV [28] and Kunjin virus [62], as well as SINV (Fig. 2), but not after peripheral infection with dengue virus [60], murine norovirus [63], or oropouche virus [64], suggesting greater importance for the response to CNS infection.
IRF3−/− mice develop fatal disease after infection with WNV, Sendai and encephalomyocarditis viruses and CNS infection with HSV, but survive CNS infection with Theiler’s murine encephalomyelitis virus (TMEV) and peripheral infection with herpes simplex, Kunjin, chikungunya and Oropouche viruses [27, 29–31, 61, 62, 64–66]. IRF3 was not required for survival after SINV CNS infection, or for the induction of IFN and ISGs, or for the early control of virus replication. However, compared to WT mice, IRF3 deficiency was associated with a higher proportion of infiltrating CD4+ T cells and B cells, and higher levels of antiviral IgG and IL1b mRNA, consistent with a role for IRF3 in regulating B cell responses and microglial expression of proinflammatory genes [11, 67, 68]. IRF3 also plays a role in the differentiation of T cells by influencing the production of IFN by antigen-presenting cells and the induction of Th1 and Th17 cells, and by suppressing the production of IL-17, granzyme B and IFN-γ and inhibiting the establishment of T cell memory [66, 69, 70]. These effects on T cell function have been implicated in persistent signs of neurological impairment after TMEV infection associated with failure to clear viral RNA [66]. However, IFN-γ responses are robust (Fig. 4a-c) and SINV persistence is not greater in IRF3−/− than WT mice (Fig. 3b). Nevertheless, Th17 cells can play an immunopathogenic role in SINV infection of the CNS [71], so an in-depth functional characterization of the inflammatory cells in IRF3−/− mice early and late after infection will be of interest.
A function of IRF3, in addition to its role as a transcription factor for the induction of IFNα/β and ISGs, is as a pro-apoptotic factor [72]. IRF3 has a BH3 domain and can be activated by a distinct ubiquitination pathway to interact with the proapoptotic protein Bax, translocate to mitochondria and trigger apoptotic cell death, thus inhibiting virus replication and preventing persistence [65, 73–75]. When ubiquitinated, IRF3 can also induce the death of virus-infected cells through the RIG-I-like receptor-induced apoptosis pathway [65]. The importance of these mechanisms in the CNS is not clear. Previous studies have shown that, independent of type I IFN, IRF3 protects neurons from damage induced by infection with western equine encephalitis virus [76] and both cell death and virus clearance in the CNS of IRF3−/− mice infected with SINV were similar to those observed in WT mice (Figs 3 and 8).
IRF7 is a multifunctional regulator that can be both a transcriptional activator and repressor. In the cytoplasm IRF7 is associated with MyD88, TRAF6 and IKK and can be activated through both TLR engagement and MyD88-independent viral signalling pathways [29]. IRF7 is not only important for the induction of a subset of IFNα genes [15], but also for the expression of several mitochondrial and DNA repair genes and the generation of the anti-viral effector CD8+ T cell repertoire [77, 78]. Importantly, IRF7 also negatively regulates NF-κB signalling by binding IκBβ [79, 80]. Genes (e.g. Ccl2, Tnf, Cxcl13) with continued increases during the late response to infection are NF-κB-induced genes [81], suggesting that an important consequence of the lack of IRF7 may be failure to control NF-κB signalling in the CNS.
It is not clear why IRF7 is required for the survival of mice with SINV CNS infection, as well as other neurotropic virus infections, including WNV, encephalomyocarditis virus and HSV, but not for systemic infection with murine norovirus, lymphocytic choriomeningitis virus (LCMV), human metapneumovirus, Oropouche virus or CHIKV [27, 29, 32, 61, 63, 64, 77, 82, 83]. In WT mice infected with SINV by intracerebral inoculation, IFN is induced in the CNS, but not in peripheral tissue, which is consistent with limited replication outside the CNS. In IRF7-deficient mice, the levels of IFNα, but not IFNβ, were decreased in brain and spinal cord, and, as previously observed after CNS infection with LCMV [84], the induction of ISGs was the same or higher compared to that for WT mice, and initial control of virus replication was similar. Furthermore, previous studies have shown that the inhibition of SINV clearance alone does not lead to neuronal death or fatal outcome [45, 85–87], indicating a role for IRF7 in regulating neuronal cell death by autonomous or non-cell autonomous processes. Although neurons do not produce IFN in response to alphavirus infection in vitro, the expression of functionally mature IRF7 increases with neuronal maturation [88]. Knock down of IRF7 expression in EBV-transformed B cells increases apoptosis, so IRF7 deficiency may increase the likelihood of SINV-induced neuronal cell death [89]. IRF7 deficiency may also increase susceptibility to neurotoxic factors such as TNFα produced by glial cells or inflammatory cells infiltrating the CNS from the periphery. TNFα-deficient mice have improved survival after infection with a virulent neuroadapted SINV (NSV), [90] suggesting a role for this cytokine in determining outcome.
TNFα can be produced by astrocytes, microglia and neurons, in addition to infiltrating immune cells, and production is highly regulated [91–93]. TNFα protein levels were substantially higher in both the brain and particularly the spinal cord of SINV-infected IRF7−/− mice compared to WT or IRF3−/− mice, without evidence of downregulation of mRNA production (Fig. 6). TNFα can lead to neuronal cell death both directly and indirectly, with multiple levels of regulation [94–97]. TNFα signals through either of two receptors, TNFR1 and TNFR2, both of which can lead to NF-κB activation to promote inflammation [98]. TNFR1 is ubiquitously expressed, while TNFR2 is restricted to immune cells, endothelial cells and some neurons, including motor neurons [99]. Shut off of host protein synthesis in SINV-infected neurons may increase susceptibility to TNFα-mediated cell death by decreasing levels of c-FLIP, an inhibitor of caspase-8 activation [100, 101]. TNFα also decreases astrocyte production of the glutamate transporter GLT-1 necessary for preventing toxic increases in parenchymal glutamate after synaptic release [90]. IFIT1, which is also highly expressed by IRF7−/− mice (Fig. 5a), can inhibit TNFα-mediated apoptosis in the liver and the induction of TNFα in macrophages [102, 103], but its role in the CNS is unknown. Failure to control TNFα production in the spinal cord, where it is directly neurotoxic and also potentiates glutamate excitotoxicity for motor neurons [97], may lead to the rapidly progressive paralysis observed in IRF7−/− mice, despite the production of IFIT1, and it is a focus of future investigation.
TNFα also induces the neuronal production of chemokines, including CCL2 [104, 105]. Ccl2 is typically upregulated during CNS virus infections and is a potent chemoattractant for Ly6Chi CCR2-bearing inflammatory monocytes [106–109] that can differentiate during viral encephalomyelitis into M1-type pro-inflammatory macrophages that express TNFα [108, 110]. Transgenic mice expressing CCL2 in the CNS infected with an attenuated strain of mouse hepatitis virus develop fatal encephalomyelitis associated with increased numbers of MI/M2-type macrophages, regulatory T cells and delayed virus clearance [111]. Therefore, increased production of CCL2 may contribute to the fatal outcome in SINV-infected IRF7−/− mice by promoting a neurotoxic glial response that includes the production of TNFα.
CXCL13 is a lymphoid chemokine that is induced in follicular dendritic cells by TNFα and attracts B and T cells expressing CXCR5 during the development of secondary lymphoid tissue and the formation of germinal centres [112, 113]. CXCL13 is also expressed in multiple non-lymphoid organs, including the CNS, where it sustains and is produced by pathogenic CD4+ T cells differentiated in the presence of transforming growth factor β during autoimmune disease [114–116], In the CNS, CXCL13 is produced by spinal cord neurons after nerve ligation and by microglial cells and macrophages during inflammation, and can be downregulated by IFN [104]. Expression during viral infection promotes the accumulation of memory B cells and antibody-secreting cells [117–119], but has not been associated with more CNS B cells or antibody production during the acute phase of SINV infection. Increased levels of CXCL13 compared to WT mice have previously been reported in IRF7−/− mice infected with NSV, but a direct neurotoxic role for this chemokine was not identified and CXCL13 deficiency did not improve outcome [120].
In summary, fatal outcome in SINV-infected IRF7−/− mice was not due to the failure to produce type I IFN or induce an antiviral response in the CNS. Uncontrolled production of TNFα during the inflammatory phase of infection is postulated to lead to rapidly progressive paralysis associated with extensive cell death throughout the CNS.
Methods
Mice, virus and infection
C57BL/6J (B6) WT mice were purchased from Jackson Laboratories. IRF3−/− [16], IRF7−/− [29], IRF3/7−/− [28] and IRF3/5/7−/− [24] mice on a B6 background were obtained from Michael Diamond (Washington University, St Louis, MO, USA). IFNAR−/− mice [121] on a B6 background were obtained from Howard Young (National Cancer Institute, Frederick, MD, USA). All mice were bred in-house in specific pathogen-free facilities.
The TE strain of SINV [122] was grown and assayed by plaque formation in BHK-21 cells. For infection, 4–6-week-old male and female mice were inoculated intracranially with 103p.f.u. TE in 20 µl phosphate-buffered saline (PBS). Mice were monitored daily for weight change, evidence of neurological disease and death. The disease scoring system used was: 0, no signs of disease; 1, abnormal hindlimb and tail posture, ruffled fur and/or hunched back; 2, unilateral hind-limb paralysis; 3, bilateral hind limb or full body paralysis; 4, dead.
For tissue collection, mice were anaesthetized with isoflurane, bled by cardiocentesis and perfused with ice-cold PBS. Brains, spinal cords and cervical lymph nodes were collected, snap frozen and stored at −80°C. Tissues were homogenized in ice-cold PBS to make 10–20 % (wt/vol) homogenates and clarified by centrifugation. Infectious virus was assayed by plaque formation on BHK-21 cells and the data are presented as the mean of the log10 value of p.f.u. g−1 of tissue ±SEM. For the purposes of graphing and statistics, samples without detectable infectious virus were assigned a value halfway between the limit of detection and 0. All experiments were performed according to protocols approved by the Johns Hopkins University Institutional Animal Care and Use Committee.
Gene expression analysis using real-time PCR
RNA was isolated from frozen tissue homogenates using the RNeasy Lipid Mini RNA Isolation kit (Qiagen) and quantified using a nanodrop spectrophotometer. Residual genomic DNA was removed (TURBO DNA-free kit; Ambion) prior to cDNA synthesis for analysis of Ifnb, Ifna1 and Isg15 gene expression. For the analysis of IFN, cytokine and chemokine gene expression cDNA was synthesized using the High Capacity cDNA Reverse Transcription kit (Life Technologies), 0.5–2.5 µg RNA and random primers according to the manufacturer’s instructions. Quantitative real-time PCR was performed using 2.5 µl cDNA, PrimeTime gene expression assays (Integrated DNA Technologies) and 2× EagleTaq PCR Mix (Roche). Gapdh mRNA levels were determined using the rodent primer and probe set (Applied Biosystems). All reactions were run on an Applied Biosystems 7500 real-time PCR machine with the following conditions: 50°C for 2 min, 95°C for 10 min, 95°C for 15 s and 60°C for 1 min for 50 cycles. The target gene Ct value was normalized to the Ct value of Gapdh. This normalized value was used to calculate the gene expression level relative to the average of the uninfected WTB6 control value.
SINV RNA copies were measured by qRT-PCR using TaqMan Universal PCR Master Mix (Roche), TaqMan probe [5′–6-carboxyfluorescein (FAM)-CGCATACAGACTTCCGC CCAGT–6-carboxytetramethylrhodamine (TAMRA)-3′] (Applied Biosystems) and primers to the SINV E2 gene (forward, 5′-TGGGACGAAGCGGACGATAA-3′; reverse, 5′-CTGCTCCGCTTTGGTCGTAT-3′) on a 7500 Fast Real-Time PCR System for 50 cycles. The results were analysed using Sequence Detector software, version 1.4 using a standard curve made from 10-fold dilutions of the SINV subgenomic region RNA derived from a pGEM-3Z plasmid and normalized to endogenous mouse Gapdh.
Enzyme immunoassays (EIAs)
Commercial EIAs were used to measure IFNα and IFNβ (Verikine ELISA kits, PBL Interferon Source, Piscataway, NJ, USA) and TNFα and IFNγ (Mouse Ready-SET-Go! kits, Ebioscience, San Diego, CA, USA) following the manufacturers’ instructions. Sera and tissue homogenates were tested undiluted (IFNα, IFNβ and TNFα) or at 1 : 2 dilution (IFNγ) in duplicate. The sensitivity was 125–4000 pg g−1 for IFNα, 156–10 000 pg g−1 for IFNβ, 313–20 000 pg g−1 for IFNγ and 39–5000 pg g−1 for TNFα.
An in-house EIA for SINV-specific IgM and IgG was performed as previously described [33]. Briefly, Maxisorp (Nunc) 96-well plates were coated with 106p.f.u. PEG-concentrated TE in 50 µl coating buffer (50 mM NaHCO3 pH 9.6) at 4 °C overnight, washed and blocked with 10 % FBS in PBS with 0.05 % Tween20. Samples diluted in blocking buffer (serum, 1 : 100; brain homogenate, 1 : 8; spinal cord homogenate, 1 : 4) were incubated for 2 h and developed with horseradish peroxidase-conjugated anti-mouse IgG or IgM (1 : 1000; Southern Biotech) and TMB substrate (BD Biosciences). The optical density (OD) was read at 450 nm on a Multiskan MCC (Thermo Scientific).
Histology
Uninfected (day 0) and TE-infected mice at 5 and 7 d after infection were perfused with ice-cold PBS followed by 4 % (wt/vol) paraformaldehyde (PFA). Brains were removed, cut into 2 mm coronal slices with an adult Mouse Brain Slicer (Zivic Instruments) and postfixed in 4 % PFA for 18–24 h at 4°C. Spinal columns were trimmed of excess soft tissue, fixed overnight in 4 % PFA at 4 °C, and then decalcified on a rotator for 24 to 36 h in a 10 % sodium citrate/22 % formic acid solution. Spinal columns were washed in ice-cold PBS and cut to isolate the L4–L6 regions. Tissues were embedded in paraffin for sectioning and staining.
H and E-stained sections (10 µm) of brain and spinal cord were coded and scored for inflammation. Brain sections were scored using a 0–3/4 scale as previously described [46]: 0, no detectable inflammation; 1, one to two small inflammatory foci per slide; 2, moderate inflammatory foci in ≤50 % of 10× fields; and 3, moderate to large inflammatory foci in >50 % of 10× fields. An additional point was given for excessive parenchymal cellularity, allowing for a maximum score of 4. Spinal cord sections were scored using a modified 0–2/3 scale as previously described [45]: 0, no detectable inflammation; 1, one to two small inflammatory foci; 2, greater than two inflammatory foci per spinal cord or moderate to marked inflammatory foci. An additional point was given for excessive parenchymal cellularity, allowing for a maximum score of 3.
For TUNEL staining, sections (10 µm) were rehydrated and treated with 1 mg ml−1 proteinase K for 30 min for antigen retrieval. Endogenous peroxidases were quenched in methanol+3 % H2O2 for 5 min, and after immersion in TdT Labeling Buffer for 5 min, sections were stained with TdT Labeling Reaction mix for 60 min at 37 °C. To stop the reaction, slides were immersed in TdT Stop Buffer for 5 min and incubated with streptavidin–HRP for 10 min (TACS 2 TdT kit, Trevigen, Inc). Tissues were developed with 3,3′-diaminobenzidine (Vector Labs) for 8 min, counterstained with haematoxylin, dehydrated and mounted with Permount (Fisher Scientific). Slides were coded, and the whole visible hippocampus on one brain section per mouse or the entire spinal cord cross section was outlined to determine the tissue area using a Nikon Eclipse E600 microscope and StereoInvestigator software (MBF Bioscience). All TUNEL-positive cells were counted within the outlined area to determine TUNEL-positive cells per mm2 tissue.
Flow cytometry
Single-cell suspensions were made from brain and spinal cord tissue homogenized in RPMI+1 % FBS, 1 mg ml−1 collagenase (Roche) and 0.1 mg ml−1 DNase (Roche) using the GentleMACS system (Miltenyi) and isolated on a percoll gradient as previously described [71]. Then 1−2×106 cells were stained with the violet Live/Dead Fixable Cell Stain kit (Invitrogen) in PBS+2 mM EDTA, blocked with rat anti-mouse CD16/CD32 (BD Pharmingen), diluted in PBS+2 mM EDTA, surface-stained for 30 min on ice and resuspended in 200 µl PBS+2 mM EDTA +0.5 % BSA. All antibodies were from BD Pharmingen or eBioscience and were specific for: CD45 (clone 30-F11), CD3 (clone 17A2), CD4 (clone RM4-5), CD8 (clone 53–6.7), and CD19 (clone 1D3). Data were acquired with a BD FACS Canto II using FACS Diva software (version 6.0) and analysed using FlowJo 8.8.7 (TreeStar, Inc.).
Statistics
Statistical analyses were performed using GraphPad Prism 6 software. Time-course studies were analysed by two-way analysis of variance (ANOVA) with Tukey’s multiple comparison post-test comparing WT, IRF3−/− and IRF7−/− mice 0–7 days p.i. For time points later than 7 days p.i., data were analysed by two-way ANOVA with Bonferroni’s post-test, comparing WT and IRF3−/− mice. Survival was assessed using Kaplan–Meier curves and a log-rank test. A P value of <0.05 was considered significant for all analyses.
Funding information
These studies were supported by research grants R01 NS038932 (D. E. G.), R01 NS087539 (D. E. G.), T32 AI007247 (K. L. W. S.), T32 AI007417 (E. M. T.), T32 OD011089 (V. K. B.) and F31 NS101775 (E. M. T.) from the US National Institutes of Health.
Acknowledgements
The authors wish to thank Jane Yeh for assistance with cell harvesting and Debra Hauer for technical assistance.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: B6, C57Bl/6; CNS, central nervous system; IFN, interferon; IFNAR, IFN receptor; IRF, IFN regulatory factor; LCMV, lymphocytic choriomeningitis virus; NSV, neuroadapted Sindbis virus; SINV, Sindbis virus; TLR, toll-like receptor; TMEV, Theiler’s murine encephalomyelitis virus; TNF, tumour necrosis factor; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling; WNV, West Nile virus; WT, wild-type.
References
- 1.Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330–342. doi: 10.1016/S0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- 2.Griffin DE. Emergence and re-emergence of viral diseases of the central nervous system. Prog Neurobiol. 2010;91:95–101. doi: 10.1016/j.pneurobio.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin DE. Role of the immune response in age-dependent resistance of mice to encephalitis due to Sindbis virus. J Infect Dis. 1976;133:456–464. doi: 10.1093/infdis/133.4.456. [DOI] [PubMed] [Google Scholar]
- 4.Jackson AC, Moench TR, Trapp BD, Griffin DE. Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest. 1988;58:503–509. [PubMed] [Google Scholar]
- 5.Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. 2010;207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryman KD, Meier KC, Gardner CL, Adegboyega PA, Klimstra WB. Non-pathogenic Sindbis virus causes hemorrhagic fever in the absence of alpha/beta and gamma interferons. Virology. 2007;368:273–285. doi: 10.1016/j.virol.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 7.Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74:3366–3378. doi: 10.1128/JVI.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fragkoudis R, Breakwell L, Mckimmie C, Boyd A, Barry G, et al. The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J Gen Virol. 2007;88:3373–3384. doi: 10.1099/vir.0.83191-0. [DOI] [PubMed] [Google Scholar]
- 9.Byrnes AP, Durbin JE, Griffin DE. Control of Sindbis virus infection by antibody in interferon-deficient mice. J Virol. 2000;74:3905–3908. doi: 10.1128/JVI.74.8.3905-3908.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. 2013;78:105–116. doi: 10.1101/sqb.2013.78.020321. [DOI] [PubMed] [Google Scholar]
- 11.Tarassishin L, Bauman A, Suh HS, Lee SC. Anti-viral and anti-inflammatory mechanisms of the innate immune transcription factor interferon regulatory factor 3: relevance to human CNS diseases. J Neuroimmune Pharmacol. 2013;8:132–144. doi: 10.1007/s11481-012-9360-5. [DOI] [PubMed] [Google Scholar]
- 12.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, et al. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andersen J, Vanscoy S, Cheng TF, Gomez D, Reich NC. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 2008;9:168–175. doi: 10.1038/sj.gene.6364449. [DOI] [PubMed] [Google Scholar]
- 14.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 15.Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity. 2000;13:539–548. doi: 10.1016/S1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 17.Osterlund PI, Pietilä TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-λ) genes. J Immunol. 2007;179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 18.Prakash A, Smith E, Lee CK, Levy DE. Tissue-specific positive feedback requirements for production of type I interferon following virus infection. J Biol Chem. 2005;280:18651–18657. doi: 10.1074/jbc.M501289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, et al. Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 20.Ning S, Huye LE, Pagano JS. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J Biol Chem. 2005;280:12262–12270. doi: 10.1074/jbc.M404260200. [DOI] [PubMed] [Google Scholar]
- 21.Marié I, Smith E, Prakash A, Levy DE. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol Cell Biol. 2000;20:8803–8814. doi: 10.1128/MCB.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Au WC, Moore PA, Lafleur DW, Tombal B, Pitha PM. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J Biol Chem. 1998;273:29210–29217. doi: 10.1074/jbc.273.44.29210. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Pagano JS. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol Cell Biol. 1997;17:5748–5757. doi: 10.1128/MCB.17.10.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013;9:e1003118. doi: 10.1371/journal.ppat.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proenca-Modena JL, Hyde JL, Sesti-Costa R, Lucas T, Pinto AK, et al. Interferon-regulatory factor 5-dependent signaling restricts orthobunyavirus dissemination to the central nervous system. J Virol. 2015;90:189–205. doi: 10.1128/JVI.02276-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gardner J, Rudd PA, Prow NA, Belarbi E, Roques P, et al. Infectious Chikungunya virus in the saliva of mice, monkeys and humans. PLoS One. 2015;10:e0139481. doi: 10.1371/journal.pone.0139481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, et al. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J Virol. 2012;86:9888–9898. doi: 10.1128/JVI.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daffis S, Suthar MS, Szretter KJ, Gale M, Diamond MS. Induction of IFN-β and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 2009;5:e1000607. doi: 10.1371/journal.ppat.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 30.Menachery VD, Pasieka TJ, Leib DA. Interferon regulatory factor 3-dependent pathways are critical for control of herpes simplex virus type 1 central nervous system infection. J Virol. 2010;84:9685–9694. doi: 10.1128/JVI.00706-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daffis S, Samuel MA, Keller BC, Gale M, Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007;3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, et al. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol. 2008;82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nilaratanakul V, Chen J, Tran O, Baxter VK, Troisi EM, et al. Germ line IgM is sufficient, but not required, for antibody-mediated alphavirus clearance from the central nervous system. J Virol. 2018;92:e02081-17. doi: 10.1128/JVI.02081-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baxter VK, Troisi EM, Pate NM, Zhao JN, Griffin DE. Death and gastrointestinal bleeding complicate encephalomyelitis in mice with delayed appearance of CNS IgM after intranasal alphavirus infection. J Gen Virol. 2018 doi: 10.1099/jgv.0.001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakaya T, Sato M, Hata N, Asagiri M, Suemori H, et al. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem Biophys Res Commun. 2001;283:1150–1156. doi: 10.1006/bbrc.2001.4913. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 2002;297:2063–2066. doi: 10.1126/science.1074900. [DOI] [PubMed] [Google Scholar]
- 37.der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bréhin AC, Casadémont I, Frenkiel MP, Julier C, Sakuntabhai A, et al. The large form of human 2',5'-Oligoadenylate Synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology. 2009;384:216–222. doi: 10.1016/j.virol.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 39.Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O'Guin AK, et al. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo . J Virol. 2005;79:13974–13983. doi: 10.1128/JVI.79.22.13974-13983.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo X, Ma J, Sun J, Gao G. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc Natl Acad Sci USA. 2007;104:151–156. doi: 10.1073/pnas.0607063104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Burke CW, Ryman KD, Klimstra WB. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J Virol. 2007;81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barry G, Breakwell L, Fragkoudis R, Attarzadeh-Yazdi G, Rodriguez-Andres J, et al. PKR acts early in infection to suppress Semliki Forest virus production and strongly enhances the type I interferon response. J Gen Virol. 2009;90:1382–1391. doi: 10.1099/vir.0.007336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bosco A, Wiehler S, Proud D. Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection. BMC Genomics. 2016;17:76. doi: 10.1186/s12864-016-2405-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. 2010;30:459–473. doi: 10.1038/jcbfm.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baxter VK, Glowinski R, Braxton AM, Potter MC, Slusher BS, et al. Glutamine antagonist-mediated immune suppression decreases pathology but delays virus clearance in mice during nonfatal alphavirus encephalomyelitis. Virology. 2017;508:134–149. doi: 10.1016/j.virol.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rowell JF, Griffin DE. The inflammatory response to nonfatal Sindbis virus infection of the nervous system is more severe in SJL than in BALB/c mice and is associated with low levels of IL-4 mRNA and high levels of IL-10-producing CD4+ T cells. J Immunol. 1999;162:1624–1632. [PubMed] [Google Scholar]
- 47.Marsili G, Perrotti E, Remoli AL, Acchioni C, Sgarbanti M, et al. IFN regulatory factors and antiviral innate immunity: how viruses can get better. J Interferon Cytokine Res. 2016;36:414–432. doi: 10.1089/jir.2016.0002. [DOI] [PubMed] [Google Scholar]
- 48.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 49.Ousman SS, Wang J, Campbell IL. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J Virol. 2005;79:7514–7527. doi: 10.1128/JVI.79.12.7514-7527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delhaye S, Paul S, Blakqori G, Minet M, Weber F, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen M, Matcovitch O, David E, Barnett-Itzhaki Z, Keren-Shaul H, et al. Chronic exposure to TGFβ1 regulates myeloid cell inflammatory response in an IRF7-dependent manner. EMBO J. 2014;33:2906–2921. doi: 10.15252/embj.201489293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagemeyer N, Prinz M. Burning down the house: IRF7 makes the difference for microglia. EMBO J. 2014;33:2885–2886. doi: 10.15252/embj.201490345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juang YT, Lowther W, Kellum M, Au WC, Lin R, et al. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc Natl Acad Sci USA. 1998;95:9837–9842. doi: 10.1073/pnas.95.17.9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 56.Sharma S, Tenoever BR, Grandvaux N, Zhou GP, Lin R, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 57.Servant MJ, Grandvaux N, Hiscott J. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol. 2002;64:985–992. doi: 10.1016/S0006-2952(02)01165-6. [DOI] [PubMed] [Google Scholar]
- 58.Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/S1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 59.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe. 2016;19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen HW, King K, Tu J, Sanchez M, Luster AD, et al. The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J Immunol. 2013;191:4194–4201. doi: 10.4049/jimmunol.1300799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, et al. Cutting edge: independent roles for IRF-3 and IRF-7 in hematopoietic and nonhematopoietic cells during host response to Chikungunya infection. J Immunol. 2012;188:2967–2971. doi: 10.4049/jimmunol.1103185. [DOI] [PubMed] [Google Scholar]
- 62.Daffis S, Lazear HM, Liu WJ, Audsley M, Engle M, et al. The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol. 2011;85:5664–5668. doi: 10.1128/JVI.00232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thackray LB, Duan E, Lazear HM, Kambal A, Schreiber RD, et al. Critical role for interferon regulatory factor 3 (IRF-3) and IRF-7 in type I interferon-mediated control of murine norovirus replication. J Virol. 2012;86:13515–13523. doi: 10.1128/JVI.01824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Proenca-Modena JL, Sesti-Costa R, Pinto AK, Richner JM, Lazear HM, et al. Oropouche virus infection and pathogenesis are restricted by MAVS, IRF-3, IRF-7, and type I interferon signaling pathways in nonmyeloid cells. J Virol. 2015;89:4720–4737. doi: 10.1128/JVI.00077-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC. Ubiquitination of the transcription factor IRF-3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity. 2016;44:1151–1161. doi: 10.1016/j.immuni.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moore TC, Vogel AJ, Petro TM, Brown DM. IRF3 deficiency impacts granzyme B expression and maintenance of memory T cell function in response to viral infection. Microbes Infect. 2015;17:426–439. doi: 10.1016/j.micinf.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tarassishin L, Suh HS, Lee SC. Interferon regulatory factor 3 plays an anti-inflammatory role in microglia by activating the PI3K/Akt pathway. J Neuroinflammation. 2011;8:187. doi: 10.1186/1742-2094-8-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology. 2018;223:101–111. doi: 10.1016/j.imbio.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Ysebrant de Lendonck L, Martinet V, Goriely S. Interferon regulatory factor 3 in adaptive immune responses. Cell Mol Life Sci. 2014;71:3873–3883. doi: 10.1007/s00018-014-1653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fitzgerald DC, O'Brien K, Young A, Fonseca-Kelly Z, Rostami A, et al. Interferon regulatory factor (IRF) 3 is critical for the development of experimental autoimmune encephalomyelitis. J Neuroinflammation. 2014;11:130. doi: 10.1186/1742-2094-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kulcsar KA, Baxter VK, Greene IP, Griffin DE. Interleukin 10 modulation of pathogenic Th17 cells during fatal alphavirus encephalomyelitis. Proc Natl Acad Sci USA. 2014;111:16053–16058. doi: 10.1073/pnas.1418966111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010;29:1762–1773. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Yamashita M, et al. Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J Virol. 2013;87:16–24. doi: 10.1128/JVI.01853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol. 2011;85:3708–3716. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peters K, Chattopadhyay S, Sen GC. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol. 2008;82:3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peltier DC, Lazear HM, Farmer JR, Diamond MS, Miller DJ. Neurotropic arboviruses induce interferon regulatory factor 3-mediated neuronal responses that are cytoprotective, interferon independent, and inhibited by Western equine encephalitis virus capsid. J Virol. 2013;87:1821–1833. doi: 10.1128/JVI.02858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou S, Cerny AM, Fitzgerald KA, Kurt-Jones EA, Finberg RW. Role of interferon regulatory factor 7 in T cell responses during acute lymphocytic choriomeningitis virus infection. J Virol. 2012;86:11254–11265. doi: 10.1128/JVI.00576-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, et al. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem. 2004;279:45194–45207. doi: 10.1074/jbc.M400726200. [DOI] [PubMed] [Google Scholar]
- 79.Jiang DS, Liu Y, Zhou H, Zhang Y, Zhang XD, et al. Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension. 2014;63:713–722. doi: 10.1161/HYPERTENSIONAHA.113.02653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rollenhagen C, Rollenhage C, Macura SL, Lathrop MJ, Mackenzie TA, et al. Enhancing interferon regulatory factor 7 mediated antiviral responses and decreasing nuclear factor κB expression limit HIV-1 replication in cervical tissues. PLoS One. 2015;10:e0131919. doi: 10.1371/journal.pone.0131919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Freter RR, Alberta JA, Hwang GY, Wrentmore AL, Stiles CD. Platelet-derived growth factor induction of the immediate-early gene MCP-1 is mediated by NF-κB and a 90-kDa phosphoprotein coactivator. J Biol Chem. 1996;271:17417–17424. doi: 10.1074/jbc.271.29.17417. [DOI] [PubMed] [Google Scholar]
- 82.Spann KM, Loh Z, Lynch JP, Ullah A, Zhang V, et al. IRF-3, IRF-7, and IPS-1 promote host defense against acute human metapneumovirus infection in neonatal mice. Am J Pathol. 2014;184:1795–1806. doi: 10.1016/j.ajpath.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 83.Li W, Hofer MJ, Jung SR, Lim SL, Campbell IL. IRF7-dependent type I interferon production induces lethal immune-mediated disease in STAT1 knockout mice infected with lymphocytic choriomeningitis virus. J Virol. 2014;88:7578–7588. doi: 10.1128/JVI.03117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Christensen JE, Fenger C, Issazadeh-Navikas S, Krug A, Liljestrøm P, et al. Differential impact of interferon regulatory factor 7 in initiation of the type I interferon response in the lymphocytic choriomeningitis virus-infected central nervous system versus the periphery. J Virol. 2012;86:7384–7392. doi: 10.1128/JVI.07090-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nargi-Aizenman JL, Havert MB, Zhang M, Irani DN, Rothstein JD, et al. Glutamate receptor antagonists protect from virus-induced neural degeneration. Ann Neurol. 2004;55:541–549. doi: 10.1002/ana.20033. [DOI] [PubMed] [Google Scholar]
- 86.Manivannan S, Baxter VK, Schultz KL, Slusher BS, Griffin DE. Protective effects of glutamine antagonist 6-diazo-5-oxo-l-norleucine in mice with alphavirus encephalomyelitis. J Virol. 2016;90:9251–9262. doi: 10.1128/JVI.01045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Greene IP, Lee EY, Prow N, Ngwang B, Griffin DE. Protection from fatal viral encephalomyelitis: AMPA receptor antagonists have a direct effect on the inflammatory response to infection. Proc Natl Acad Sci USA. 2008;105:3575–3580. doi: 10.1073/pnas.0712390105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schultz KL, Vernon PS, Griffin DE. Differentiation of neurons restricts Arbovirus replication and increases expression of the alpha isoform of IRF-7. J Virol. 2015;89:48–60. doi: 10.1128/JVI.02394-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu D, Zhang Y, Zhao L, Cao M, Lingel A, et al. Interferon regulatory factor 7 is involved in the growth of Epstein-Barr virus-transformed human B lymphocytes. Virus Res. 2015;195:112–118. doi: 10.1016/j.virusres.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 90.Carmen J, Rothstein JD, Kerr DA. Tumor necrosis factor-alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res. 2009;1263:143–154. doi: 10.1016/j.brainres.2009.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kraft AD, McPherson CA, Harry GJ. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. Neurotoxicology. 2009;30:785–793. doi: 10.1016/j.neuro.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 93.Park KM, Bowers WJ. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal. 2010;22:977–983. doi: 10.1016/j.cellsig.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu Z, Cheng G, Wen X, Wu GD, Lee WT, et al. Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NFκB-dependent mechanism. Neurobiol Dis. 2002;11:199–213. doi: 10.1006/nbdi.2002.0530. [DOI] [PubMed] [Google Scholar]
- 95.Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, et al. Cell death after spinal cord injury is exacerbated by rapid TNF α-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosci. 2000;23:175–180. doi: 10.1016/S0166-2236(99)01533-7. [DOI] [PubMed] [Google Scholar]
- 97.Tolosa L, Caraballo-Miralles V, Olmos G, Lladó J. TNF-α potentiates glutamate-induced spinal cord motoneuron death via NF-κB. Mol Cell Neurosci. 2011;46:176–186. doi: 10.1016/j.mcn.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 98.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 99.Tortarolo M, Vallarola A, Lidonnici D, Battaglia E, Gensano F, et al. Lack of TNF-α receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J Neurochem. 2015;135:109–124. doi: 10.1111/jnc.13154. [DOI] [PubMed] [Google Scholar]
- 100.Toribio R, Ventoso I. Inhibition of host translation by virus infection in vivo . Proc Natl Acad Sci USA. 2010;107:9837–9842. doi: 10.1073/pnas.1004110107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 102.Chang A, Chen Y, Shen W, Gao R, Zhou W, et al. Ifit1 protects against lipopolysaccharide and d-galactosamine-induced fatal hepatitis by inhibiting activation of the JNK pathway. J Infect Dis. 2015;212:1509–1520. doi: 10.1093/infdis/jiv221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.John SP, Sun J, Carlson RJ, Cao B, Bradfield CJ, et al. IFIT1 exerts opposing regulatory effects on the inflammatory and interferon gene programs in LPS-activated human macrophages. Cell Rep. 2018;25:95–106. doi: 10.1016/j.celrep.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci. 2017;74:3275–3291. doi: 10.1007/s00018-017-2513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karrer M, Lopez MA, Meier D, Mikhail C, Ogunshola OO, et al. Cytokine-induced sleep: neurons respond to TNF with production of chemokines and increased expression of Homer1a in vitro. Brain Behav Immun. 2015;47:186–192. doi: 10.1016/j.bbi.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 106.Bennett JL, Elhofy A, Canto MC, Tani M, Ransohoff RM, et al. CCL2 transgene expression in the central nervous system directs diffuse infiltration of CD45(high)CD11b(+) monocytes and enhanced Theiler's murine encephalomyelitis virus-induced demyelinating disease. J Neurovirol. 2003;9:623–636. doi: 10.1080/13550280390247551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Terry RL, Getts DR, Deffrasnes C, van Vreden C, Campbell IL, et al. Inflammatory monocytes and the pathogenesis of viral encephalitis. J Neuroinflammation. 2012;9:270. doi: 10.1186/1742-2094-9-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Getts DR, Terry RL, Getts MT, Müller M, Rana S, et al. Ly6c+ "inflammatory monocytes" are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lane TE, Asensio VC, Yu N, Paoletti AD, Campbell IL, et al. Dynamic regulation of alpha- and β-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J Immunol. 1998;160:970–978. [PubMed] [Google Scholar]
- 110.Templeton SP, Kim TS, O'Malley K, Perlman S. Maturation and localization of macrophages and microglia during infection with a neurotropic murine coronavirus. Brain Pathol. 2008;18:40–51. doi: 10.1111/j.1750-3639.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trujillo JA, Fleming EL, Perlman S. Transgenic CCL2 expression in the central nervous system results in a dysregulated immune response and enhanced lethality after coronavirus infection. J Virol. 2013;87:2376–2389. doi: 10.1128/JVI.03089-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor alpha, and CCR7 ligands in lymph node development. J Exp Med. 2003;197:1191–1198. doi: 10.1084/jem.20021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi K, Hayashida K, Kaneko M, Hashimoto J, Tomita T, et al. Lymphoid chemokine B cell-attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J Immunol. 2001;166:650–655. doi: 10.4049/jimmunol.166.1.650. [DOI] [PubMed] [Google Scholar]
- 114.Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. 2006;176:7676–7685. doi: 10.4049/jimmunol.176.12.7676. [DOI] [PubMed] [Google Scholar]
- 115.Aloisi F, Columba-Cabezas S, Franciotta D, Rosicarelli B, Magliozzi R, et al. Lymphoid chemokines in chronic neuroinflammation. J Neuroimmunol. 2008;198:106–112. doi: 10.1016/j.jneuroim.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kobayashi S, Watanabe T, Suzuki R, Furu M, Ito H, et al. TGF-β induces the differentiation of human CXCL13-producing CD4(+) T cells. Eur J Immunol. 2016;46:360–371. doi: 10.1002/eji.201546043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rainey-Barger EK, Rumble JM, Lalor SJ, Esen N, Segal BM, et al. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav Immun. 2011;25:922–931. doi: 10.1016/j.bbi.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol. 2013;87:2420–2429. doi: 10.1128/JVI.01769-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Phares TW, Disano KD, Stohlman SA, Segal BM, Bergmann CC. CXCL13 promotes isotype-switched B cell accumulation to the central nervous system during viral encephalomyelitis. Brain Behav Immun. 2016;54:128–139. doi: 10.1016/j.bbi.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Esen N, Blakely PK, Rainey-Barger EK, Irani DN. Complexity of the microglial activation pathways that drive innate host responses during lethal alphavirus encephalitis in mice. ASN Neuro. 2012;4:207–221. doi: 10.1042/AN20120016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 122.Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, et al. Molecular basis of Sindbis virus neurovirulence in mice. J Virol. 1988;62:2329–2336. doi: 10.1128/jvi.62.7.2329-2336.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]