

Abstract
Background
Dilated cardiomyopathy may be heritable but shows extensive genetic heterogeneity. The utility of whole exome sequencing as a first‐line genetic test for patients with dilated cardiomyopathy in a contemporary “real‐world” setting has not been specifically established. Using whole exome sequencing with rigorous, evidence‐based variant interpretation, we aimed to identify the prevalence of a molecular diagnosis in patients with dilated cardiomyopathy in a clinical setting.
Methods and Results
Whole exome sequencing was performed in eligible patients (n=83) with idiopathic or familial dilated cardiomyopathy. Variants were prioritized for curation in up to 247 genes and classified using American College of Medical Genetics and Genomics–based criteria. Ten (12%) had a pathogenic or likely pathogenic variant. Eight (10%) participants had truncating TTN variants classified as variants of uncertain significance. Five (6%) participants had variants of unknown significance according to strict American College of Medical Genetics and Genomics criteria but classified as either pathogenic or likely pathogenic by other clinical laboratories. Pathogenic or likely pathogenic variants were found in 8 genes (all within tier 1 genes), 2 (20%) of which are not included in a standard commercially available dilated cardiomyopathy panel. Using our bioinformatics pipeline, there was an average of 0.74 variants of uncertain significance per case with ≈0.75 person‐hours needed to interpret each of these variants.
Conclusions
Whole exome sequencing is an effective diagnostic tool for patients with dilated cardiomyopathy. With stringent classification using American College of Medical Genetics and Genomics criteria, the rate of detection of pathogenic variants is lower than previous reports. Efforts to improve adherence to these guidelines will be important to prevent erroneous misclassification of nonpathogenic variants in dilated cardiomyopathy genetic testing and inappropriate cascade screening.
Keywords: cardiomyopathy, whole exome sequencing, clinical exome, next generation sequencing
Subject Categories: Genetics, Cardiomyopathy
Clinical Perspective
What Is New?
Dilated cardiomyopathy commonly has an underlying genetic basis, with causative variants identified in >60 genes of heterogeneous ontologies.
Using whole exome sequencing with stringent criteria for classifying genomic variants, pathogenic or likely pathogenic variants were identified in 12% of the study population.
What Are the Clinical Implications?
With stringent classification of variants using American College of Medical Genetics criteria, the rate of detection of pathogenic variants is lower than in previous reports.
Despite the growing number of dilated cardiomyopathy–related genes, the incremental value of an extended gene set has not been convincingly demonstrated in the present study.
Efforts to improve adherence to the American College of Medical Genetics criteria will be important to prevent erroneous misclassification of nonpathogenic variants in dilated cardiomyopathy genetic testing and inappropriate cascade screening.
Introduction
Dilated cardiomyopathy (DCM) is a primary myocardial disorder characterized by left ventricular dilation and contractile dysfunction in the absence of abnormal loading conditions or coronary disease sufficient to cause global systolic dysfunction.1 DCM is an important cause of death and disability from heart failure and the disease course is determined by the underlying cause. DCM commonly has an underlying genetic basis, with causative variants identified in >60 genes of heterogeneous ontologies in both familial and apparently nonfamilial forms of disease.2, 3, 4, 5 Identification of a genetic diagnosis is important not only to allow prognostication and possibly personalization of management but is essential also for family screening and to predict risk of recurrence. In contrast to conditions such as hypertrophic cardiomyopathy where up to 60% of hypertrophic cardiomyopathy patients carry mutations,6 a genetic cause is less often identified in DCM, such that the role of routine genetic testing in DCM is less established and requires further study.
There is increasing evidence that clinical whole exome sequencing (WES) demonstrates a high diagnostic yield in achieving a molecular diagnosis of rare Mendelian disorders.7, 8 In contrast to panel testing, which involves testing a limited number of genes, WES provides genomic data on all protein coding sequences. In DCM it may have a role in reducing the number of unsolved cases without the additional costs of retesting as new genes are discovered. It is important to note, however, that broad analytical approaches in the evaluation of cardiomyopathies also have potential disadvantages. The higher number of genes tested allow an increase in diagnostic yield at the expense of longer turnaround time from sequencing and analytical processing with higher identification of variants of unknown significance, which increases clinical uncertainty.9 Furthermore, WES‐based analysis is associated with increased costs compared with targeted next generation sequencing approaches.9
Concerningly, up to 31% of all variants associated with DCM were subsequently identified in a recently published population exome browser, ExAC, questioning the pathogenic validity of many variants and/or genes previously thought to cause DCM.10 As the estimated prevalence of DCM is 1:2500 in the general population, this genotype prevalence was up to 400‐fold higher than the expected phenotypic prevalence of DCM.10 Additionally, the reported diagnostic yield from previous research studies2, 11, 12 comprising large multiplex families is expected to be higher than that can be achieved in a clinical setting. To date, the utility of WES as a first‐line genetic test for patients with DCM in a contemporary “real‐world” setting alongside routine clinical care has not been specifically established. Furthermore, the incremental value of an inclusive approach with the analysis of newer, lower‐priority genes with indirect links to cardiac or skeletal myopathies is unknown.
We thus aimed to evaluate the rate of molecular diagnosis in a prospectively recruited cohort of patients with DCM using a WES approach.
Subjects and Methods
Study Design and Participants
Eligible participants were prospectively recruited from April 6, 2016 to September 24, 2017 from outpatient services of 4 tertiary cardiovascular centers in Melbourne Australia (Austin Health, Melbourne Health, Monash Health, and the Royal Children's Hospitals). The authors declare that all supporting data are available within the article and its online supplementary files.
The study cohort comprised patients for whom it was considered clinically appropriate to offer genetic testing in addition to their routine clinical care. We recruited patients with idiopathic DCM who were either (1) diagnosed under the age of 40 years, or (2) had a family history (≥2 members per family including the proband) of DCM and/or early (<35 years) sudden unexplained death. Family history was extensively investigated by trained genetic counselors in centers with expertise in genetic cardiomyopathies and a ≥3‐generation pedigree was constructed with available information. Patients with known nongenetic causes or patients who had undergone prior genetic testing involving cardiomyopathy‐related genes were excluded from the study.
Children (age <18 years) were only included after exclusion of metabolic, mitochondrial, and infective causes of cardiomyopathy. For patients suspected of having an infectious cause of cardiomyopathy (eg, viral myocarditis), viral cultures and/or titers were performed for the most common causes (eg, enterovirus, respiratory viruses, influenzae, adenovirus, parvovirus B19, Epstein‐Barr virus, cytomegalovirus, and human herpes 6). Children with suspected mitochondrial disorders presenting with distinct clinical syndromes including skeletal myopathies, neurological, and developmental symptoms were subjected to an extensive evaluation with involvement of a multidisciplinary team including geneticists and neurologists and by testing for mitochondrial mutations. Given the increased risks from endomyocardial biopsy, it was performed selectively, with the decision assessed individually for each patient based on the risk and benefit of the procedure. Endomyocardial biopsy was reserved for patients who required catheterization for another reason and/or those needing advanced heart failure management. A typical metabolic screen included assessment of serum amino acids, urine organic acids, acylcarnitine profile, lactate, pyruvate, electrolytes, glucose, and creatinine phosphokinase.
Potential participants were discussed by a panel of study investigators and the treating clinicians (JR, MW, IM, EL, SL, RW, TT, AT, DZ, JV, DH, and PJ) to determine eligibility before obtaining signed informed consent. All patients and/or parents provided written informed consent after genetic counseling regarding the test. To determine whether there were further affected members, family history was revisited in all patients during a mandated genetic counseling follow‐up consultation.
The study was part of the Melbourne Genomics Health Alliance project (http://www.melbournegenomics.org.au).13 The study received Human Research Ethics Committee approval (HREC/13/MH/326) from the Melbourne Health Human Research Committee. The investigation conforms with the principles outlined in the Declaration of Helsinki. Participants or parents provided written informed consent after genetic counseling.
WES, Variant Detection, and Variant Filtering
We performed WES, variant detection, and filtering at the Victorian Clinical Genetics Services Laboratory as described previously.8, 14 The Nextera Rapid Capture Exome kit by Illumina (San Diego, CA) was used. Variants were characterized using the Melbourne Genomics Health Alliance shared bioinformatics pipeline, Cpipe14 version 2.3. Cpipe is a clinically oriented pipeline built on Bpipe,15 and includes the Burrows‐Wheeler Aligner (BWA‐0.7.5a‐r405)16 and the GenomeAnalysis ToolKit (GATK version 3.6)17 using HaplotypeCaller for variant calling.
Variants were assessed using the Melbourne Genomics variant curation database, a modification of the Leiden Open Variation Database between April 6, 2016 and September 24, 2017.18 Variants were prioritized based on a predetermined gene list for all participants and on predicted effect (Variant Prioritisation Index). The gene list was selected following extensive review of literature, online databases, and expert opinion. The list (Table S1) comprised high‐priority or tier 1 clinical cardiomyopathy genes (n=74) and lower‐priority or tier 2 genes that comprised general myopathy, mitochondrial genes, and other new or recently reported cardiomyopathy genes (n=173). If no pathogenic or likely pathogenic (P/LP) variants were identified following analysis of tier 1 genes, then variants in the tier 2 gene list were curated. Variants were rigorously classified based on principles outlined by the American College of Medical Genetics and Genomics (ACMG) standards for interpretation of sequence variants.19 The in silico tools used were Polyphen2,20 SIFT,21 and Mutation Taster22 and CADD.23 Allele frequency was obtained from the Genome Aggregation Database (gnomADbrowser: http://gnomad.broadinstitute.org; accessed January 15, 2019). The disease database used was ClinVar (http://www.ncbi.nlm.nih.gov/clinvar; accessed January 15, 2019). Variant classifications were reviewed in a multidisciplinary team meeting attended by clinical geneticists, cardiologists, genetic counselors, scientists, nurses, and bioinformaticians. Families with variant of uncertain significance (VUS) considered of probable clinical relevance were all invited for cosegregation testing. To avoid delays with initial release of results, our study protocol did not mandate that segregation testing be performed before initial variant adjudication. Variants were therefore adjudicated without results of segregation testing except in 1 individual with a suspected de novo variant, where segregation studies could be promptly performed. The processes for participant recruitment and data analysis are summarized (Figure 1).
Figure 1.
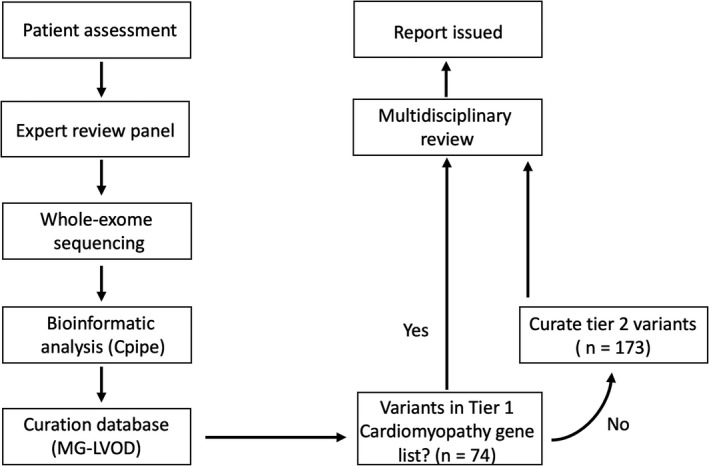
Processes of patient enrollment and variant curation. LP∕P indicates likely pathogenic or pathogenic; MG‐LOVD, Melbourne Genomics‐Leiden Open Variation Database.
Exome sequencing results emanating from this laboratory have previously been validated across a wide spectrum of clinical disorders and are accredited by the Australian National Association of Testing Authorities (NATA) for routine clinical use, without any mandate of orthogonal validation of results.8, 24, 25, 26, 27
Challenges exist in assigning pathogenicity to rare truncating Titin (TTN) variants given the prevalence in the general population. On the basis of a recent, comprehensive study of the TTN gene and its transcripts, nonsense, frameshift, and canonical splice site TTN variants, particularly those that truncate both principal isoforms of TTN and/or reside towards the C terminus, were considered to have a higher likelihood of pathology.28 In contrast, truncations that occur in novex‐specific exons or other infrequently used TTN exons were considered less likely to be deleterious.28
Other approaches including single‐nucleotide polymorphism microarray analysis and/or multiplex ligation‐dependent probe amplification were performed as needed either before recruitment or following review by the multidisciplinary team (N=5). Dedicated Sanger sequencing was used when thought to be clinically appropriate to genotype further family members to observe whether the variant cosegregated with disease. Formal quantification of cosegregation was not made.
Statistical Analysis
Statistical analysis was performed using STATA, version 15.1 (Statacorp., College Station, TX). Normally distributed continuous variables are expressed as mean±SD and non‐normally distributed data are expressed as the median and interquartile range. Student t test or the Mann Whitney U test (for non‐normally distributed data) was used to assess differences in continuous variables. Categorical variables are expressed as counts and percentages and compared using Fisher exact or χ2 tests.
The main outcome of the study was the proportion of patients with P/LP variants. Adverse clinical outcomes such as heart failure hospitalization, heart transplantation, and death were collected by experienced investigators via medical records review and by contacting each patient and/or the nominated general practitioner for additional information.
Results
Of 90 eligible patients evaluated, we enrolled 83 unrelated probands with DCM. The 7 patients excluded had a probable nongenetic cause for their condition. The clinical characteristics of the probands are presented in Table 1. The majority of participants were adults (≥18 years, n=72, 87%). The overall cohort comprised 57% males (n=47) with a mean±SD age of 36±19 years. Most had a family history of cardiomyopathy and/or early sudden unexplained death (n=60, 72%). Forty‐eight of 83 probands (58%) had a family history of only cardiomyopathy and 6 of 83 (7%) had a family history of only sudden death. The remaining participants (n=23, 28%) did not have a known family history of cardiomyopathy or sudden death affecting first‐ and second‐degree relatives and met study inclusion criteria on the basis of a diagnosis of cardiomyopathy under the age of 40 years. Most families with familial DCM/sudden cardiac death had 1 additional family member (33 of 60, 55%) with DCM/sudden cardiac death, with 15 (25%) having 2 and 12 (20%), respectively, having >2. There was a high incidence of atrial (n=18, 22%) and ventricular arrhythmias (n=10, 12%). With regard to pharmacological therapy, of the entire cohort, 72 (87%) were on angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers, 69 (83%) on β‐blockers, and 38 (46%) on mineralocorticoid receptor antagonists.
Table 1.
Proband Characteristics
| Whole Cohort (n=83) | Isolated (n=23) | Familial (n=60) | P Value | |
|---|---|---|---|---|
| Age at diagnosis (y) | 37±19 | 25±14 | 42±18 | <0.001a |
| Male | 47 (57%) | 11 (48%) | 36 (60%) | 0.317 |
| LVEF at time of diagnosis, % | 31±13 | 30±14 | 31±13 | 0.868 |
| Number of first‐degree family relatives | 4 (3–6) | 4 (3–5) | 5 (4–6) | 0.131 |
| Number of second‐ or higher‐degree relatives | 15 (10–20) | 15 (10–26) | 15 (11–20) | 0.983 |
| Comorbidities/therapy at time of recruitment | ||||
| Prior heart failure admission | 51 (61%) | 14 (61%) | 37 (62%) | 0.947 |
| Atrial fibrillation | 18 (22%) | 5 (22%) | 13 (22%) | 0.977 |
| Ventricular tachycardia | 10 (12%) | 5 (22%) | 5 (8%) | 0.106 |
| Diabetes mellitus | 10 (12%) | 3 (13%) | 7 (12%) | 0.883 |
| β‐Blocker | 69 (83%) | 18 (78%) | 51 (85%) | 0.269 |
| ACEi/ARB | 72 (87%) | 19 (83%) | 53 (88%) | 0.257 |
| Mineralocorticoid receptor antagonists | 38 (46%) | 9 (39%) | 29 (48%) | 0.377 |
| Implantable cardiac defibrillator | 22 (27%) | 4 (17%) | 18 (30%) | 0.244 |
| Cardiac resynchronization therapy | 14 (17%) | 5 (22%) | 9 (15%) | 0.463 |
Values are mean±SD or median (interquartile range) or n (%). ACEi indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; LVEF, left ventricular ejection fraction.
Denotes p < 0.05.
Genetic Diagnosis
We generated high‐coverage WES data with 97% of tier 1 and tier 2 genes covered by >20 reads. Genes without 100% of coverage of >20 reads are listed in Table S2. Of the 83 probands, 10 (12.0%) had a pathogenic (n=1) or likely pathogenic (n=9) variants (Table 2 and Figure 2).29, 30, 31, 32, 33 There was a nominally higher rate of genetic diagnosis in pediatric compared with adult cases (18% versus 11%). Among the pediatric probands (n =11), there was no significant difference in the rate of P/LP variants in those with (1 of 5, 20%) or without a family history of DCM or sudden cardiac death (1 of 6, 17%). Of the adult cases (n =72), there was a nominally higher number of patients with P/LP variants in those with a family history (7 of 55, 13%) compared with isolated cases (1 of 17, 6%), though this did not reach statistical significance (P=0.433). Each ACMG rule activated to classify P/LP variants as well as rare VUS is detailed in Tables S3 and S4. Eight of 83 patients (9.6%) had a truncating TTN VUS (Table 3). Taken together, P/LP variants were found in 8 genes (all within tier 1 genes), 2 (20%) of which are not included in a standard commercially available DCM panel (Table S5). However, as the 2 patients involved had arrhythmogenic phenotypes, it is likely that they would have received a comprehensive cardiac panel comprising these 2 genes (DSP and RYR2).
Table 2.
Characteristics of Patients With Likely Pathogenic and Pathogenic Variants
| Subject, Sex | Age(y) | Phenotype | Gene (Ref Seq Access number): Variant | Predicted Canonical Protein Isoform | Variant Co‐Segregationa | In Silico Prediction Tools | Previously Reported Pathogenic Variant | Functional Data |
|---|---|---|---|---|---|---|---|---|
| Pathogenic | ||||||||
| 2, F | 65 | LV dysfunction diagnosed following VF arrest. ECG: Left anterior fascicular block, septal Q waves. | NM_001001432.2(TNNT2):c.517C>T | NP_001001432.1(TNNT2):p.(Arg173Trp) | Not performed | Consistently pathogenic | Yes30, 32 | Yes33 |
| Likely pathogenic | ||||||||
| 4, M | 53 | Moderate to severe LV dysfunction; ECG: Leftward axis, mild QT prolongation. | NM_003319.4(TTN):c.69855dupA | NP_001254479.1TTN):p.(Glu32351Argfs*6) | Not performed | Consistently pathogenic | No, other truncating variants in region reported as pathogenic | No |
| 6, F | 29 | Severe biventricular dysfunction. ECG: rightward axis, deep widespread T wave inversion and QT prolongation. | NM_003319.4(TTN):c.21991G>T | NP_001254479.1(TTN):p.(Glu16396*) | Not performed | Consistently pathogenic | No. Downstream variants reported as pathogenic (ClinVar) | No |
| 8, F | 52 | Moderate LV dysfunction. ECG: left anterior fascicular block. | NM_003319.4(TTN):c.44430_44449del | NP_001254479.1(TTN):p.(Val23876Argfs*16) | 1/0/0 | Consistently pathogenic | No | No |
| 9, F | 49 | Familial dilated cardiomyopathy, ECG: atrial fibrillation | NM_133378.4(TTN):c.10361‐3042T>A | NP_001254479.1(TTN):p.(Leu3944*) | 1/0/0 | Consistently pathogenic | No | No |
| 1, F | 23 | Mild LV dysfunction; Cardiac arrest because of ventricular fibrillation (VF). Family history of SCD. Recurrent ventricular tachycardia. ECG: low voltage R waves. | NM_004415.2(DSP):c.2638dupG | NP_004406.2(DSP):p.(Asp880Glyfs*14) | 1/0/0 | Consistently pathogenic | No. Downstream variants reported as pathogenic (ClinVar) | No |
| 3, M | 12 | Biventricular dysfunction diagnosed in the setting of multifocal atrial tachycardia. Multiple foci in the left and right atria on EPS. | NM_001035.2(RYR2):c.14570T>Ab | NP_001026.2(RYR2):p.(Ile4857Asn) | 0/0/0 | Consistently pathogenic | No | No |
| 5, M | 31 | Recurrent atrial and ventricular arrhythmias. Biventricular dysfunction. Family history of SCD. ECG: RBBB. | NM_170707.3(LMNA):c.1608+1G>A | NP_733821.1(LMNA):p.? | 1/0/0 | Predicted to cause aberrant splicing | Yes29 | No |
| 7, F | 57 | Moderate LV dysfunction. ECG: left anterior fascicular block, widespread T wave inversion. | NM_004281.3(BAG3):c.108G>A | NP_004272.2(BAG3):p.(Trp36*) | Not performed | Consistently pathogenic | No. Downstream variants reported as pathogenic (ClinVar) | No |
| 10, M | 5 | LV noncompaction with mild LV dilatation and normal LV systolic function. | NM_005159.4(ACTC1):c.998C>T | NP_005150.1(ACTC1):p.(Ala333Val) | 1/0/0 | Consistently deleterious | Yes31 | No |
No allele frequencies were available as all were absent from GnomAD. References are shown as superscripts. EPS indicates electrophysiology studies; LV, left ventricular; RBBB, right bundle branch block; SCD, sudden cardiac death; VF, ventricular fibrillation.
Data presented as A/B/C where A=number of affected family members (excluding the proband) who carry the variant, B=number of affected members who do not carry the variant, C=number of unaffected members who carry the variant.
Subsequent testing of parental samples indicated that this variant is likely because of a de novo event.
Figure 2.
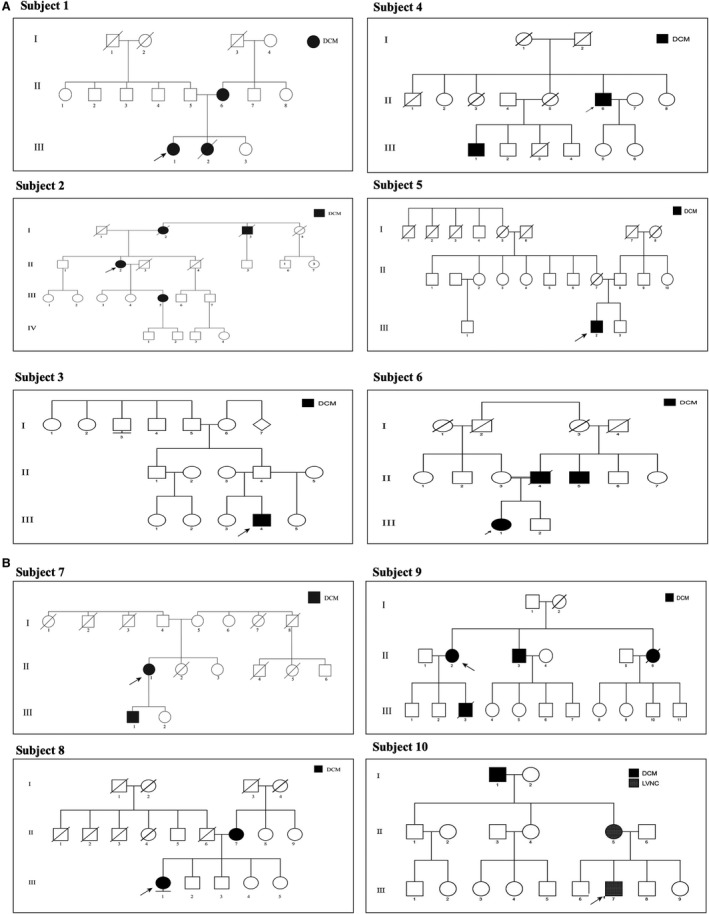
A and B, Pedigrees for patients with pathogenic and likely pathogenic variants. Arrow denotes proband. DCM indicates dilated cardiomyopathy; LVNC, Left ventricular non‐compaction.
Table 3.
TTN Variants of Uncertain Significance
| Subject, Sex | Exon Number | Canonical Isoform Nomenclature | Predicted Canonical Protein Isoform | Affecting N2BA & N2B | Protein Region | In Silico Tools | PSI | Population Database |
|---|---|---|---|---|---|---|---|---|
| 14, M | 29 | NM_001267550.1:c.6825del | NP_001254479.1(TTN):p.(Asp2275Glufs*2) | Both | I‐band | Pathogenic | 1.0 | Absent |
| 15, M | 154 | NM_001267550.1:c.80174_80202dup | NP_001254479.1(TTN):p.(Ser26735Glnfs*9) | Both | A‐band | Pathogenic | 1.0 | Absent |
| 16, M | Splice‐site (Intron 54) | NM_001267550.1:c.41609‐2A>G | NP_001254479.1(TTN):p.? | Both | N/A | Aberrant splicing | 1.0 | Absent |
| 17, M | 94 | NM_001267550.1:c.50170C>T | NP_001254479.1(TTN):p.(Arg16724*) | Both | A‐band | Pathogenic | 0.60 | Absent |
| 18, F | 29 | NM_001267550.1:c.60961_60962dupAA | NP_001254479.1(TTN):p.(Asn20321Lysfs*9) | Both | A‐band | Pathogenic | 1.0 | Absent |
| 19, M | 153 | NM_001267550.1:c.69422_69426delinsAAAAGGACCC | NP_001254479.1(TTN):p.(Gly23141Glufs*38) | Both | A‐band | Pathogenic | 0.48 | 0.04% |
| 20, F | 186 | NM_001267550.1:c.101107C>T | NP_001254479.1(TTN):p.(Arg33703*) | Both | M‐band | Pathogenic | 0.07 | Absent |
| 21, M | 9 | NM_001267550.1:c.50296C>T | NP_001254479.1(TTN):p.(Arg16766*) | Both | A‐band | Pathogenic | 0.64 | <0.00041% |
N∕A indicates not applicable; PSI, percentage spliced in.
Of note, 59 variants of uncertain significance were identified in tier 1 genes and 3 variants of uncertain significance were identified in tier 2 genes. On average there were 0.74 (61/83) variants of uncertain significance per case. Time to curate each variant was approximated to be 0.75 person‐hours. Five of 83 (6%) participants had variants of unknown significance according to strict ACMG criteria but classified as either P/LP by other clinical laboratories. One individual was found to have 1p36.32 deletion on chromosome SNP microarray testing.
Impact of Genetic Diagnosis on Patient Management
Of the 10 patients with a P/LP result, clinical management was altered following genetic testing in 1 individual (10%). One patient (Subject 5, Table 2) underwent implantable cardiac defibrillator implantation for primary prevention of sudden cardiac death following identification of a LMNA variant.
Clinical Outcomes
Median follow‐up time was 501 days. Over the follow‐up period, there was no significant difference in the incidence of heart failure–related admissions (2 of 10 [20%] versus 6 of 73 [8%], P=0.236), treatment with left ventricular assist device, or cardiac transplantation (0 [0%] versus 3 of 73 [4%], P=0.514) or death (1 of 10 [10%] versus 2 of 73 [3%], P=0.249) between those with or without P/LP variants.
Discussion
We performed singleton WES as a first‐line genomic test in a prospectively enrolled cohort of patients with DCM enrolled from routine clinical practice. The major finding of the present study was that WES identified a likely P/LP variant in 12% of patients with DCM in a “real‐world” clinical setting. A further 10% of patients had truncating TTN variants classified as VUS. No variants in the tier 2 (extended gene set data) met clinical criteria for pathogenicity. Concerningly, 5 (6%) participants had variants of unknown significance according to strict ACMG criteria, but such variants were classified as either P/LP by other clinical laboratories.
Our data indicate that with stringent classification according to contemporary ACMG guidelines, the rate of detection of pathogenic variants in the present study performed in a clinical setting is lower than previously reported.2, 11 Indeed, concerningly, several lines of evidence suggest previous overreporting of many rare protein‐altering variants as pathogenic, with such variants subsequently being detected in the general population, raising doubts with regard to the reliability of previously released genetic results.34, 35, 36, 37, 38 Assigning pathogenicity of rare genetic variants is particularly challenging with the enormous TTN gene that encodes titin, the largest human protein spanning the entire sarcomere of heart and skeletal muscle tissue.35 Truncating TTN variants have been shown to occur in up to 3% of apparently healthy individuals.35, 39 It is therefore imperative to distinguish truncating TTN variants that cause disease from those that do not. We note that other laboratories11 classify many such variants as pathogenic and hence comparatively this alone would account for the lower rate of pathogenic diagnoses in the present study.
Detailed exon use data from the human myocardium demonstrate that I‐band titin contained a high proportion of symmetrical exons with a very low “percentage spliced‐in” score in expressed exons.28 Variants that truncate exons consistently expressed in both of the dominant N2BA and N2B isoforms have an increased likelihood of pathogenicity with truncations involving the C‐terminal portion of the protein, encoding the A‐band titin, being the predominant class of pathogenic variants.28 Use of the aforementioned criteria help resolve the majority of truncating TTN variants, but strikingly truncating TTN variants that fit the criteria for pathogenicity can still be seen in ≈1% of apparently normal individuals.40. It is nonetheless important to emphasize that DCM may develop at any age and therefore population databases have incomplete ascertainment of cardiovascular outcomes. It is therefore possible that although the high frequency of some of these TTN variants preclude them from being the driving causative factors, lower‐frequency variants may be making a significant contribution to phenotype. Indeed, in a detailed analysis of population data involving >71 000 individuals, Haggerty et al41 recently reported the association between high percentage spliced‐in truncating TTN variants and preclinical cardiac abnormalities even in those without a DCM diagnosis.
At the present time, we have chosen to conservatively classify all apart from the most likely to be pathogenic truncating TTN variants as VUS (Table 3) until further evidence is gathered. Of the 8 variants in this cohort listed in Table 3, 4 do not have a high percentage spliced‐in (subjects 17, 19, 20, and 21) and 2 (subjects 14 and 16) do not encode the A‐band, rendering the pathogenicity of these variants uncertain. The remaining 2 rare variants have a higher likelihood of pathogenicity (subjects 15 and 18) as they involve the A‐band and have high percentage spliced‐in scores and hence familial cosegregation studies are planned to further establish their pathogenicity.
Other reasons for a lower yield of identifying P/LP variants in this study include the careful interpretation of ACMG criteria in the interpretation of loss of function variants. This criterion is the only one assigned PVS1 (very strong evidence for pathogenicity) and hence extra caution was used in the present analysis to prevent overestimation of variant impact and subsequent classification. Use of recent recommendations to interpret loss of function variants from ClinGen's Sequence Variant Interpretation working group will help promote consistent interpretation of loss of function variants among laboratories.42 In DCM, further refinement of these criteria will be required by integrating gene‐level and variant‐level characteristics.34
Interestingly, there was no significant difference with regard to family history or age of onset between those with and without a molecular diagnosis. These findings highlight that lack of family history of cardiomyopathy and later age of disease onset do not exclude a genetic cause, especially as certain environmental factors may interact to induce pathology at a later stage in life.39
Despite the growing number of DCM‐related genes, the incremental value of an extended gene set, beyond which would be included in a comprehensive cardiac panel, has not been convincingly demonstrated in the present study. Not unsurprisingly, no variants in the extended (tier 2) gene set met clinical criteria for pathogenicity using stringent criteria of the ACMG. By definition, genes included in the extended data set included those that were of lower priority and/or newly reported, and thus one of the challenges in evaluating variants in the extended gene set is paucity of genetic and functional effects for DCM association. These findings are in keeping with a recent study of whole genome sequencing in DCM that included 406 cardiac‐enriched genes but found that no variants in the extended gene set met clinical criteria.12 Importantly, however, as evidence of disease associations build in the extended gene list, data from exome sequencing, much of which cannot be obtained from panel sequencing, can be reanalyzed in the future.
Our results indicate that with the use of our Melbourne Genomics Health Alliance shared bioinformatics pipeline, WES with a targeted analysis was time efficient with an average of 0.74 variants of uncertain significance per case and 0.75 person‐hours needed to interpret each of these variants. The manpower costs of expert clinical reporting would therefore be expected to be equivalent for panel testing and WES.
The strengths of this study include its demonstration of the utility of singleton, genomic testing in “real‐life” care of patients with DCM. Only ≈70% of patients had a family history of cardiomyopathy, and in fact most of the probands only had 1 other first‐ or second‐degree affected relative and therefore the likelihood of finding pathogenic variants are unsurprisingly substantially lower than in large multiplex families typically observed in research settings. Some important limitations should be acknowledged. Although family history was extensively investigated and every effort was made to invite first‐ and second‐degree relatives of probands for further cardiovascular evaluation, this was not comprehensively adhered to. Thus, the presence of a family history of DCM may have been underrecognized in some probands. WES has limited sensitivity for the detection of insertion/deletion variants over 10 bp in length, copy number variation, translocations, chromosomal rearrangements, and repeat expansions. Other limitations of exome sequencing include the potential of missing variants because of variability in breadth of coverage involving certain genes such as LMNA and low mappability of certain sarcomeric genes. Comprehensive cardiac gene panels with associated higher coverage and lower costs may have achieved similar findings. However, WES has added the advantage for future re‐analysis of data as new DCM‐related genes are identified.
In conclusion, WES with targeted gene analysis is an effective diagnostic tool for patients with DCM in a clinical setting. The data from this study indicate that with stringent classification, according to current evidence, the rate of detection of pathogenic variants in DCM is lower than previously reported. Ongoing efforts to improve adherence to these guidelines may prevent erroneous misclassification of nonpathogenic variants in DCM genetic testing and cascade screening.
Sources of Funding
Melbourne Genomics Health Alliance is funded by the State Government of Victoria (Department of Health and Human Services) and the 10 members.
Disclosures
None.
Supporting information
Table S1. Gene List
Table S2. Genes Without 100% of Coverage of >20 Reads
Table S3. ACMG Criteria Used to Guide Classifying Likely Pathogenic and Pathogenic Variants
Table S4. ACMG Criteria Used to Guide Classifying Rare Variants of Unknown Significance
Table S5. List of Genes With Pathogenic or Likely Pathogenic Variants and Coverage by Commercially Available Gene Panels in Victoria, Australia (October 2018)
Acknowledgments
We thank Ms Fran Maher for constructing the pedigrees. We are grateful to the patients and their families for participation in the study. We also thank the Genetic counselors and referring cardiologists at Austin Health, Melbourne Health, Monash Health, and the Royal Children's Hospital.
(J Am Heart Assoc. 2020;9:e013346 DOI: 10.1161/JAHA.119.013346.)
References
- 1. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. [DOI] [PubMed] [Google Scholar]
- 2. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 3. Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. 2016;67:2996–3010. [DOI] [PubMed] [Google Scholar]
- 4. Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez‐Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Spaendonck‐Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, de Boer RA, Jongbloed JD, Pinto YM, van Tintelen JP. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur J Heart Fail. 2013;15:628–636. [DOI] [PubMed] [Google Scholar]
- 6. Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–715. [DOI] [PubMed] [Google Scholar]
- 7. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole‐exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, Yeung A, Peters H, Mordaunt D, Cowie S, Amor DJ, Savarirayan R, McGillivray G, Downie L, Ekert PG, Theda C, James PA, Yaplito‐Lee J, Ryan MM, Leventer RJ, Creed E, Macciocca I, Bell KM, Oshlack A, Sadedin S, Georgeson P, Anderson C, Thorne N; Melbourne Genomics Health A , Gaff C, White SM. A prospective evaluation of whole‐exome sequencing as a first‐tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–1096. [DOI] [PubMed] [Google Scholar]
- 9. Herkert JC, Abbott KM, Birnie E, Meems‐Veldhuis MT, Boven LG, Benjamins M, du Marchie Sarvaas GJ, Barge‐Schaapveld D, van Tintelen JP, van der Zwaag PA, Vos YJ, Sinke RJ, van den Berg MP, van Langen IM, Jongbloed JDH. Toward an effective exome‐based genetic testing strategy in pediatric dilated cardiomyopathy. Genet Med. 2018;20:1374–1386. [DOI] [PubMed] [Google Scholar]
- 10. Nouhravesh N, Ahlberg G, Ghouse J, Andreasen C, Svendsen JH, Haunso S, Bundgaard H, Weeke PE, Olesen MS. Analyses of more than 60,000 exomes questions the role of numerous genes previously associated with dilated cardiomyopathy. Mol Genet Genomic Med. 2016;4:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, Postma AV, Bronk M, van Rijsingen IA, de Haij S, Biagini E, van Haelst PL, van Wijngaarden J, van den Berg MP, Wilde AA, Mannens MM, de Boer RA, van Spaendonck‐Zwarts KY, van Tintelen JP, Pinto YM. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail. 2017;19:512–521. [DOI] [PubMed] [Google Scholar]
- 12. Minoche AE, Horvat C, Johnson R, Gayevskiy V, Morton SU, Drew AP, Woo K, Statham AL, Lundie B, Bagnall RD, Ingles J, Semsarian C, Seidman JG, Seidman CE, Dinger ME, Cowley MJ, Fatkin D. Genome sequencing as a first‐line genetic test in familial dilated cardiomyopathy. Genet Med. 2019;2:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaff CL, I MW, S MF, D PH, Clark J, P MW, South M, A HS. Preparing for genomic medicine: a real world demonstration of health system change. NPJ Genom Med. 2017;2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sadedin SP, Dashnow H, James PA, Bahlo M, Bauer DC, Lonie A, Lunke S, Macciocca I, Ross JP, Siemering KR, Stark Z, White SM, Melbourne Genomics Health A, Taylor G, Gaff C, Oshlack A, Thorne NP. Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med. 2015;7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sadedin SP, Pope B, Oshlack A. Bpipe: a tool for running and managing bioinformatics pipelines. Bioinformatics. 2012;28:1525–1526. [DOI] [PubMed] [Google Scholar]
- 16. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics. 2010;26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy‐Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD vol 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563. [DOI] [PubMed] [Google Scholar]
- 19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adabag AS, Maron BJ, Appelbaum E, Harrigan CJ, Buros JL, Gibson CM, Lesser JR, Hanna CA, Udelson JE, Manning WJ, Maron MS. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51:1369–1374. [DOI] [PubMed] [Google Scholar]
- 21. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 23. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, Chong B, Phelan D, Brett GR, Creed E, Jarmolowicz A, Yap P, Walsh M, Downie L, Amor DJ, Savarirayan R, McGillivray G, Yeung A, Peters H, Robertson SJ, Robinson AJ, Macciocca I, Sadedin S, Bell K, Oshlack A, Georgeson P, Thorne N, Gaff C, White SM. Diagnostic impact and cost‐effectiveness of whole‐exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 2017;171:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Melbourne Genomics Health A, Gaff C, White SM, Tan TY. Exome sequencing has higher diagnostic yield compared to simulated disease‐specific panels in children with suspected monogenic disorders. Eur J Hum Genet. 2018;26:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walsh M, Bell KM, Chong B, Creed E, Brett GR, Pope K, Thorne NP, Sadedin S, Georgeson P, Phelan DG, Day T, Taylor JA, Sexton A, Lockhart PJ, Kiers L, Fahey M, Macciocca I, Gaff CL, Oshlack A, Yiu EM, James PA, Stark Z, Ryan MM; Melbourne Genomics Health Alliance . Diagnostic and cost utility of whole exome sequencing in peripheral neuropathy. Ann Clin Transl Neurol. 2017;4:318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stark Z, Schofield D, Alam K, Wilson W, Mupfeki N, Macciocca I, Shrestha R, White SM, Gaff C. Prospective comparison of the cost‐effectiveness of clinical whole‐exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19:867–874. [DOI] [PubMed] [Google Scholar]
- 28. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O'Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O'Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ambrosi P, Mouly‐Bandini A, Attarian S, Habib G. Heart transplantation in 7 patients from a single family with limb‐girdle muscular dystrophy caused by lamin A/C mutation. Int J Cardiol. 2009;137:e75–e76. [DOI] [PubMed] [Google Scholar]
- 30. Campbell N, Sinagra G, Jones KL, Slavov D, Gowan K, Merlo M, Carniel E, Fain PR, Aragona P, Di Lenarda A, Mestroni L, Taylor MR. Whole exome sequencing identifies a troponin T mutation hot spot in familial dilated cardiomyopathy. PLoS One. 2013;9:e78104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–1694. [DOI] [PubMed] [Google Scholar]
- 32. Sommese RF, Nag S, Sutton S, Miller SM, Spudich JA, Ruppel KM. Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross‐bridge kinetics of human beta‐cardiac myosin. PLoS One. 2013;9:e83403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun N, Yazawa M, Liu J, Han L, Sanchez‐Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC. Patient‐specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Horvat C, Johnson R, Lam L, Munro J, Mazzarotto F, Roberts AM, Herman DS, Parfenov M, Haghighi A, McDonough B, DePalma SR, Keogh AM, Macdonald PS, Hayward CS, Roberts A, Barton PJR, Felkin LE, Giannoulatou E, Cook SA, Seidman JG, Seidman CE, Fatkin D. A gene‐centric strategy for identifying disease‐causing rare variants in dilated cardiomyopathy. Genet Med. 2019;21:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pan S, Caleshu CA, Dunn KE, Foti MJ, Moran MK, Soyinka O, Ashley EA. Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation. Circ Cardiovasc Genet. 2012;5:602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Norton N, Robertson PD, Rieder MJ, Zuchner S, Rampersaud E, Martin E, Li D, Nickerson DA, Hershberger RE; National Heart, Lung and Blood Institute GO Exome Sequencing Project . Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet. 2012;5:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave‐Castillo LM, Wolfgeher D, McNally EM. Population‐based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haggerty CM, James CA, Calkins H, Tichnell C, Leader JB, Hartzel DN, Nevius CD, Pendergrass SA, Person TN, Schwartz M, Ritchie MD, Carey DJ, Ledbetter DH, Williams MS, Dewey FE, Lopez A, Penn J, Overton JD, Reid JG, Lebo M, Mason‐Suares H, Austin‐Tse C, Rehm HL, Delisle BP, Makowski DJ, Mehra VC, Murray MF, Fornwalt BK. Electronic health record phenotype in subjects with genetic variants associated with arrhythmogenic right ventricular cardiomyopathy: a study of 30,716 subjects with exome sequencing. Genet Med. 2017;19:1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ware JS, Amor‐Salamanca A, Tayal U, Govind R, Serrano I, Salazar‐Mendiguchia J, Garcia‐Pinilla JM, Pascual‐Figal DA, Nunez J, Guzzo‐Merello G, Gonzalez‐Vioque E, Bardaji A, Manito N, Lopez‐Garrido MA, Padron‐Barthe L, Edwards E, Whiffin N, Walsh R, Buchan RJ, Midwinter W, Wilk A, Prasad S, Pantazis A, Baski J, O'Regan DP, Alonso‐Pulpon L, Cook SA, Lara‐Pezzi E, Barton PJ, Garcia‐Pavia P. Genetic etiology for alcohol‐induced cardiac toxicity. J Am Coll Cardiol. 2018;71:2293–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watkins H. Tackling the achilles’ heel of genetic testing. Sci Transl Med. 2015;7:270 fs1. [DOI] [PubMed] [Google Scholar]
- 41. Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, Hu Y, Judy R, Kelly MA, Kember RL, Lester Kirchner H, Leader JB, Liang L, McDermott‐Roe C, Babu A, Morley M, Nealy Z, Person TN, Pulenthiran A, Small A, Smelser DT, Stahl RC, Sturm AC, Williams H, Baras A, Margulies KB, Cappola TP, Dewey FE, Verma A, Zhang X, Correa A, Hall ME, Wilson JG, Ritchie MD, Rader DJ, Murray MF, Fornwalt BK, Arany Z. Genomics‐first evaluation of heart disease associated with titin‐truncating variants. Circulation. 2019;140:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) . Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene List
Table S2. Genes Without 100% of Coverage of >20 Reads
Table S3. ACMG Criteria Used to Guide Classifying Likely Pathogenic and Pathogenic Variants
Table S4. ACMG Criteria Used to Guide Classifying Rare Variants of Unknown Significance
Table S5. List of Genes With Pathogenic or Likely Pathogenic Variants and Coverage by Commercially Available Gene Panels in Victoria, Australia (October 2018)