

Introduction: Pazopanib is an oral protein kinase inhibitor (PKI) that targets vascular endothelial growth factor (VEGF) receptors, fibroblastic growth factor receptors, platelet-derived growth factor receptors, and stem cell factor that inhibits VEGF-induced cellular proliferation. Pazopanib is approved for use in advanced renal cell carcinoma and subtypes of advanced soft-tissue sarcoma (Deguchi et al., 2018). Major adverse drug reactions of pazopanib include hypertension, high-grade hyponatremia and posterior reversible encephalopathy syndrome (PRES) (Berardi et al., 2016; Deguchi et al., 2018). In clinical trials, few investigations have been conducted to determine the aetiology of PKI-associated hyponatremia, the mechanism remains therefore unknown. Only rare cases of PKI-induced syndrome of inappropriate secretion of antidiuretic hormone (SIADH) (Largeau et al., 2019), and none with pazopanib, have been reported. PRES is a clinical and radiological entity where a bilateral white matter oedema, occurring predominantly in the posterior occipital and parietal lobes, is associated with several neurologic symptoms. Interestingly, a recent review suggests that SIADH could be a symptom of PRES (Largeau et al., 2019). To our knowledge, this is the first case published where pazopanib-induced PRES occurs contemporaneously with possible SIADH.
Case presentation: This report was prepared in accordance with the CAse REport (CARE) guidelines (Riley et al., 2017). A 73-year-old woman presented with high blood pressure and frontal headache to the oncology unit. Her medical history was significant for stage IV clear cell renal carcinoma, chronic hypertension on irbesartan/hydrochlorothiazide, which were prescribed at the same dosing for more than a year. She was recently started on pazopanib at 600 mg/d for metastatic clear cell renal carcinoma then was reduced to 400 mg/d on day 7 due to high blood pressure (Figure 1). On day 12 after pazopanib initiation, the patient developed severe frontal headache, nausea and high blood pressure (210/100 mmHg), leading to amlodipine therapy on day 14. The following day the patient was admitted in the oncology unit and reported headache, her blood pressure was 229/112 mmHg and heart rate was 90 beats per minute. Except a psychomotor retardation, neurological examination was without abnormality and the patient had no visual impairment.
Figure 1.
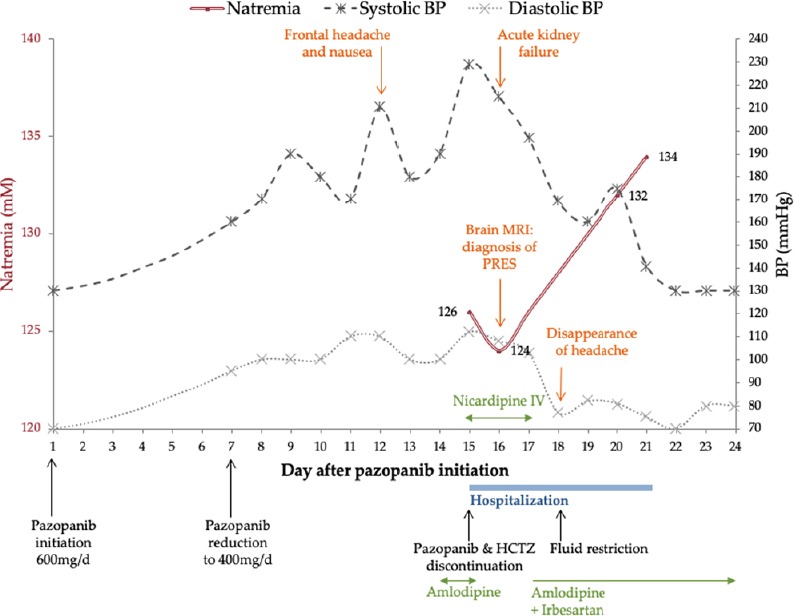
Clinical and biological time course.
BP: Blood pressure.
At admission (day 15) initial serum biochemistry was significant for a sodium of 126 mM, potassium of 2.9 mM, albumin of 46.5 g/L and 88 giga/L thrombocytopenia. Serum magnesium was not measured. Urinary electrolytes and serum osmolality were obtained to establish the aetiology of the patient’s euvolemic hyponatraemia. Plasma osmolality was 261.7 mOsm/kg. Urinary labs showed sodium of 51 mM, osmolality of 215 mOsm/kg and 2.6 g/L proteinuria. Thyroid stimulating hormone and serum cortisol were within normal limits, ruling out hypothyroidism and glucocorticoid deficiency, respectively. On day 16, the patient developed acute kidney failure where serum creatinine experienced a 1.4-fold increase from admission baseline. Brain magnetic resonance imaging (MRI) performed on day 16 showed typical imaging features of PRES with vasogenic oedema characterized by parieto-occipital hyperintense signal within the posterior white matter (Figure 2A and B). Brain MRI did not reveal central progression of the cancer, nor infarction, nor hemorrhage.
Figure 2.
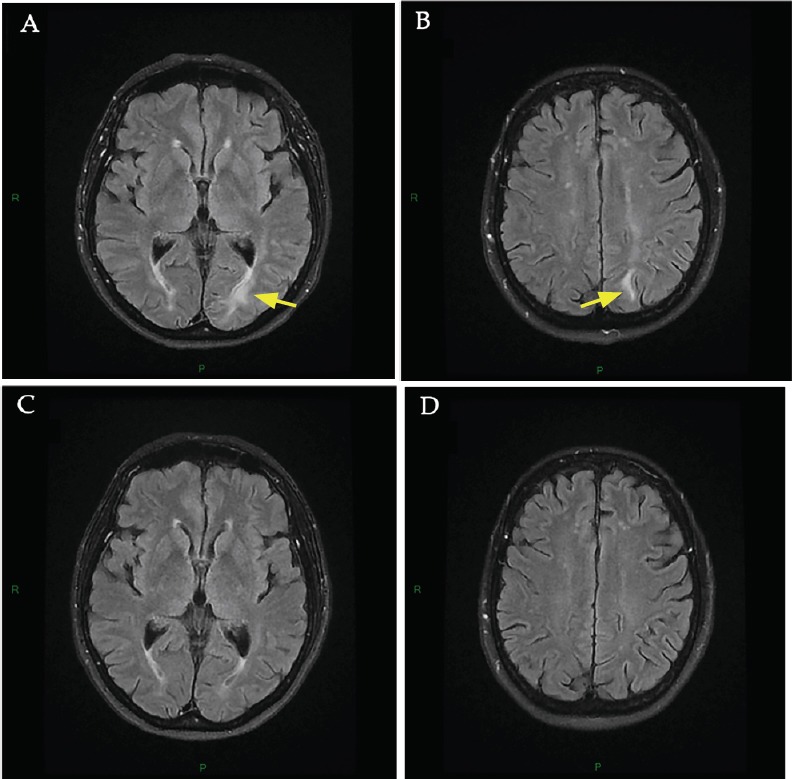
Brain MRI at onset of neurological disturbances (day 16 after pazopanib initiation) and 3-month follow-up.
(A, B) Brain MRI at onset of neurological disturbances. MRI at onset of PRES showed hyperintensities in the left occipital (A, arrow) and the left parietal (B, arrow) regions involving the white matter in T2-FLAIR sequence. No diffusion abnormalities were found in the diffusion weighted imaging sequence and apparent diffusion coefficient was increased. These lesions were consistent with a vasogenic edema of PRES. Major differential diagnoses were excluded including posterior reversible vasoconstriction syndrome (time-of-flight MRI), cerebral bleeding (T2* MRI) and stroke. (C, D) 3-month follow-up brain MRI. New brain MRI 3 months later showing complete resolution of the lesions of PRES in the left occipital (C) and the left parietal regions (D).
Treatments included pazopanib, amlodipine, irbesartan and hydrochlorothiazide discontinuation, administration of intravenous nicardipine (day 15) and fluid restriction (day 18). Antihypertensive therapy was switched to oral irbesartan and amlodipine on day 17. Despite this combination of antihypertensive agents, the patient’s blood pressure remained high until the normalization of natremia on day 21 (Figure 1), 6 days after pazopanib discontinuation.
The psychomotor retardation, headache and renal failure resolved on day 18. The patient was discharged 6 days after admission (day 21), with a serum sodium of 134 mM. One month after discharge, blood pressure and natremia were still normal. Hydrochlorothiazide was not reintroduced. After 2 months of drug discontinuation, pazopanib was restarted at 200 mg/d. A new brain MRI performed 3 months after discharge showed complete resolution of the PRES lesions (Figure 2C and D). The patient no longer had hyponatremia, headache or other iterative neurological recurrence.
Discussion: Drug-causality assessment in drug-induced PRES is difficult due to the fact that (1) the underlying diseases are also strongly linked to PRES (e.g., transplantation, active cancers, autoimmune disorders); (2) various drugs, often used in combination, can cause PRES; (3) delays of occurrence are extremely variable; and (4) incriminated drugs can be reintroduced without iterative PRES recurrence (Largeau et al., 2019). Nevertheless, the close temporal relationship (i.e., onset and improvement) between high blood pressure, hyponatremia, PRES and pazopanib treatment is consistent with the role of this drug. The role of hydrochlorothiazide in hyponatremia, administered and well tolerated for a long time (i.e., natremia of 136 mM before pazopanib initiation), is less evocative. The patient’s hyponatremia was consistent with drug-induced SIADH diagnostic criteria (i.e., euvolemic hyponatraemia with urine sodium > 40 mM and urine osmolality > 100 mOsm/kg, recovery after drug discontinuation and fluid restriction) whereas the recent use of the diuretic agent cannot allow to confirm this diagnosis (Ellison and Berl, 2007), without excluding it. Furthermore, other aetiology such as paraneoplastic syndrome, neurological and pulmonary disorders were ruled out.
Pazopanib has been associated with both hyponatremia (Berardi et al., 2016) and PRES (Deguchi et al., 2018) but PRES with pazopanib-induced SIADH has to date never been reported. PRES associated with pazopanib is supposed to be precipitated by endothelial dysfunction and high blood pressure induced by anti-VEGF therapy (Deguchi et al., 2018). The mechanism of hyponatremia associated with pazopanib is unclear but the role of VEGF pathway in sodium homeostasis has been suggested (Berardi et al., 2016). Another hypothesis could be a SIADH mechanism. SIADH has been associated with other PKI (Largeau et al., 2019) and a pathophysiologic link between PRES and SIADH may explain the association of these two syndromes.
Evidence for anti-VEGF therapy induced arginine vasopressin hypersecretion: Arginine vasopressin (AVP), also known as the antidiuretic hormone, is involved in the regulation of renal water reabsorption and urine protein excretion through renal V2 receptors. AVP also regulates arterial blood pressure and renal blood flow through vasoconstriction induced by V1a receptors activation (Largeau et al., 2019). SIADH, where hypersecretion of AVP occurs without osmotic stimulus, is characterized by hypotonic hyponatremia. The use of anti-VEGF therapy for 6 days in mice significantly increased the density of AVP-immunoreactive axonal terminals that were away from the vasculature (Furube et al., 2014). In addition, a 6-week treatment by anti-VEGF increased serum copeptin, a stable precursor of AVP, in a cohort of patients with metastatic colorectal cancer (Hagman et al., 2017). Given the fact that AVP is known to induce VEGF secretion (Tahara et al., 2011), anti-VEGF therapy induced AVP secretion could be considered as a positive feedback loop (Figure 3). This control loop could explain the safety profile of anti-VEGF drugs (i.e., pre-eclampsia like syndrome with kidney failure and high blood pressure) with renal dysfunction/proteinuria and hypertension induced by the action of supraphysiologic concentration of AVP on V2 and V1a receptors, respectively (Largeau et al., 2019). These effects of anti-VEGF therapy on AVP axis could also explain the very high prevalence of hyponatremia with antiangiogenic PKI (32% with pazopanib (Berardi et al., 2016)) probably by SIADH mechanism, as in our case.
Figure 3.
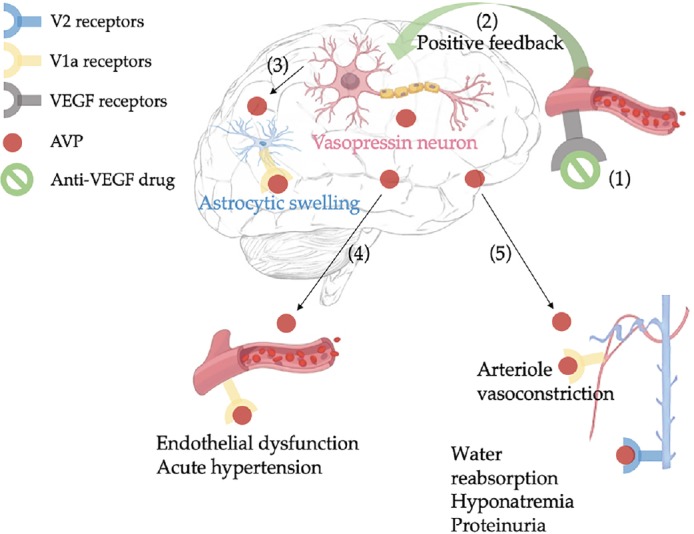
Possible mechanism involved in anti-VEGF therapy-induced posterior reversible encephalopathy syndrome.
Anti-VEGF therapy (1) leads to vasopressin neurons stimulation through a positive feedback loop (2); AVP release or direct V1a receptors activation leads to constriction of cerebral vessels and increased sympathetic tone, causing both endothelial dysfunction and cerebral ischemia; combination of these effects promotes dysregulation of ionic/water transglial and subsequent brain edema (3). In the periphery, AVP can induce endothelial dysfunction and acute hypertension (4); stimulation of V1a and V2 receptors leads to acute kidney failure and dilutional hyponatremia (5). AVP: Arginine vasopressin; VEGF: vascular epithelial growth factor.
AVP as a possible trigger of anti-VEGF therapy induced PRES: PRES has been largely reported with the use of anti-VEGF agents (Shah, 2017). The mechanism by which antiangiogenics drugs lead to PRES remains elusive, although it is suggested that they induce endothelial dysfunction and high blood pressure. These effects could promote cerebrovascular autoregulation breakdown, leading to blood-brain barrier disruption and subsequent brain oedema. Endothelial dysfunction, defined as impaired vasodilatation phenotype and proinflammatory state of the endothelium, is an on-target effect of anti-VEGF drugs. The vasoconstrictive response to VEGF inhibitors is related to both reduced levels of the vasodilator nitric oxide and increase of vasoactive peptides (e.g., endothelin and AVP) (Hagman et al., 2017; Touyz et al., 2017). Nevertheless, this endothelial/hypertensive theory is challenged by the absence of hypertension in a substantial proportion of patients with PRES (Largeau et al., 2019), including anti-VEGF therapy-induced PRES (Shah, 2017).
A recent review highlighted that AVP overstimulation seems to be involved in PRES development and subsequent symptoms, in particular because of both its pathophysiologic role in brain oedema formation and its involvement in most of PRES aetiologies (Largeau et al., 2019). AVP hypersecretion, known to up-regulate sodium–proton exchangers, Na+-K+-Cl– cotransporters and aquaporin 4, could be the trigger of PRES brain oedema through a dysregulation of ionic/water transglial flux induced by astrocytic ion channels dysfunction (i.e., astrocytic swelling due to the increase of sodium, chloride and water glial influx) (Largeau et al., 2019). In the periphery, AVP receptors stimulation could be responsible of symptoms usually reported in PRES such as acute hypertension (75–80%) and impaired renal function (55%) (Largeau et al., 2019) (Figure 3). Interestingly, in our case, acute kidney failure occurred at the nadir of hyponatremia and blood pressure normalization occurred simultaneously with natremia normalization, supporting the central pathophysiological role of AVP in PRES symptoms. In 6 cases of pazopanib-induced PRES, duration from starting pazopanib to onset of PRES ranged from 9 days to 2 months (Deguchi et al., 2018). Interestingly, hyponatremia (Deguchi et al., 2018) and acute kidney failure (Asaithambi et al., 2013; Miaris et al., 2017) have also been described in these cases.
The fact that pazopanib can be reintroduced without recurrence (Deguchi et al., 2018) suggests that other parameters, exogenous or endogenous, must be present to cause an increase in AVP beyond the threshold of PRES development. Another hypothesis, that cannot be ruled out, is that this adverse event is concentration-dependent, which would explain the absence of recurrence at reduced dosage.
Potential relationship between PRES and SIADH: Taken together, overstimulation of the AVP axis occurs in SIADH and probably in PRES, suggesting a close connection between these two syndromes. PRES may be caused by the convergence of various processes involved in AVP release (e.g., underlying disease, drugs, nausea) associated with risk factors for endothelial dysfunction and high blood pressure. Therefore, AVP increase can stimulate its effectors both through central receptors (i.e., V1 receptors) and peripheral ones (i.e., V1 and V2 receptors). First, cerebrovascular stimulation of V1a receptors induces ionic/water transglial flux disruption through astrocytic ion channels dysfunction, leading to the brain edema of PRES. Second, elevated AVP levels can activate vascular V1a receptors and renal V2 receptors, leading to hypertension and dilutional hyponatremia, respectively (Figure 3). Schematically, central activation of V1 receptors appears to be involved in the genesis of brain edema, while peripheral V1 and V2 receptors are more likely to be responsible for PRES symptoms. The predominant role of V1 receptors compared to V2 receptors would explain why not all occurrences of PRES are complicated by SIADH (Largeau et al., 2019).
Conclusion: In pazopanib-induced hyponatremia, a SIADH mechanism should be considered. AVP could be the trigger of pazopanib-induced PRES. Concurrent SIADH in drug-induced PRES should be considered as a symptom. If this AVP theory is confirmed, a promising therapeutic approach would be to prevent the action of AVP on its effectors in PRES patients. Suppression of AVP hypersecretion with corticosteroids (potent inhibitors of central AVP release) and/or its pharmacologic effects by antagonizing AVP receptors with conivaptan (a dual V1a and V2 receptors antagonist) (Largeau et al., 2019), may deserve to be evaluated in the management of patients with PRES.
Footnotes
Conflicts of interest: The authors declare no conflict of interest.
Financial support: No commercial organizations had any role in the completion or publication of this study. This article was completed without any external funding.
Declaration of patient consent: The authors certify that they have obtained the appropriate patient consent forms. In the form the patient has given her consent for her images and other clinical information to be reported in the journal.
Reporting statement: This manuscript was prepared in accordance with the CAse REport (CARE) guidelines.
Biostatistics statement: No statistical method was used in this study.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Li CH; T-Editor: Jia Y
References
- 1.Asaithambi G, Peters BR, Hurliman E, Moran BP, Khan AS, Taylor RA. Posterior reversible encephalopathy syndrome induced by pazopanib for renal cell carcinoma. J Clin Pharm Ther. 2013;38:175–176. doi: 10.1111/jcpt.12031. [DOI] [PubMed] [Google Scholar]
- 2.Berardi R, Santoni M, Rinaldi S, Nunzi E, Smerilli A, Caramanti M, Morgese F, Torniai M, Savini A, Fiordoliva I, Onofri A, Pistelli M, Taccaliti A, Cascinu S. Risk of hyponatraemia in cancer patients treated with targeted therapies: a systematic review and meta-analysis of clinical trials. PLoS One. 2016;11:e0152079. doi: 10.1371/journal.pone.0152079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deguchi S, Mitsuya K, Nakasu Y, Hayashi N, Katagiri H, Murata H, Wasa J, Takahashi M, Endo M. Posterior reversible encephalopathy syndrome (PRES) induced by pazopanib, a multi-targeting tyrosine kinase inhibitor, in a patient with soft-tissue sarcoma: case report and review of the literature. Invest New Drugs. 2018;36:346–349. doi: 10.1007/s10637-017-0521-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–2072. doi: 10.1056/NEJMcp066837. [DOI] [PubMed] [Google Scholar]
- 5.Furube E, Mannari T, Morita S, Nishikawa K, Yoshida A, Itoh M, Miyata S. VEGF-dependent and PDGF-dependent dynamic neurovascular reconstruction in the neurohypophysis of adult mice. J Endocrinol. 2014;222:161–179. doi: 10.1530/JOE-14-0075. [DOI] [PubMed] [Google Scholar]
- 6.Hagman H, Bendahl PO, Melander O, Sundberg J, Johnsson A, Belting M. Vasoactive peptides associate with treatment outcome ofbevacizumab-containing therapy in metastatic colorectal cancer. Acta Oncol. 2017;56:653–660. doi: 10.1080/0284186X.2017.1302098. [DOI] [PubMed] [Google Scholar]
- 7.Largeau B, Le Tilly O, Sautenet B, Salmon Gandonnière C, Barin-Le Guellec C, Ehrmann S. Arginine vasopressin and posterior reversible encephalopathy syndrome pathophysiology: the missing link? Mol Neurobiol. 2019;56:6792–6806. doi: 10.1007/s12035-019-1553-y. [DOI] [PubMed] [Google Scholar]
- 8.Miaris N, Maltezou M, Papaxoinis G, Visvikis A, Samantas E. Posterior reversible encephalopathy syndrome with concurrent nephrotic syndrome in a patient treated with pazopanib for metastatic renal cell carcinoma: case report and review of the literature. Clin Genitourin Cancer. 2017;15:e99–103. doi: 10.1016/j.clgc.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Riley DS, Barber MS, Kienle GS, Aronson JK, von Schoen-Angerer T, Tugwell P, Kiene H, Helfand M, Altman DG, Sox H, Werthmann PG, Moher D, Rison RA, Shamseer L, Koch CA, Sun GH, Hanaway P, Sudak NL, Kaszkin-Bettag M, Carpenter JE, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol. 2017;89:218–235. doi: 10.1016/j.jclinepi.2017.04.026. [DOI] [PubMed] [Google Scholar]
- 10.Shah RR. Anti-angiogenic tyrosine kinase inhibitors and reversible posterior leukoencephalopathy syndrome: could hypomagnesaemia be the trigger? Drug Saf. 2017;40:373–386. doi: 10.1007/s40264-017-0508-3. [DOI] [PubMed] [Google Scholar]
- 11.Tahara A, Tsukada J, Tomura Y, Yatsu T, Shibasaki M. Vasopressin induces human mesangial cell growth via induction of vascular endothelial growth factor secretion. Neuropeptides. 2011;45:105–111. doi: 10.1016/j.npep.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Touyz RM, Lang NN, Herrmann J, van den Meiracker AH, Danser AHJ. Recent advances in hypertension and cardiovascular toxicities with vascular endothelial growth factor inhibition. Hypertension. 2017;70:220–226. doi: 10.1161/HYPERTENSIONAHA.117.08856. [DOI] [PMC free article] [PubMed] [Google Scholar]