

ABSTRACT
The TLK1/Nek1 axis contributes to cell cycle arrest and implementation of the DDR to mediate survival upon DNA damage. However, when the damage is too severe, the cells typically are forced into apoptosis, and the contribution of TLKs in this process has not been investigated. In contrast, it is known that Nek1 may play a role by phosphorylating VDAC1 maintaining proper opening and closure of the channel and thus mitochondrial integrity. We now show that the activating phosphorylation of Nek1-T141 by TLK1 contributes to the phosphorylation and stability of VDAC1 and thereby to mitochondrial permeability and integrity. Treatment of three different cell lines model that overexpress Nek1-T141A mutant with doxorubicin showed exquisite sensitivity to the drug, with implementation of rapid accumulation of cells with subG1 DNA content (apoptotic) and other alterations in the cell cycle. In addition, these cells displayed reduced oxygen consumption under normal conditions and less reliance on mitochondria and more dependence on glycolysis for energy production. Consistent with greater apoptosis, upon treatment with low doses of doxorubicin, cells overexpressing Nek1-T141A displayed leakage of Cyt-C into the cytoplasmic fraction. This suggests that inhibiting the TLK1/Nek1/VDAC1 nexus could sensitize cancer cells to apoptotic killing in combination with an appropriate DNA damaging agent. We in fact have previously reported that Nek1 expression is elevated in advanced Prostate Cancer (PCa) and we now report that VDAC1 expression is elevated and correlated with disease stage, thereby making the TLK1/Nek1/VDAC1 nexus a very attractive target for PCa.
KEYWORDS: Tousled Like Kinase, Nek1, VDAC1, Mitochondrial Integrity, Apoptosis, Prostate Cancer
Introduction
Mitochondria play a major role in different processes beyond their critical bioenergetics function of supplying ATP; these include cell signaling, interorganellar communication, aging, cell proliferation, and importantly: apoptosis, which is one of the main natural mechanisms protecting against cancer development by removing genetically altered cells. Apoptosis is often triggered when functional integrity of mitochondria is compromised via the intrinsic as well as extrinsic apoptotic pathway. Functional integrity requires appropriate communication and transfer of metabolites between mitochondria and cytosol. The translocator protein (TSPO), located at the outer mitochondrial membrane (OMM), serves multiple functions and contributes to numerous processes, including cholesterol import, mitochondrial metabolism, apoptosis, Ca2+ signaling, oxidative stress, and inflammation. TSPO functions in a complex with the voltage-dependent anion channel (VDAC), a protein that mediates the flux of ions, including Ca2+, nucleotides, and metabolites across the OMM [1].
VDAC is a small pore-forming protein found in the OMM of all eukaryotes, where it functions as a point of entry and exit of metabolites. Hence, VDAC occupies a crucial position in the cell, by mediating the flux of ions, nucleotides and other metabolites across the OMM. Correspondingly, down-regulation of VDAC1 expression results in reduced metabolite exchange between mitochondria and the cytosol, leading to mitochondrial dysfunction and cell growth arrest [2]. Moreover, its location at the boundary between the mitochondria and the cytosol enables VDAC to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities, and it can contribute to the release of Cyt-C from the IMS, triggering APAF1-mediated activation of the caspases cascade during apoptosis. While several models have been proposed to explain how VDAC rearranges so that a relatively large molecule such as Cyt-C can leak out, in one mechanism, VDAC hetero-oligomerizes with the pore forming pro-apoptotic protein BAX to form large enough channels [3].
VDAC was found to interact with many proteins, including several pro- and anti-apoptotic components, and some kinases (rev. in [4]). Key to our work, Yumai Chen and colleagues discovered (initially with a two-hybrid screen) that the protein kinase Nek1 interacts with, and regulates the activity of VDAC [5]. In addition, they presented evidence that one key mechanism by which depletion of Nek1 exacerbates cell death in response to DNA damage [6] is because of the resulting drop in phosphorylation of VDAC and its consequent loss of activity, resulting in mitochondrial dysfunction and apoptosis. Specifically, they also reported that Nek1 phosphorylates a specific residue (S193) of VDAC1, and that this is a critical determinant of cell survival following DNA-damaging treatment. Since we recently showed that the activity of Nek1 is regulated by TLK1, and that there is a regulated wave of TLK1 loss and regain of function following the induction of DNA damage (by IR) [7], we wanted to study in greater depth if the proposed regulatory pathway (TLK1> Nek1> VDAC1) is an important element in preserving mitochondrial function. Our working hypothesis is that the TLK1> Nek1> VDAC1 axis is a delicate balancing act from the DNA Damage Response (DDR) to mitochondrial leakage. It has been clearly established that a close relation exists between the activation of the DDR and a mitochondrial function that is intimately involved in the decision to implement the apoptotic pathway if the damage sustained is too severe for repair. While several proteins have been identified in this process – e.g. p53, ATM/ATR and their downstream relay kinases Chk1/Chk2, as well as pro- and anti-apoptotic regulators – the pathways linking directly the DNA damage signal and the subsequent mitochondrial dysfunction are not clearly defined. We propose that the newly discovered roles of the TLK1/Nek1 axis in both the DDR and as regulators of VDAC1 may be a key link between irreparable DNA damage and mitochondria-mediated apoptosis. In the simplest model we conceive, the recruitment/relocalization of TLK1 and Nek1 to sites of DNA damage (particularly DSBs), which we [8] and others [9] have observed by IF microscopy, must occur at the expense of a reduced localization and function of Nek1 at the mitochondria. This, in turn, will result in VDAC1 dephosphorylation and consequent mitochondrial permeability leakage, mitochondrial dysfunction with diminished respiratory spare capacity, and finally leakage of Cyt-C and activation of the intrinsic apoptotic relay. Indeed, it was reported that the dissociation of Nek1 and VDAC1 is genotoxic dependent, where prolonged Nek1/VDAC1 dissociation will lead to VDAC1 dephosphorylation and initiate apoptosis [10].
Regarding the possible involvement of VDAC in cancer development and progression, it is well known that in cancer cells, high glycolytic rates and net formation of pyruvate and lactate persist despite adequate oxygenation, accounting for up the 40% of the ATP generated, instead of ~5% for normal cells that use primarily oxidative phosphorylation. VDAC likely contributes to cancer metabolism via the transport of various metabolites, mediating ATP/ADP exchange across the OMM and the binding and channeling of mitochondrial ATP directly to hexokinase (HK) [4]. And this HK-VDAC1 association appears to protect tumor cells from cell death [4]. Several studies have reported marked differences in the expression of VDAC between cancer cells and normal tissues [11] and elevated VDAC is a predictor of poor outcome [12] and is associated with decreased sensitivity to DNA damaging treatment [10]. The Involvement of VDAC in prostate cancer (PCa) progression and its association with apoptotic pathway has been reported [13,14] and it was shown that siRNA-mediate reduction of VDAC inhibited the growth, migration, and invasive properties of several cancer lines, including PC3 PCa cells, but the underlying mechanisms of VDAC1 functioning was not explored in PCa. Here we expand on some of the roles on the TLK1> Nek1> VDAC in controlling mitochondrial function and in triggering apoptosis, with some emphasis on PCa.
Materials and methods
Doxorubicin hydrochloride (Catalog No. D1515) and G418 disulfate salt (Catalog No. A1720) were obtained from Sigma. Hek293 and LNCaP cells were recently purchased form ATCC. HSG (normal human salivary gland cells) were a kind gift of Dr. Gulshan Sunavala Dossabhoy. Dulbecco’s modified Eagle’s medium (DMEM) for HEK293 cells and RPMI for LNCaP cells were purchased from Sigma Aldrich and Caisson labs. Antibodies used in this study were VDAC1 total (Catalog No. ab14734) Abcam, phospho-VDAC1 (a generous gift from Dr. Yumai Chen, UCI), Cyclin A (Catalog No. sc-271645), Cyclin B (Catalog No. sc-245), Cytochrome C (Catalog No. ab65311) Abcam, Complex COX IV (Catalog No. sc-376731), actin (Catalog No. sc-8432), GAPDH (Catalog No. 2118S) CST, Cleaved Caspase-3Asp (175) (Catalog No. 9661) CST, Alpha rabbit IgG-HRP (Catalog No. 7074S) CST. Alpha -mouse IgG-HRP (Catalog No.7076) CST.
Plasmids and transfections
The Nek1 mammalian expression vector was purchased from Origene (MR216282). The NEK1-T141A mutated plasmids were obtained by site-directed mutagenesis using QuickChange (Agilent) and used to transfect and generate stable cell lines of LNCaP, Hek293 and HSG cells. Hek293 and LNCaP expressing Nek1 (wt and T141A mutant) were already described in [8,15], respectively. The HSG cells are described for the first time here.
Clonogenic assay
For each doxorubicin concentration (100–400 nM), aliquots of serially diluted cells were plated on 6-well plates in triplicate. After 2 days, the medium was replaced w/o drug, and after 2 weeks of growth, the wells were rinsed with PBS, stained with crystal violet, and colonies counted. The results were expressed as the fraction of colonies surviving after treatment compared to number of colonies in control samples (plating efficiency).
Cell cycle analysis
We performed cell cycle analysis as described earlier [8] with LNCaP and Hek293 cells with Nek1 OE and Nek1 mutant with Doxorubicin treatment.
Western blot analysis
Western blot analysis of different proteins: phospho-VDAC1, Cyclin A, Cyclin B, Cytochrome C, Complex COX IV, Actin, GAPDH, and Cleaved Caspase-3 from control and cells treated with Doxorubicin (200 nM and 400 nM) for different times were carried out as described in [15].
Assessing mitochondrial dysfunction
To determine effects of Nek1 on mitochondrial function, we evaluated mitochondrial oxygen consumption rate in wild type or T141A mutant Nek1 transfected prostate cancer cells, LNCaP and HSG cells, using the Seahorse Bioscience XF24 Extracellular Flux Analyzer. In brief, 25,000 parental and stably overexpressing Nek1-wild type (Nek1 WT) and Nek1-T141A mutant LNCaP cells were plated, and at preset time points, they were treated with specific inhibitors of the respiratory chain. The samples were measured for basal OCR at 2, 10, 18 mins. Next, oligomycin was added at 20 mins, and OCR was measured at 25, 33, 42 mins. At 45 mins, FCCP was added and measures were taken at 48, 56, 63 mins. Finally, Rotenone was added at 65 minutes and OCR was measured at 71, 79, 86 minutes. The results shown are average of 6 independent well measures. The results of parental and transfected LNCaP and HSG cells were calculated. Basal: the value in untreated cells; ATP-linked: value after oligomycin treatment; Maximal capacity: value obtained after FCCP; Spare Capacity: Basal value – FCCP value.
Cytochrome C apoptosis assay kit
The Cyt-C Releasing Apoptosis Assay Kit ab65311 (Abcam) were used for detecting Cyt-C translocation from mitochondria to cytosol during apoptosis. Mitochondrial and cytoplasmic Cyt-C was measured by immunoblotting.
Data analysis for VDAC1
Data analysis for VDAC1 expression was performed as described in [16] from TCGA datasets. Human Protein Atlas were used for VDAC1 immunohistochemistry images:
(https://www.proteinatlas.org/ENSG00000213585-VDAC1/pathology/prostate+cancer#img) [17].
Results
Overexpression of the hypoactive Nek1-T141A mutant that cannot be activated by TLK promotes cell death in combination with low dose doxorubicin likely via destabilization of VDAC and consequent mitochondrial dysfunction
We previously reported that several phenothiazine antipsychotics that were found to be potent inhibitors of TLKs, strongly synergized in promoting cell death when the cells were exposed to increasing concentrations of doxorubicin, which induces formation of Double Strand Breaks. (DSBs). We attributed this effect to inhibition of the critical roles of TLKs in DSB repair and in the DNA Damage Response (DDR), but precise mechanistic aspects of this remained elusive. More recently, we identified the proteome targets of TLK1 [8] and among these the protein kinase Nek1, which was previously implicated as a key mediator of the DDR [6] upstream of ATR and Chk1 [18]. And in fact we reported that either the administration of Thioridazine (THD, inhibitor of TLK1) or overexpression of Nek1-T141A, results in abolishment of the DDR via reduced ATR and Chk1 activation, and bypass of the checkpoint induced by H2O2 or Hydroxyuruea [8]. However, this seemed insufficient to explain the rapid and extensive induction of apoptosis that we observed in these Hek293 cells overexpressing Nek1-T141A. However, Chen et al. uncovered that one key target of Nek1 is the OMM protein VDAC, and presented evidence that when VDAC fails to be phosphorylated by Nek1, apoptosis ensues [5], while the common increased expression of Nek1 in RCC is associated with decreased sensitivity to killing by DNA damaging agents [10]. It then seemed logical to test if cells overexpressing Nek1-T141A could be more sensitive to killing by a low concentration of doxorubicin (200–400 nM). To test this, we used two cell lines that we previously described overexpressing Nek1-T141A: Hek293 [8] and LNCaP (human PCa cells) [15]. We first determined the survivability of cells after two-day exposure to doxorubicin (200 nM) by clonogenic assays. In control Hek293 cells, doxorubicin treatment resulted in a ~ 90% loss of colony formation ability (Figure 1(a)), whereas the LNCaP cells were significantly more resistant with a 50% drop in clonogenic ability (Figure 1(b)). Importantly, in both cell lines, overexpression of wt-Nek1 resulted in nearly complete resistance to a low-dose doxorubicin, consistent with the work by Chen et al. in RCC [10]. But further supporting the critical role of Nek1, the dominant hypoactive Nek1-T141A mutant that cannot be activated by TLK1 [8] (herein labeled N5) was unable to promote resistance to doxorubicin in both cell lines (Figure 1(a,b)).
Figure 1.
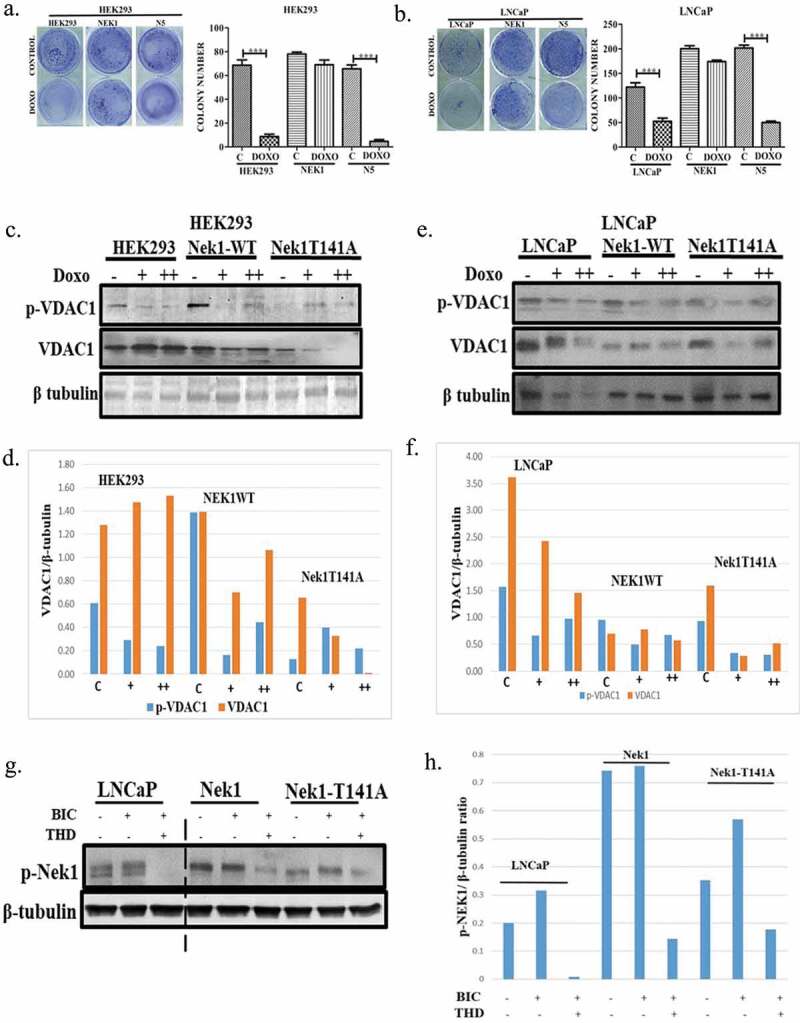
Overexpression of the hypoactive Nek1-T141A promotes cell death in combination with low dose doxorubicin likely via destabilization of VDAC. (a, b) Hek293 and LNCaP cells were stably transfected to overexpress Nek1-wt or T141A mutant [8,15], and clonogenic assays were carried after 48 h exposure to 200 nM doxorubicin. (c–f) Western blots and corresponding quantitation of VDAC1 and phospho-VDAC1(S193) in Hek293 and LNCaP cells overexpressing wt-Nek1 or T141A mutant after treatment with 200 (+) or 400 (++) nM doxorubicin. (g–h) Phosphorylation of Nek1 in parental LNCaP cells and overexpressing Nek1 (wt) and T141A mutant after incubation with bicalutamide +/− THD (a kinase inhibitor of TLK1B).
We wanted to study more mechanistically how Nek1 activity would impact VDAC level and phosphorylation state (S193, the residue phosphorylated by Nek1). Our working hypothesis is that the accumulation of DSBs induced by doxorubicin would be met by accumulation of Nek1 in the nucleus at sites of damage, and a corresponding loss of its function at the mitochondria. Consistent with this expectation, the phosphorylation of VDAC was reduced in cells treated with 200 (+) or 400 (++) nM doxorubicin, whereas total VDAC changed little in control or cell overexpressing wt-Nek1 (Figure 1(c,e), with quantitations in (d,f)). However, in cell overexpressing the Nek1-T141A mutant, not only was the phosphorylation of VDAC reduced with doxorubicin, but also the total level of VDAC. This suggests that when the endogenous wt-Nek1 is outcompeted by expression of hypoactive Nek1-T141A, the inability to phosphorylate VDAC is compounded also by a loss of this critical protein in maintaining OMM integrity, particularly in the Hek293 cell line. Furthermore, treatment with Thioridazine [THD] (a rather specific TLK1 inhibitor) completely suppressed p-Nek1 in LNCaP control cells (Figure 1(g)), in contrast to its increase when cells were incubated with Bicalutamide (BIC), which induces the expression of the kinase variant TLK1B [15]. LNaCaP cells that overexpress wt-Nek1 showed a corresponding increase in p-Nek1, which could still be inhibited by THD), whereas cells expressing the Nek1-T141A mutant showed weak signal for p-Nek1, all attributable to the endogenous p-Nek1 protein (Figure 1(g,h)).
Reduced proliferation rates and increased apoptosis are the reason for increased doxorubicin sensitivity
Clonogenic assays are unable to distinguish the effects of a DNA damaging agent, which can result in either an impaired or delayed resumption of growth (cell division) or loss of viability due to increased killing of the initial population. To better distinguish between these two possibilities, we measured the change in cell doubling (MTT assay) over a week period after a 12 h exposure to doxorubicin (200 nM). In control Hek293, treatment with doxorubicin resulted in a ~ 25% loss of doubling, which can simply be attributed to a lag in resuming proliferation. In contrast, in control LNCaP cells, there appeared to be a significant loss in viability, resulting in an apparent loss of doubling of 50%, which can be attributed to a loss of cells. Overexpression of wt-Nek1 in both Hek and LNCaP resulted in reduced doubling times respect to control cells. Especially it resulted in significant protection against doxorubicin, possibly by avoiding a delay in resumption of growth, but also by preventing loss of viable cells through impeding apoptosis. In contrast, cells overexpressing Nek1-T141A mutant (N5) after treatment with doxorubicin displayed reduced doubling times, suggesting either a delay in resumption of growth, or actual loss of viable cells. To establish if cells exposed to doxorubicin displayed an increase in apoptosis, we then determined the presence of cleaved Caspase 3 (c-Cas 3) by WB. In control Hek293 and LNCaP, there was a dose-dependent increase in c-Cas 3, but there was a difference between Hek293 and LNCaP cells when wt-Nek1 was overexpressed. In Hek293, there was still a dose dependent increase in cleaved Cas 3 in doxorubicin treated cells. In contrast, in LNCaP cells overexpressing wt-Nek1, there was a strong reduction in c-Cas 3, and in fact complete resistance to doxorubicin-induced apoptotic activation. Confirming the importance of Nek1 in this process, and the role of TLK1 as a positive regulator of Nek1 function, Hek and LNCaP cells overexpressing Nek1-T141A mutant displayed elevated c-Cas 3 even in the absence of doxorubicin, while LNCaP cells show very high level of c-Cas 3 in a dose response to doxorubicin (Figure 2(c,e), with quantitation in (d,f)).
Figure 2.
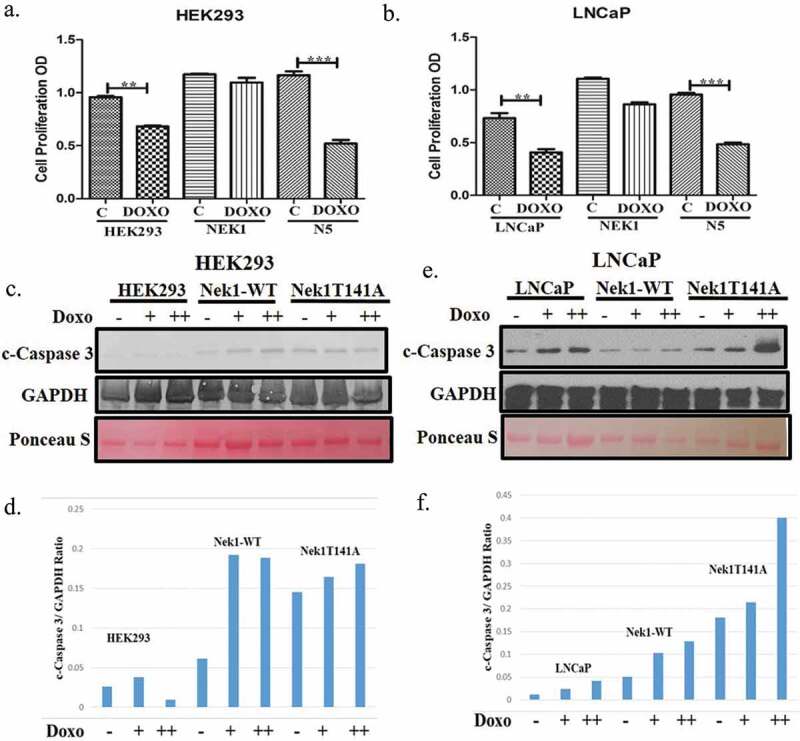
Reduced proliferation rates and increased apoptosis in cell overexpressing Nek1-T141A mutant after treatment with doxorubicin. (a, b) Proliferation rates were determined for Hek293 and LNCaP cells during a week recovery from 12 h exposure to doxorubicin (200 nM). Results are expressed in relation to untreated “parental” cells. (c–f). Cells overexpressing Nek1-T141A show elevated activated Caspase 3 when treated with low doses [200 (+) or 400 (++) nM] doxorubicin.
To better establish if the reduced cell doubling and viability (clonogenic assays) was due to an initial loss of cells due to apoptosis, we studied the cell cycle profiles of these cells 24 h after treatment with doxorubicin (Figure 3). Control Hek293cells displayed a cell cycle block in two positions: S and G2 – in Hek cells 4% in G1 36% in S, 35% in G2 and 8% in subG1. In contrast, LNCaP treated with doxorubicin displayed arrests largely in G1 and S – 43% in G1, 32% in S, 8% in G2, 17% in subG1. Hek293 overexpressing wt-Nek1, after treatment with doxorubicin displayed primarily a block in G2, and less so in S – 16% in G1, 22% in S, 58% in G2, 4% in subG1. LNCaP cells overexpressing wt-Nek1 arrested primarily in G1 (and not S) – 42% in G1, 8% in S, 24% in G2 and 26% in subG1. Hek293 overexpressing Nek1-T141A, while attempting to arrest in G2, displayed in addition a high percentage of apoptotic cells (subG1) – 10% G1, 14% in S, 52% in G2, 24% in subG1. LNCaP cells overexpressing Nek-T141A, while attempting to arrest in G1/S, displayed a very strong increase in subG1 cells (apoptotic) – 43% in G1, 6% in S, 14% in G2, 37% in subG1. To recapitulate, the most striking effect of the overexpression of the hypoactive Nek1-T141A mutant in cells exposed to a low dose of doxorubicin was a pronounced increase in apoptotic cells (subG1) in comparison to controls or cells overexpressing wt-Nek1. However, particularly for Hek293 cells but to a lesser degree in LNCaP, another difference seemed to lay in the cell cycle arrest position of parental cells vs. Nek1-T141A overexpressing. Parental cells treated with doxorubicin arrested in S and G2, whereas, particularly Hek293 cells arrested only in G2. To better establish this, we determined the expression of cyclin A (expressed mostly in S phase) and cyclin B (expressed mostly in G2/M). Particularly, parental Hek293 cells (but also LNCaP cells) displayed an increase in cells expressing Cyclin A and Cyclin B after treatment with doxorubicin (Figure 3(c,d)), consistent with their accumulation in S and G2. In contrast, Hek293 cells overexpressing Nek1-T141A, showed a minor increase in Cyclin A after doxorubicin treatment and no significant increase in Cyclin B, indicating that they were arrested in early G2 and not late G2 or G2/M. For parental LNCaP, there was a similar increase in Cyclin A and Cycling B (Figure 3(d)), consistent with their arrest in S and G2. In contrast, LNCaP cells overexpressing Nek1-T141A (untreated) showed a 3-4-fold increase in Cyclin A and B (Figure 3(d–f)), consistent with a higher fraction of cells in G2 compared to parental LNCaP cells (compare in Figure 3(b)). After treatment with doxorubicin Nek1-T141A overexpressing cells did not show a strong increase in Cyclin A or B (Figure 3(d)) consistent with the fact that rather than arresting in S or G2 (like the parental cells), they underwent massive apoptosis.
Figure 3.
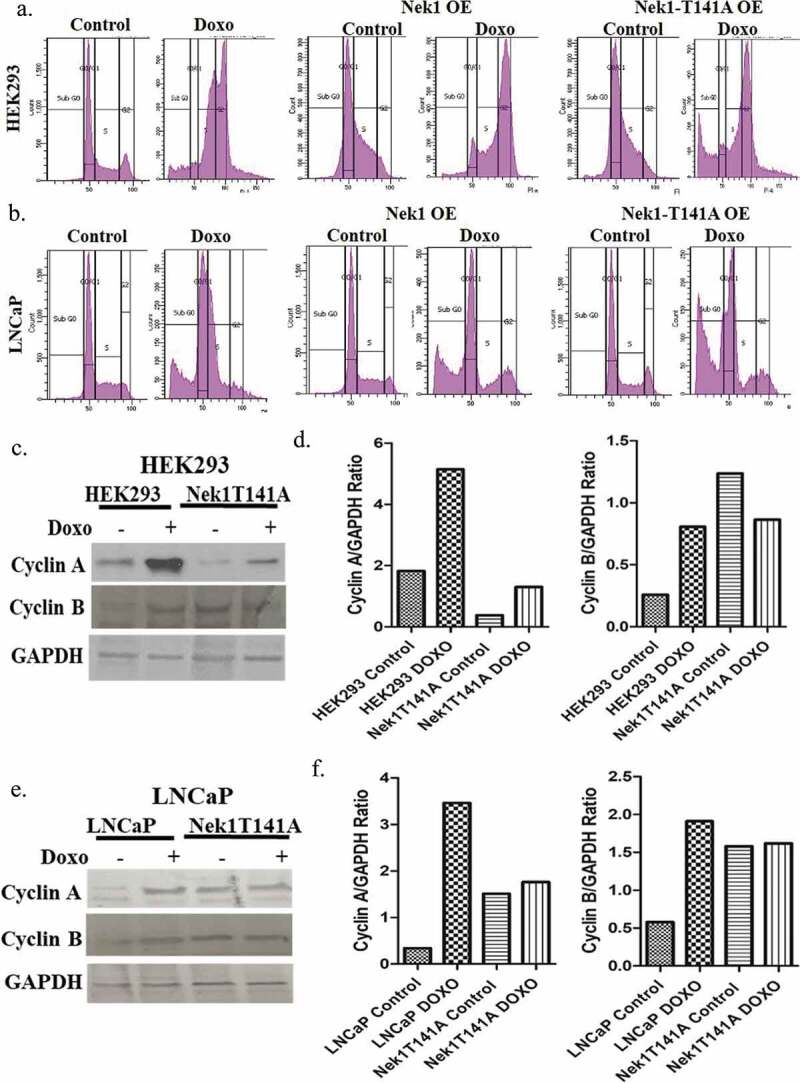
Cell cycle profiles and cycling markers in Hek293 and LNCaP overexpressing Nek1 or T141A mutant. Hek293 (a) and LNCaP (b) cells overexpressing Nek1 or T141A mutant were analyzed by PI/FACS after 24 h treatment with 200 nM doxorubicin. (c–f) Western blots and quantitations of the expression of cyclin A (marker of S phase) and cyclin B (G2/M) after treatment with doxorubicin.
Assessment of mitochondrial dysfunction and release of Cyt-C
To determine effects of Nek1 on mitochondrial function, we evaluated mitochondrial oxygen consumption rate in wild type or T141A mutant Nek1 transfected LNCaP cells. In addition, we also wanted to study this in a “normal” cell line, thinking ahead for a possible selective treatment modality given that Hek293 are actually tumorigenic in SCID mice. So, we stably transfected HSG cells (normal epithelial salivary gland cells) with wt and Nek1-T141A to generate “normal” overexpressing cells (Figure 4(e)). We measured the oxygen consumption rate (OCR) to assess basal respiration and mitochondrial functionality using the Seahorse Bioscience XF24 Extracellular Flux Analyzer. The results of parental and LNCaP transfected cells showed that the basal rate of oxygen consumption in the Nek1 transfected cells was increased relative to non-transfected (Figure 4(a,b) – One-way ANOVA/Bonferroni, p < 0.001). The basal rate of oxygen consumption in the Nek1-T141A overexpressing cells was also significantly decreased compared to the parental and wt Nek1 (p < 0.001). Spare capacity is the difference in rates before and after the addition of FCCP, a mitochondrial uncoupler that opens pores between the matrix and the interspace of the mitochondria to reorganize the proton gradient. The uncoupling, therefore, results in the continuous transport of protons and a maximal rate of O2 consumption. As expected, FCCP treatment induced an acute increase in the oxygen consumption rate in parental and Nek1 cells. However, FCCP-induced increase in the Nek1-T141A cells was severely diminished compared to the other groups (p < 0.001) and resulted in total oxygen consumption that was no higher than the basal rate. These results indicated an impairment of oxidative phosphorylation and mitochondrial function in Nek1-T141A-transfected cultures, which is contrasted by improved mitochondrial function in the wild-type Nek1 overexpressing cells compared to parental. These effects were to some extent reproduced in HSG cells in that wt-Nek1 overexpression strongly increase basal OCR and reserve capacity (Figure 4(c)). However, the overexpression of Nek1-T141A resulted actually in a modest increase in basal respiration and reserve capacity. We cannot make any definitive conclusions when comparing only one PCa cell line vs. one non-tumorigenic, but if these results could be extended to more examples, it would suggest that cancer cells (or at least LNCaP) rely more heavily on the TLK1> Nek1> VDAC axis to maintain OMM integrity and avoid leakage of protons, and possibly other metabolites. In contrast, normal cells appear unaffected by reduced Nek1 phosphorylation/activation by TLK1, and this could constitute a therapeutic point of discrimination, since specific TLK inhibitors may preferentially affect the mitochondrial integrity of cancer cells.
Figure 4.
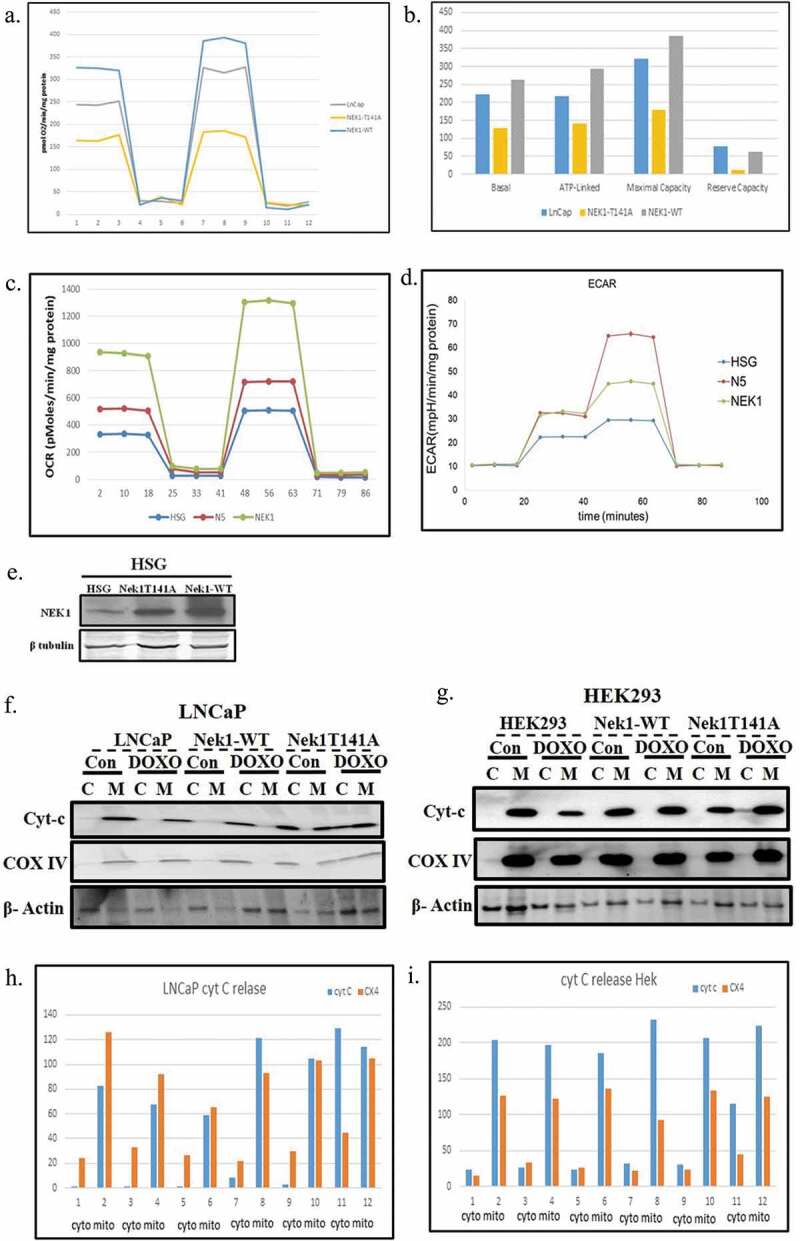
Assessment of mitochondrial dysfunction and release of Cyt-C. (a–d) LNCaP cells and normal HSG salivary cells overexpressing Nek1 or T141A mutant were monitored for OCR and ECAR with a Seahorse analyzer according to the manufacturer instructions. Cells in A-C were sequentially treated with oligomycin, FCCP, and Antymycin A/Rotenone. Cells in D were sequentially treated with Glucose after 3 h starvation, oligomycin, and finally 2-deoxyglucose (2-DG). (e) Expression of wt or T141A mutant Nek1 in HSG cells, since these cells were not previously characterized. (f–i) Western blots of Cyt-C in cytoplasmic and mitochondrial fractions of Nek1 and t14A mutant overexpressing cells with and without treatment with doxorubicin (200 nM) for 12 h.
The reduced ATP production from oxidative phosphorylation in Nek1-T141A overexpressing cells must be compensated for by an increased glycolytic flux, resulting in accumulation of pyruvate and lactate and thus acidification of the conditioned medium. This can also be measured in the Seahorse Analyzer under different conditions – sequential addition of glucose after 3 h starvation, oligomycin, and finally 2DG to stop glycolysis (Figure 4(d)). This analysis showed that both NEK1 and Nek1-T141A cells (N5) are capable of greater rates of glycolysis when provided with glucose following 3 h of glucose starvation. The HSG cells overexpressing Nek-T141A (N5) showed the highest glycolytic reserve, as shown by the increase in ECAR following the disruption of mitochondrial ATP production by oligomycin (a Complex V inhibitor that inhibits ATP production). This suggests that the N5 cells may be metabolically more adapted to rely on glycolysis rather than mitochondrial production of ATP, as VDAC phosphorylation is expected to be decreased leaving the channel in open position and creating an ion leak.
The evidence of altered mitochondrial function, as seen by increased aerobic glycolysis and reduced oxygen consumption, combined with evidence of increased apoptosis from low exposure to doxorubicin in cells overexpressing Nek1-T141A, suggested that these cells might implement the intrinsic apoptotic axis via leakage of Cyt-C from a defective VDAC pore function. To determine this, we analyzed the distribution of Cyt-C in cytoplasmic and mitochondrial fractions after treatment with 200 nM doxorubicin. As expected, in parental LNCaP and Hek293 cells and in cells overexpressing wt-Nek1, the distribution of Cyt-C was predominantly in the mitochondrial fraction, even after exposure to a low dose of doxorubicin (Figure 4(e–i)). In contrast, in Hek293 and LNCaP cells overexpressing Nek1-T141A, the distribution of Cyt-C was increased in the cytoplasmic fraction after treatment with doxorubicin. To validate the fractionation, the same blots were probed for complex IV (exclusively mitochondrial) and β-actin (which is found also in association with mitochondria [19]). In summary, reduced Nek1 functionality resulted in increased pro-apoptotic sensitization to doxorubicin and Cyt-C release, possibly due to reduced specific phosphorylation of VDAC and resulting in a permanently open channel in the OMM [5].
VDAC1 expression is high in PCa
As our lab is focused on prostate cancer (PCa), and VDAC has been associated with cancer development/progression and with the well-known Warburg effect (rev in [1]), so we wanted to probe if there is a role of VDAC1 in PCa. To study this, we interrogated the TCGA data for VDAC1 mRNA levels in patients diagnosed with prostate cancer vs. normal prostate samples (Figure 5). On analyzing the mRNA levels for VDAC1 gene in TCGA dataset we found a high levels of VDAC1 in PCa samples (p = 2.24E-07). Analysis of VDAC1 mRNA levels was carried out also in patients divided according to Gleason score pathology. When the Gleason scores were compared with normal samples based on VDAC1 mRNA expression levels, we observed an increase in levels of VDAC1 mRNA with higher Gleason scores such as Gleason 7 (p = 2.99E-04, n = 247), Gleason 8 (p = 6.42E-04, n = 64), Gleason 9 (p = 1.35E-10, n = 136) and Gleason 10 (p = 1.76E-04, n = 4) advising the significance of VDAC1 expression in PCa progression and disease etiology. This conclusion was further verified by IHC image analysis of VDAC1 protein from Human Protein Atlas, done on the PCa samples with low-grade and high-grade PCa tumor. The expression of the VDAC1 protein was analyzed in the different PCa tissues with low and high grade tumor samples, which showed that high-grade PCa tumor patients have more VDAC1 protein levels as compared to low-grade patients that is in-line with mRNA expression data for VDAC1 and disease aggressiveness. Taking these data together, the current analysis of VDAC1 expression (mRNA expression and protein levels) reveals a pronounced expression of VDAC1 in distinct PCa cancer types.
Figure 5.
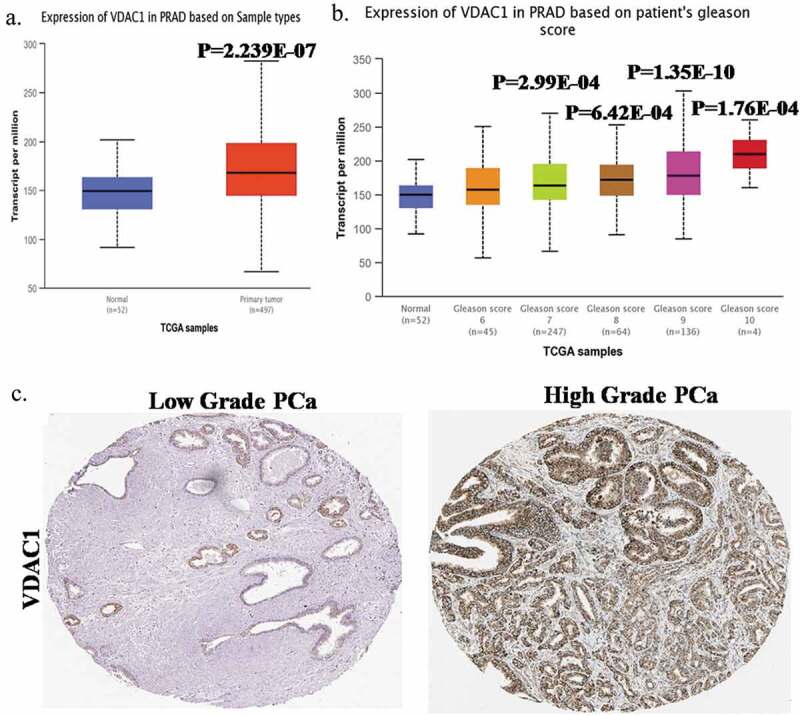
VDAC1 expression is elevated in PCa.(a) mRNA expression data of VDAC1 in PCa patients and in normal prostate extracted from TCGA dataset.(b) VDAC1 mRNA expression based on Gleason score from TCGA dataset.(c) Protein levels of VDAC1 in low-grade and high-grade PCa tumor patients analyzed through IHC images of VDAC1 protein from The Human Protein Atlas.
Discussion
The Tousled Like kinases (TLK1 and TLK2) are involved in several processes including chromatin assembly, replication, transcription, DNA repair, and chromosome segregation (rev. in [20,21]). However, the TLKs have not been implicated directly in processes of intrinsic apoptotic enactment, although their chemical inhibition was already known to enhance cell death when combined with doxorubicin [22]; but previously this was attributed to their critical activity on DNA repair and the DDR [15,22]. Now, largely based on a key paper by Chen et al [5] combined with our more recent discovery of the critical role of TLK1 in activating the dual-specificity protein kinase Nek1 [8], we have explored the possibility that the TLK1/Nek1 axis may be involved in regulating the apoptotic pathway through controlling the phosphorylation of VDAC1 and thus OMM permeability/leakage. Specifically, our working hypothesis is that the recruitment of TLK1 in association with Nek1 to the nuclear sites of DNA damage [8,9] occurs at the expense of its reduced functioning in the mitochondria. Nek1 was previously found to control VDAC1 phosphorylation and channel opening/closing [23], the loss of which results in proton leakage, loss of mitochondrial membrane potential, and finally implementation of the intrinsic apoptotic pathway through Cyt-C release. Indeed, we showed that the cells overexpressing Nek1-T141A mutant (the site normally phosphorylated by TLK1) show much depressed oxygen consumption under normal conditions (with reduced spare capacity – FCCP-mediated ATP uncoupling), compensated for by elevated glycolytic flux. Furthermore, both LNCaP PCa cells and Hek293 overexpressing Nek1-T141A showed exquisite sensitivity to low-dose killing with doxorubicin, which was manifested also by leakage of Cyt-C to the cytoplasm that is a hallmark of early stages of the apoptotic enactment. These results thus establish the involvement of the TLK1> Nek1> VDAC1 axis in implementing the intrinsic apoptotic cascade upon an elevated DNA damage that cannot be repaired efficiently.
Our lab is currently working on identifying more specific and potent TLK inhibitors than some of our previously identified phenothiazines that have been used extensively as anti-psychotics [22] and which have some undesirable side effects. Identification of such compounds could therefore become very useful for the treatment of various cancers in combination with selective DNA damaging agents. In particular, we have recently reported that in several models of androgen-dependent PCa cells, ADT often promotes a cell cycle arrest due to activation of the DDR, and that suppression of the TLK1/Nek1 axis then promotes apoptosis [15,24]. Therefore, there is a prima faciae urgency to identify some novel inhibitors of TLK1 for the adjuvant treatment of PCa. However, there is now an even more compelling rationale due to the expression of Nek1 and VDAC1 in the progression of PCa. We found an increase in VDAC1 mRNA levels in PCa patients and this is associated with PCa disease stage at both mRNA and protein levels (as high grade patients have higher levels of VDAC1 as compare to normal/low grade samples), suggesting that VDAC1 can act as maker for aggressive PCa; but to reach out this conclusion further study in large cohort samples is highly recommended. A potential role of VDAC in PCa has been reported in an earlier study that showed VDAC inhibition leads to the reduced cell growth, migration, and invasive properties of Androgen Independent PC3 PCa cells [13,14]. If this can be generalized to most PCa cases, it would suggest that targeting the TLK1> Nek1> VDAC1 axis could greatly improve the response to appropriate DNA damaging agents. Interestingly, while this paper was undergoing review, an important paper was published, which showed the Mitochondrial Fission Factors (MFF1 and MFF2) formed homo- and heterodimeric complexes with VDAC1 [25]. A cell permeable MFF Ser223-Leu243 D-enantiomeric peptidomimetic disrupted the MFF-VDAC1 complex and acutely depolarized mitochondria triggering apoptosis in several tumors types and in xerografts, but had no effect on normal cells. Among the cell lines they investigated, they included several PCa lines: LNCaP, C4-2, C4-2B, PC3, and DU145 [25]. Therefore, there is already some indirect evidence that targeting VDAC1 activity, through its phosphorylation by activated Nek1, via inhibition of TLK1 activity could be a very useful means to enhancing PCa cells killing with focused XRT or doxorubicin.
Funding Statement
This work was supported primarily by DoD-PCRP grant [W81XWH-17-1-0417] to A. De Benedetti.
Acknowledgments
We wish to thank Dr. Yumai Chen (UCI) for the kind gift of the phospho-VDAC1 antiserum. We thank Dr. Sumitra Miriyala for help with the Seahorse analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Shoshan-Barmatz V, Pittala S, Mizrachi D.. VDAC1 and the TSPO: expression, interactions, and associated functions in health and disease states. Int J Mol Sci. 2019;20(13):(pii):ijms20133348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Abu-Hamad S, Sivan S, Shoshan-Barmatz V.. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci U S A. 2006;103(15):5787–5792. Epub 0600102006 Apr 0600103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Banerjee J, Ghosh S. Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem Biophys Res Commun. 2004;323(1):310–314. [DOI] [PubMed] [Google Scholar]
- [4].Shoshan-Barmatz V, Golan M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr Med Chem. 2012;19(5):714–735. [DOI] [PubMed] [Google Scholar]
- [5].Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle (Georgetown, Tex). 2009;8(2):257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen Y, Chen CF, Riley DJ, et al. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle. 2011;10(4):655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sunavala-Dossabhoy G, De Benedetti A. Tousled homolog, TLK1, binds and phosphorylates Rad9; tlk1 acts as a molecular chaperone in DNA repair. DNA Repair (Amst). 2009;8:87–102. [DOI] [PubMed] [Google Scholar]
- [8].Singh V, Connelly ZM, Shen X, et al. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle. 2017;16(10):915–926. Epub 15382017 Apr 15384120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen Y, Chen PL, Chen CF, et al. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle. 2008;7(20):3194–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen Y, Chen CF, Polci R, et al. Increased Nek1 expression in renal cell carcinoma cells is associated with decreased sensitivity to DNA-damaging treatment. Oncotarget. 2014;5(12):4283–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Simamura E, Hirai K, Shimada H, et al. Furanonaphthoquinones cause apoptosis of cancer cells by inducing the production of reactive oxygen species by the mitochondrial voltage-dependent anion channel. Cancer Biol Ther. 2006;5(11):1523–1529. Epub 2006 Nov 1519. [DOI] [PubMed] [Google Scholar]
- [12].Grills C, Jithesh PV, Blayney J, et al. Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS One. 2011;6(1):e14635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thinnes FP. Human type-1 VDAC, a cisplatin target involved in either apoptotic pathway. Mol Genet Metab. 2009;97(2):163. Epub 2009 Feb 1013. [DOI] [PubMed] [Google Scholar]
- [14].Arif T, Vasilkovsky L, Refaely Y, et al. Silencing VDAC1 expression by siRNA inhibits cancer cell proliferation and tumor growth in vivo. Mol Ther Nucleic Acids. 2014;3:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Singh V, Jaiswal P, Ghosh I, et al. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. Int J Cancer. 2019;145:1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jaiswal PK, Koul S, Shanmugam PST, et al. Eukaryotic translation initiation Factor 4 Gamma 1 (eIF4G1) is upregulated during prostate cancer progression and modulates cell growth and metastasis. Sci Rep. 2018;8(1):7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Thul PJ, Akesson L, Wiking M, et al. A subcellular map of the human proteome. Science. 2017;356(6340):(pii): science.aal3321. Epub 2017 May 3311. [DOI] [PubMed] [Google Scholar]
- [18].Liu S, Ho CK, Ouyang J, et al. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci U S A. 2013;110(6):2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Reyes A, He J, Mao CC, et al. Actin and myosin contribute to mammalian mitochondrial DNA maintenance. Nucleic Acids Res. 2011;39(12):5098–5108. Epub 2011 Mar 5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].De Benedetti A. The Tousled-like-kinases as guardians of genome integrity. ISRN Mol Biol. 2012;2012:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Segura-Bayona S, Stracker TH. The Tousled-like kinases regulate genome and epigenome stability: implications in development and disease. Cell Mol Life Sci. 2019;76(19):3827–3841. Epub 02019 Jul 00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ronald S, Awate S, Rath A, et al. Phenothiazine inhibitors of TLKs affect double-strand break repair and DNA damage response recovery and potentiate tumor killing with radiomimetic therapy. Genes Cancer. 2013;4(1–2):39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen Y, Gaczynska M, Osmulski P, et al. Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1. Biochem Biophys Res Commun. 2010;394(3):798–803. Epub 2010 Mar 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Singh V, Jaiswal PK, Ghosh I, et al. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019;453:131–141. Epub 2019 Mar 1027. [DOI] [PubMed] [Google Scholar]
- [25].Seo JH, Chae YC, Kossenkov AV, et al. MFF regulation of mitochondrial cell death is a therapeutic target in cancer. Cancer Res. 2019;79(24):6215–6226. Epub 2019 Oct 6213. [DOI] [PMC free article] [PubMed] [Google Scholar]