
Figure 1.
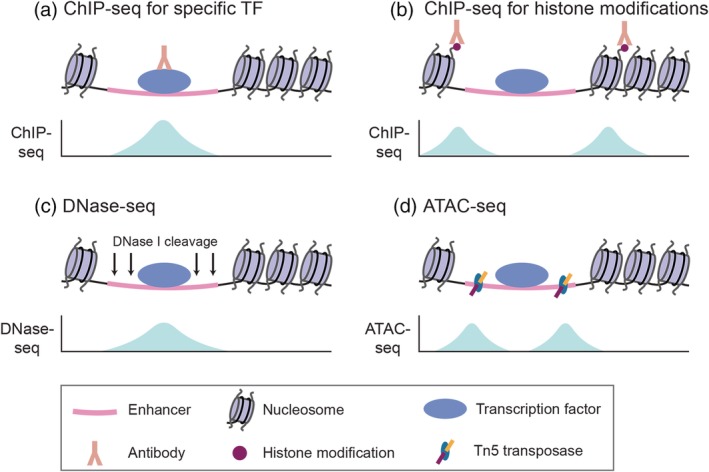
Genome‐wide methods to identify putative enhancers. The basic principle of each experimental approach and the corresponding data readout is shown for each method. (a) Chromatin immunoprecipitation followed by high‐throughput sequencing (ChIP‐seq) uses antibodies targeting a specific TF to determine the location of its binding sites genome‐wide. The broad peaks generated by ChIP‐seq cannot determine precise footprints of TF binding, but this can be achieved by adding a 5′ to 3′ exonuclease digestion step to the protocol (ChIP‐exo). (b) Nucleosomes flanking active enhancers often carry stereotypical histone modifications (e.g., H3K4me1 and H3K27ac) that can be detected with specific antibodies by ChIP‐seq. (c) Active enhancers and other cis‐regulatory elements are found within open chromatin that is depleted of nucleosomes. Accessible chromatin can be detected by DNase I digestion followed by high‐throughput sequencing (DNase‐seq). (d) Accessible chromatin can also be detected by assay for transposase‐accessible chromatin using sequencing (ATAC‐seq), where Tn5 transposase simultaneously fragments and tags accessible DNA prior to sequencing